

Abstract
Multidrug-resistant Enterobacteriaceae are emerging as a serious infectious disease challenge. These strains can accumulate many antibiotic resistance genes though horizontal transfer of genetic elements, those for β-lactamases being of particular concern. Some β-lactamases are active on a broad spectrum of β-lactams including the last-resort carbapenems. The gene for the broad-spectrum and carbapenem-active metallo-β-lactamase NDM-1 is rapidly spreading. We present the complete genome of Klebsiella pneumoniae ATCC BAA-2146, the first U.S. isolate found to encode NDM-1, and describe its repertoire of antibiotic-resistance genes and mutations, including genes for eight β-lactamases and 15 additional antibiotic-resistance enzymes. To elucidate the evolution of this rich repertoire, the mobile elements of the genome were characterized, including four plasmids with varying degrees of conservation and mosaicism and eleven chromosomal genomic islands. One island was identified by a novel phylogenomic approach, that further indicated the cps-lps polysaccharide synthesis locus, where operon translocation and fusion was noted. Unique plasmid segments and mosaic junctions were identified. Plasmid-borne bla CTX-M-15 was transposed recently to the chromosome by ISEcp1. None of the eleven full copies of IS26, the most frequent IS element in the genome, had the expected 8-bp direct repeat of the integration target sequence, suggesting that each copy underwent homologous recombination subsequent to its last transposition event. Comparative analysis likewise indicates IS26 as a frequent recombinational junction between plasmid ancestors, and also indicates a resolvase site. In one novel use of high-throughput sequencing, homologously recombinant subpopulations of the bacterial culture were detected. In a second novel use, circular transposition intermediates were detected for the novel insertion sequence ISKpn21 of the ISNCY family, suggesting that it uses the two-step transposition mechanism of IS3. Robust genome-based phylogeny showed that a unified Klebsiella cluster contains Enterobacter aerogenes and Raoultella, suggesting the latter genus should be abandoned.
Introduction
Carbapenems are one of few antimicrobials that have been effective against multidrug-resistant bacteria, but their utility is threatened by the emergence of carbapenem-resistant Enterobacteriaceae (CRE). Klebsiella pneumoniae is the most common CRE species in the United States, typically encountered as a hospital-acquired infection with high morbidity and mortality, and resistant to nearly all available antibiotics [1]–[4]. Enzymes that inactivate carbapenems are a major mechanism of resistance. The serine β-lactamase KPC, known since 2001, has become the most common carbapenemase in the U.S. and other countries [1]. A more recent concern is the carbapenem-active metallo-β-lactamase NDM-1, first identified in a K. pneumoniae isolate from 2008 [5]. Alarmingly, bla NDM-1 is often found on large conjugative plasmids along with additional antibiotic resistance determinants [6]. In some settings the gene region can form tandem repeats, elevating copy number [7]. The recent spread of bla NDM-1 both among different species and across a large geographic area has been remarkable and well documented [5]–[11].
Non-carbapenemase mechanisms of carbapenem resistance are also known. These include increasing efflux pump activity [10] and altering the profile of outer membrane porins that control access of carbapenems to the cell wall [12], [13].
K. pneumoniae strain ATCC BAA-2146 (Kpn2146) was the first U.S isolate found to encode NDM-1 together with a wide variety of additional antibiotic resistance determinants [14]. Susceptibility testing performed at ATCC found Kpn2146 to be resistant to every one of the 34 antimicrobial and antimicrobial/inhibitor combinations tested. While Kpn2146 resistance genes have been analyzed by both microarray [15] and (incomplete) genome sequencing [16], [17], neither approach fully elucidated the complex Kpn2146 antibiotic resistance gene repertoire. For example some Kpn2146 antibiotic resistance genes were unrecognized in the previous work, and duplicated genes were counted only once by microarray and on one contig in the incomplete genome. Even when an incomplete genome does deliver the complete gene list, the question of how a pathogen accumulates such large collections of resistance genes requires the contextual information that comes from completing the genome. The complete genome is required to reveal gene duplication events, to determine plasmid vs. chromosomal gene location, and to apply phylogenomic methods to understand the evolution of the genome. In this study, we present the completed Kpn2146 genome, identifying four plasmids, and enabling a detailed survey of its antibiotic-resistance determinants that fully explains its resistance profile. These determinants include 23 primarily plasmid-borne genes encoding antibiotic-resistance enzymes, eight of which are β-lactamase genes. It is crucial to understand how such richly endowed pathogens arise, which requires analysis of the mobile fraction of the genome. Accordingly, we surveyed genomic islands in the chromosome, mosaicism in the plasmids, and transposable elements throughout the genome.
Materials and Methods
DNA preparation and sequencing
Klebsiella pneumoniae ATCC BAA-2146 (Kpn2146) was isolated in 2010 from the urine of a U.S. hospital patient who had recently received medical care in India [14]. Genomic DNA was obtained from American Type Culture Collection (ATCC), and re-suspended in water. A previously described Illumina paired-end genomic sequence dataset from a single MiSeq run, after quality and primer sequence trimming, consisted of 3,023,757 read pairs, with reads averaging 88.3 bp [17]. A Pacific Biosciences sequence dataset (PacBio) was generated from 2 µg genomic DNA at the Yale Genome Sequencing Center, which performed the 5 kb template preparation and sequenced the library on two SMRT cells, yielding 88,073 direct reads and 1744 circular consensus sequences (size distribution: mean 2408; median 1948; range 50-18951; N50 3254 bp).
Genome assembly
As detailed in File S1, the above MiSeq and PacBio datasets were sufficient for unambiguously assembling the complex genome with no need for additional PCR-based finishing. Novel software available at http://bioinformatics.sandia.gov/software/index.html was useful for visualizing MiSeq coverage and assembly branch points in the more challenging regions (Fig. S1 in File S1).
Annotation
Protein-coding genes were initially identified and annotated using RAST [18], and RNA genes were annotated with careful attention to tRNA, tmRNA and rRNA genes; Rfam/Infernal [19] found 118 additional RNA genes and motifs that helped identify certain regulatory genes and sites, mobile elements, plasmid replication origins, and toxin/antitoxin systems. The Antimicrobial Resistance Database (ARDB) [20] was used to annotate antimicrobial resistance genes among the initially-called genes, testing that hits did not have better matches to other gene families; the more recently updated ResFinder [21] added only bla NDM-1 to this list of resistance genes. Explaining the Kpn2146 antibiotic resistance profile required the identification of additional genes not called by RAST. ISs were annotated using ISFinder [22]. Intact integrons were named according to INTEGRALL [23]. The chromosomal origin of replication oriC was identified according to [24] and PCR tests [25], [26] were adapted for in silico plasmid replicon-typing. Observations on a high-copy group II intron, insertion sequences, and the lack of a CRISPR system are presented in File S1.
Phylogenetic analysis
The Kpn2146 genome was used for phylogenetic analysis, along with the 182 other Klebsiella reference genomes that were available at NCBI on December 20, 2013, and with five additional genomes (Enterobacter cloacae SCF1, Yokenella regensburgei ATCC 43003, Raoultella ornithinolytica B6 and two Enterobacter aerogenes genomes) included because they were originally placed in Klebsiella, or because a phylogenetic tree at PATRIC [27] showed that they are the closest available outgroup or fall within the Klebsiella clade. Multilocus sequence typing (MLST) was performed using K. pneumoniae data from http://www.pasteur.fr/mlst. Preliminary results showed that the 84 genomes of sequence type (ST) 258 formed a large tight clade together with the single ST512 genome; the five most divergent members of this clade were retained while the other 80 genomes of this clade were excluded from further analysis. The 108 remaining genomes were aligned into 234,232 DNA sequence blocks using default Mugsy v1.2.3 [28]. Blocks representing all ingroup genomes were selected and processed using Gblocks v0.91b [29] with the b5 = h option to remove ambiguously aligned regions, leaving 3476 blocks with a total of 2,118,733 aligned positions averaging 99.3% occupancy, which were concatenated into a supermatrix. A maximum likelihood tree was produced with RAxML v7.2.8 [30] using the GTRGAMMA substitution model. Node support values were from a bootstrap set of 150 trees produced similarly, using the fast (-x) bootstrapping function and autoFC bootstopping.
Genomic islands
Three methods were used to find chromosomal genomic islands. i) Islander identified att sites for islands integrated into a tRNA/tmRNA gene [31]. ii) PHAST identified regions enriched for phage genes [32]. We also developed iii) a novel phylogenomic method termed Learned Phyloblocks (http://bioinformatics.sandia.gov/software/index.html), in which the genome is divided into regions of shared evolutionary history termed “phyloblocks”, and those phyloblocks that are “learned”, on the basis of their enrichment among the training set of Islander and PHAST islands, are used to indicate additional islands. The chromosomes of Kpn2146 and the 11 other complete reference Enterobacter aerogenes and Klebsiella genomes were aligned using mugsy. This alignment determined the “phylotype” for each position on the Kpn2146 chromosome, i.e., the presence/absence pattern of the nucleotide among the reference genomes. This partitioned the Kpn2146 chromosome into phyloblock intervals defined as regions of uniform phylotype. Nonbiquitous phylotypes (those in which the sequence is not present in all 11 reference genomes) account for much (47.5%) of the Kpn2146 chromosome. This suggests that gene flux is high in Klebsiella, and not entirely explained by integrative genomic islands. We reasoned that some nonubiquitous phylotypes might be more indicative than others of horizontally transferred islands, if there are particularly common “highways” of island transfer among Klebsiella strains, as have been found in broader studies of horizontal gene transfer [33]. Phylotypes were ranked by the fraction of their nucleotides in the Islander and PHAST training islands. Phylotypes whose occurrence in training islands was >25% were termed “learned phyloblocks”, and accounted for 7.6% of the chromosome.
Phylotypes were analyzed with Mowgli [34], parsimoniously counting gain/loss events required to reconcile our robust genome tree (Fig. 1) with its subtree of only the phylotype taxa. This allowed us to classify nonubiquitous phylotypes as either simple (explainable by a single gain/loss event), or complex (requiring multiple gains/losses). The complex class was significantly overrepresented among the learned phylotypes (36 of 38) relative to the remaining phylotypes (183 of 246) (one-sided χ2 test of proportions: P<0.005). Only two learned phylotypes were simple: Kpn2146-only and Kpn2146/KpnHS11286-only.
Figure 1. Klebsiella phylogeny.

Tree for 108 genomes based on a 2.93-Mbp alignment, rooted at the midpoint of the outgroup (Ecl/Yre) branch. Nodes with <30% bootstrap support were combined forming the multifurcated dashed line; otherwise support values are shown only when <100%. Brackets: Kpn multilocus sequence type (ST). Inset: enlargement of the “core Kpn” phylogeny. Kpn2146 falls in a clade containing fellow ST11 strains Kpn JM45 and Kpn HS11286 and a tight clade (circled) of ST258 and ST512 strains. The ST258/ST512 clade is heavily sequenced, and represented here with only five of its most diverse members. Bold: complete genomes used for phyloblocks analysis. Species name abbreviations: Kpn, K. pneumoniae; Ksp, K. sp.; Kpl, K. cf. planticola; Kox, K. oxytoca; Kva, K. variicola; Eae, Enterobacter aerogenes; Ecl, E. cloacae; Ror, Raoultella ornithinolytica; Yre, Yokanella regensburgei.
PCR-based analysis of ISKpn21
While abundant sequence data mapped one ISKpn21 copy to the chromosome and a second to pKpn2146c, less abundant sequence data suggested additional copies either in tandem repeat form or as free circles. PCR tests to distinguish these possibilities first re-examined each genomic locus. The chromosomal copy was amplified using primers Cf (CGGTC ATAGT GTTGA TGTGGG) and Cr (CATGT CTATT TGGTC AGAGA CGG), while the plasmid copy was amplified using Pf (GCTTC CATGA CTGGT TGCTG) and Pr (GATGC CAAGC CGGTA AAGTTC). Cross-copy PCRs (i.e., Pf/Cr and Cf/Pr) tested for artifacts. Other primers tested for circular ISKpn21: ISf (GCGGT TACAG GGCAT TTG) and ISr (GCTCT TTGAC CAGAC GATCC TG). PCR employed FailSafe enzyme mix in buffer E (Epicentre) and scheduled 2 min at 95°C, 25 cycles (15 s at 95°C, 30 s at 55°C, 3 min at 68°C), and 7 min at 68°C. Products were run on 1.2% agarose E-gels (Life Technologies).
Plasmid mosaicism
The four plasmid sequences were queried against the July 29, 2013 nt database using BLASTN in default mode (i.e., task “megablast”), hitting 899 complete natural plasmids. Each query and subject was self-concatenated (to avoid circular origin issues), and BLASTN was repeated, identifying regions unique to each plasmid. To define unique mosaic junctions, each query hit boundary was tested for other hits spanning the boundary (beyond 10-bp tolerance windows).
Accession
Raw MiSeq and PacBio reads were deposited at SRA (accessions SRR931757 and SRR1185120, respectively). Genomic sequences have GenBank accessions CP006659-CP006663 and can also be browsed at http://bioinformatics.sandia.gov/klebs/.
Results and Discussion
Genome assembly using combined MiSeq and PacBio reads
We sequenced the genome of Klebsiella pneumoniae strain ATCC BAA-2146 (Kpn2146), the first U.S. isolate found to encode the NDM-1 metallo-β-lactamase. Assembly with an Illumina dataset alone was limited by poor coverage in GC-rich regions and by ambiguity at long repeats (Table S1 in File S1). However, adding a dataset of long but low accuracy PacBio reads, together with custom software for visualizing Illumina reads (Fig. S1 in File S1), allowed unambiguous assembly into five circular replicons: a chromosome and four plasmids (Table 1).
Table 1. Replicon copy numbers.
| Replicon | Accession | Incompatibility/origin of replic. | Length (bp) | GC (%) | No. unique 21-mersa | Rel. mean coverageb | Std.Dev. |
| Chromosome | CP006659 | oriC | 5435369 | 57.29 | 5328730 | 1.00 | 0.63 |
| pNDM-US | CP006661 | IncA/C | 140825 | 51.92 | 135388 | 1.65 | 0.67 |
| pKpn2146c | CP006663 | IncFIIA,IncFIB | 117755 | 51.21 | 111911 | 1.77 | 0.93 |
| pKpn2146b | CP006662 | IncFIA,IncR | 85164 | 52.74 | 67233 | 2.00 | 1.08 |
| pKpn2146a | CP006660 | ColE1 | 2014 | 49.60 | 1994 | 19.32 | 4.28 |
Unique 21-mers were identified for each replicon, and counted among MiSeq reads.
Mean 21-mer coverages were very similar to median values, and were normalized to that of the chromosome (which had 68.8 occurrences per 21-mer).
Antibiotic resistance determinants
ATCC has reported resistance of Kpn2146 to each of the 34 antimicrobial and antimicrobial/inhibitor combinations tested, including tests for 23 β-lactams (penicillins with or without inhibitors, cephalosporins, carbapenems and aztreonam), five fluoroquinolones, three aminoglycosides (tobramycin, amikacin and gentamicin), and four others (tetracycline, tigecycline, nitrofurantoin, and trimethoprim/sulfamethoxazole); see http://www.atcc.org/~/media/BA6C8F7C7C4C4649B2AEF501E51D76B8.ashx for the full list. Kpn2146 resistance genes have also been surveyed with a combination of microarray and amplicon sequencing [15]. The genome sequence fully rationalized the resistance profile, with ample evidence for one or more mechanisms explaining each observed antibiotic-resistance, and supported the gene survey. It further identified previously untested genes (like qnrB9), allelic multiplicity (aac(6′)-Ib, sul1, bla SHV-11 and bla CTX-M-15) and location (plasmid vs. chromosome), as well as housekeeping gene mutations (Table 2). These gene duplications can increase resistance; duplication of bla SHV-11 has been shown to increase amoxicillin-resistance 16-fold [35].
Table 2. Enzymes, efflux pumps, and mutations expected to confer resistance to antibiotics of clinical relevancea.
| Enzymeb | Gene location(s) | Coordinates | Resistance phenotype |
| NDM-1 (class B) | pNDM-US Tn125 | 122191–123003 | Penicillins, cephalosporins, carbapenems, inhibitor-resistant |
| SHV-11 (class A)c | 1. pKpn2146b | 36313–37173 | Penicillins, some cephalosporins, inhibitor-sensitive |
| 2. Chromosome | 2612996–2613856 | ||
| CTX-M-15 (class A) | 1. pKpn2146b ISEcp1 | 47130–48005 | Penicillins, some cephalosporins, aztreonam, |
| 2. Chromosome ISEcp1 | 5407530–5408405 | inhibitor-sensitive | |
| TEM-1 (class A) | pKpn2146b Tn2 | 50827–51687 | Penicillins, some cephalosporins, inhibitor-sensitive |
| CMY-6 (class C) | pNDM-US ISEcp1 | 72203–73348 | Penicillins, some cephalosporins, inhibitor-resistant |
| OXA-1 (class D) | pKpn2146b ΔIn37 | 38798–39673 | Penicillins, inhibitor-resistant |
| AAC(3)-IIe | pKpn2146b | 41116–41976 | Gentamicin, tobramycin, netilmicin, sisomicin |
| AAC(6′)-Ib (43) | pNDM-US In46 | 115114–115737 | Tobramycin, amikacin, netilmicin, sisomicin |
| AAC(6′)-Ib (1) | pKpn2146b ΔInTn1331 | 82745–83350 | Tobramycin, amikacin, netilmicin, sisomicin |
| AAC(6′)-Ib-cr (29) | pKpn2146b ΔIn37 | 38113–38712 | Tobramycin, amikacin, netilmicin, sisomicin, quinolones (low-level) |
| ANT(3″)-Ia | Kpn23SapB In127 | 2297711–2298502 | Streptomycin, spectinomycin |
| APH(3″)-Ib (StrA) | pKpn2146b ISCR2 | 53244–54047 | Streptomycin |
| APH(6)-Id (StrB) | pKpn2146b ISCR2 | 52408–53238 | Streptomycin |
| Sul2 | pKpn2146b ISCR2 | 54108–54923 | Sulfonamides |
| RmtC | pNDM-US ISEcp1 | 120100–120945 | Aminoglycosides (via rRNA modification) |
| Sul1 | 1. Kpn23SapB In127 | 2299007–2299846 | Sulfonamides |
| 2. pNDM-US In46 | 116245–117084 | ||
| DfrA14 | pKpn2146b In191 | 8281–8754 | Trimethoprim |
| QnrB9 | pKpn2146b | 26074–26742 | Quinolones, fluoroquinolones |
| Mph(A) | pKpn2146c | 16503–17408 | Macrolides, Erythromycin |
| FosA | Chromosome | 667960–668379 | Fosfomycin |
| Efflux pump | Gene Location | Probable substrate(s)d | |
| AcrAB-TolC | Chromosome | 1249681–1254043 | Aminoglycosides, β-lactams, tigecycline, macrolides |
| AcrEF-TolC | Chromosome | 4936203–4940465 | Minor role |
| EefABC | Chromosome | 5354323–5329922 | Chloramphenicol, tetracyclines, ciprofloxacin |
| MacAB-TolC | Chromosome | 1857393–1860445 | Macrolides |
| MdfA | Chromosome | 1781588–1782820 | Aminoglycosides, fluoroquinolones, chloramphenicol |
| MdtG,H,K,L,M,NOP | Chromosome | e | Many possible substrates (MFS superfamily pumps) |
| OqxAB | Chromosome | 4169609–4173960 | Chloramphenicol, fluoroquinolones, trimethoprim |
| EmrAB | Chromosome | 4218886–4221612 | Nalidixic acid, hydrophobic compounds |
| TetA(A) | pKpn2146c Tn1721 | 19168–20367 | Tetracyclines |
| Gene | Mutation | Resistance phenotype | |
| gyrA Gyrase | Ser83TTC → IleATC | 3763583–3766216 | Quinolone, fluoroquinolones |
| parC Topo IV | Ser80AGC → IleATC | 4689294–4691552 | Quinolone, fluoroquinolones |
| nfsA Nitroreductase | Frameshift | 1826275–1826998 | Nitrofurantoin |
Excluding the resistance enzyme for bleomycin, an antibiotic used clinically only as an antitumor agent.
Two silent differences between the two copies.
Probable efflux substrates identified from literature sources including ARDB; the substrates list is not comprehensive and in many cases has been deduced from organisms other than K. pneumoniae.
Mdt genes are scattered over the chromosome.
Eight genes for β-lactamases representing all four Ambler classes were identified; together these explain the broad β-lactam and inhibitor resistance of Kpn2146. We further identified specific resistance genes for tetracycline, trimethoprim, sulfonamides, macrolides, and multiple aminoglycoside resistance genes [36], including three aac(6′)-Ib variants, one shown to confer additional low-level resistance to quinolones [37] in addition to the usual spectrum of aminoglycosides inactivated by AAC(6′)-Ib which includes tobramycin, amikacin, and gentamicin C1a and C2.
The complete genome also reveals certain housekeeping gene mutations that are related to drug resistances. Its GyrA Ser83>Ile and ParC Ser80>Ile combination has previously been found in K. pneumoniae isolates with high-level resistance to several fluoroquinolones [38]. QnrB9 of Kpn2146, like other plasmid-encoded quinolone resistance enzymes, confers low-level resistance to fluoroquinolones, and may facilitate selection of mutations in gyrA and parC associated with high-level resistance [39]–[42]. A frameshift mutation in the nitroreductase gene nfsA is likely responsible for the observed resistance to nitrofurantoin [43].
The above observations explain the entire known resistance profile, except the tigecycline resistance. Mechanisms previously suggested for tigecycline resistance are mutations in the gene for the ribosomal protein S10 (Kpn2146 has the wild type allele) and mutations increasing the expression of the AcrAB/TolC efflux system [44], [45]. One mutation class causing overexpression of this efflux system is inactivation of its repressor RamR; Kpn2146 has such a ramR disruption (insertion of ISKpn18) that can thereby explain the observed tigecycline resistance. Additional efflux systems (Table 2), such as the macrolide-specific efflux system MacAB/TolC [45], may contribute to the intrinsic spectrum of resistance, especially if overexpressed.
We also detected an early nonsense mutation that disrupts the porin gene ompK35, fitting with many ESBL-producing K. pneumoniae strains that lack OmpK35 [12]. We do not however observe the concomitant loss of OmpK36 that significantly decreases susceptibility for meropenem and several cephalosporin β-lactams; ompK36 and ompK37 appear to be intact [46], [47]. In a recently reported Klebsiella carbapenem resistance mode, the marR regulatory gene is inactivated and the yedS porin gene is active [13]; this mode is unlikely to pertain here since marR is intact and yedS is lacking in Kpn2146.
Class 1 integrons and integron fragments
One third of the antibiotic resistance enzyme genes listed in Table 2, including all three of the aac(6′)-Ib alleles, are associated with five scattered class 1 integrons or integron fragments (Fig. S2 in File S1). Four of these are on plasmids, often within recognizable fragments of transposons, and the fifth is within a genomic island on the chromosome. We discuss below a case of cassette swapping where comparative analysis suggests the swap may have been mediated by homologous recombination rather than class 1 integron integrase action.
Plasmid overview
Plasmid copy numbers were measured relative to the chromosome from the MiSeq reads, taking unique 21-mers; extremely small pKpn2146a was high-copy, while pKpn2146b, pKpn2146c and the bla NDM-1 plasmid, pNDM-US, were large and low-copy (Table 1). The large plasmids carry most of the antibiotic resistance enzyme genes in the genome (Table 2). Some mobile genes with currently unknown function may eventually prove to be new virulence or resistance genes; hypothetical genes are enriched in the two largest plasmids relative to the total genome (Table S2 in File S1).
Conserved bla NDM-1 plasmid pNDM-US
The pNDM-US plasmid carrying bla NDM-1 (Fig. 2) was replicon-typed as IncA/C; it bears the IncA/C rep gene and iteron region, and encodes a ParAB partitioning system. Moreover, it encodes the complete set of proteins (TraABCDEFGHIKLNUVW, TrhF, DsbC, s043, s063, 123, 234, and 345) for the F-type conjugation pilus/Type IV secretion system, of the MOBH12 mobility class [47].
Figure 2. pNDM-US.

Key, color coding of genes, mobile elements, and unique regions and juxtapositions, with additional colors for non-gene features. Inner ring, representative long matches to other plasmids. abR, antibiotic resistance.
pNDM-US (140.8 kbp) is highly similar to numerous recently-sequenced plasmids, yet unique in bearing a copy of the relatively rare IS3000 between ter and krfA. Recent insertion of IS3000 is further supported by its 5-bp direct repeat of target sequence (DR), the first clear measurement of its DR length, in agreement with its membership in the Tn3 family [48]. We describe the rather few differences, each discernable as distinct DNA mobility events, between pNDM-US and its two closest known relatives: pNDM-KN (JN157804: 162.7 kbp) [49] and pNDM102337 (JF714412: 166.0 kbp), which each in total share 137 kbp at >99.98% identity with pNDM-US. pNDM-KN has three large segments missing in pNDM-US: i) an ISEc23 insert, ii) a Tn7/restriction system segment, and iii) a 4-cassette integron in place of the single (aac(6′)-Ib) cassette integron. The second reference plasmid pNDM102337 has i) the same 1-cassette integron as pNDM-US, ii) the Tn7/restriction system segment of pNDM-KN and iii) bears a segment missing from both pNDM-US and pNDM-KN that carries additional resistance determinants and a full length ISAba125 [50].
The integron in pNDM-KN and pNDM102337 is in a fragment of Tn1696 that has IS4321 inserted in its remaining IR. The presence of different gene cassettes in pNDM-KN (In578), pNDM-US (In46), and other Tn1696 variants might suggest recent integrase activity at this integron. However an alternative explanation for integron cassette swapping is by double homologous recombination in the long cassette-flanking regions that are conserved in most integrons, namely, the upstream integrase gene (5′-CS, 1352 bp) and the downstream ΔqacE-sul1-orf5 unit (3′-CS, 1616 bp) [51]. This latter suggestion is supported by the presence of three of the very few point mutational differences between pNDM-US and pNDM-KN near the att sites in these two flanks. In the 136,910 bp shared between pNDM-US and pNDM-KN there are ten sites of small-scale indel or base-substitution; three of these are in the 5′-CS and 3′-CS, for an enrichment of (3/2968)/(7/133942) = 19.3 fold.
ISEcp1 has transposed into pNDM-US, bringing its 2832-bp flanking segment bearing bla CMY-6, and has been inserted intergenically into the transfer operon tra. The pNDM-US bla NDM-1 region is found as in pNDM-KN and in many other Klebsiella plasmids; its interpretation as an immobile derivative of the mobile Tn125 of Acinetobacter baumannii strains has been discussed [52], [53]; here Tn125 is truncated at one end by ISKpn14 and within the ISCR21 unit at the other end.
Mosaic plasmid pKpn2146c
pKpn2146c (Fig. 3) was replicon-typed as both IncFIIA and IncFIB. It was typed to IncFIIA using the copA RNA gene and copB and rep protein genes, and to IncFIB through its IncFIB iteron region and rep gene. An iteron region IncD like that of the F plasmid was also identified.
Figure 3. pKpn2146c.

Key, color coding of genes, mobile elements, and unique regions and juxtapositions, with additional colors for non-gene features. Inner ring, representative long matches to other plasmids. Innermost black arrows, recent recombination events. HR, homologous recombination; abR, antibiotic resistance.
pKpn2146c is a large mosaic plasmid, which shares much of its sequence with the bla NDM-1 containing plasmid pKPX-1, including both the large copper/arsenic resistance region and the resistance gene mph(A) region. pKpn2146c is also enriched for hypothetical genes (Table S2 in File S1). Three of the eleven IS26 copies in the Kpn2146 genome occur in this plasmid (Table S3 in File S1). Directly adjacent to the mph(A) and IS26 region is a Tn1721 [54] fragment bearing the tetA(A) resistance gene. This transposition junction is unique among plasmids in public databases. The other end of ΔTn1721 is truncated by an IS26 insertion.
Highly mosaic plasmid pKpn2146b
pKpn2146b (Fig. 4) was replicon-typed as both IncFIA (iteron unit, oriS and rep gene) and IncR. It has a largely intact IncR repeat region located 34.5 kbp apart from a locus with the rep, parA and parB genes and parS site. pKpn2146b additionally has a region of the iteron from the IncN plasmid R46 which is repeated 30.6 times, but without the IncN rep gene. apparently lost through IS26 insertion followed by homologous recombination.
Figure 4. pKpn2146b.

Key, color coding of genes, mobile elements, and unique regions and juxtapositions, with additional colors for non-gene features. Inner ring, representative long matches to other plasmids. Innermost black arrows, recent recombination events. HR, homologous recombination; abR, antibiotic resistance.
pKpn2146b is the richest of the plasmids in resistance determinants (12 determinants; Table 2), and the most highly mosaic, with the highest number (six) of IS26 copies. Comparison with other plasmids shows evidence for an illegitimate recombination at the resolution site of the plasmid-encoded resolvase ResD (see Fig. 4 at coordinate 78900), where the IncR control region joins unusual sequence found elsewhere only in pK245 (DQ449578). Comparison also shows a particular pattern that we call “IS-flank switch”; one example is marked as “HR” near coordinate 38000 on Fig. 4, where homology to one reference (plasmid pRMH712) begins precisely at one end of a long repeated region (IS26) and extends through the IS and well into one flank, while the same pattern occurs for the other flank with a second reference (plasmid pKDO1). We hypothesize that this IS-flank switch pattern resulted from homologous recombination between IS26-containing parents as proposed previously [55]. This hypothesis of homologous recombination subsequent to two independent transposition events is supported by failure to find the 8-bp target sequence direct repeat (DR) expected for a recent transposition of IS26. In fact none of the six copies of IS26 in pKpn2146b, nor any of the other five copies elsewhere in the genome, contain the DR expected for recent insertion (Table S3 in File S1), suggesting that every IS26 copy in the genome has undergone homologous recombination more recently than transposition. We find another IS-flank switch pattern (“HR” at the top of Fig. 4), that we suspect provides an explanation of how the IncN iterons lost their associated IncN rep gene.
The bla SHV-11 gene originated in situ in the K. pneumoniae chromosome, and has been transferred to plasmids at least twice, in both cases as a chromosomal fragment flanked by directly repeated IS26 copies [56], [57]. The pKpn2146b copy of bla SHV-11 is like the prototype in plasmid pKPN4 (CP000649), except that one of the IS26 copies used to transmit this segment has been truncated by insertion of IS3000, which was then uniquely interrupted by ISEc22.
pKpn2146b has much of the bla TEM-1-containing Tn2 [58], (truncated by IS26 at one end as found in other plasmids [55]), and further disrupted by a bla CTX-M-15/ISEcp1 transposition unit [59]. This pKpn2146b ISEcp1 copy has spawned a recent tranposition event moving bla CTX-M-15 to a chromosomal site. Chromosomal bla CTX-M-15 has not been identified in any complete genome, but has been reported at an undetermined locus in a different multilocus sequence type [60]. This recent transposition event from the plasmid used a different right end for the transposing unit (1618 bp flank) than did the earlier insertion into the plasmid Tn2 (1315 bp flank); the resulting chromosomal copy has 100% identity with the plasmid parent and is flanked by a 5-bp DR. A partial ISCR2 (disrupted tnp and ori) is found with its frequently associated strA, strB and sul2 genes. The mercury-resistance operon-carrying ΔTn6187 is only one arm of the full-length Tn6187, but nonetheless has the same inverted repeats at both ends as the full-length, suggesting that it alone could be a transposing element; it however lacks the expected flanking direct repeats, and thereby conforms to the IS-flank switch pattern, suggesting that its flanks may have been shuffled by homologous recombination. The integron within Tn1331 (Fig. S2 in File S1) [61] is found truncated at one end by IS26, and at the other end by ISKpn14 leaving aac(6′)-Ib as its only intact resistance gene.
Mysterious plasmid pKpn2146a
pKpn2146a (Fig. S3 in File S1) was replicon-typed as ColE, encoding RNAs I and II. The ColE1 mobilization site (bom) was determined by comparison to other ColE1 plasmids. The typical short ColE1 proteins that affect ori (Rom protein) or bom-site (Mob proteins) function could not be identified; indeed, none of its potentially encoded proteins show homology to any proteins in public databases. The most closely related known plasmid pB1021 (NC_019989), from K. pneumoniae BB1090, shares the common RNAII region and uniquely shares a second large portion of pKpn2146a. This surprisingly short (2014 bp) plasmid was supported by MiSeq coverage and verified by PCR (data not shown).
Genomic islands determined by multiple approaches
Plasmids frequently disseminate antibiotic resistance genes in Klebsiella, but genomic islands are also potential vehicles. Our program Islander [31] found six islands in tRNA/tmRNA genes, including a tandem island pair at a tRNALeu gene. PHAST [32] confirmed three of these and identified four additional prophage-like islands, one precisely within the gene for the short regulatory RNA RybB. The 10 resulting islands accounted for 6.3% of the Kpn2146 chromosome. We used these 10 Islander/PHAST islands (Table 3) as a training set for a phylogenomic approach to find additional islands, based on the principle that islands tend to occur sporadically among closely related strains. The Kpn2146 chromosome was partitioned into “phyloblocks”, which we define as DNA intervals where all positions share the same phylotype, i.e., the same presence/absence profile among a given set of closely related genomes. We selected phyloblocks that were enriched in (i.e., “learned” from) the training islands. These learned phyloblocks pointed to the island Kpn23SapB, with an integrase gene and att site pair, that was missed by Islander and Phast. Learned phyloblocks also pointed to the non-island genomic locus cps-lps, described further below. An overview of learned phyloblocks across the chromosome (Fig. 5) shows the tight mapping to cps-lps, mobile islands and ISs.
Table 3. Genomic islands.
| Island | Chromosomal Coordinatesa | Length (bp) | att b | Kpn HSc | RNA gene target | Gene contentd | Additional supporte | Notef |
| Kpn21L | 583102–604583 | 21482 | Yes | No | tRNA-Leu | Restriction | Islander | Tandem with Kpn11L |
| Kpn11L | 604583–615770 | 11188 | Yes | Yesg | tRNA-Leu | Phage | Islander, PHAST | Damage; Tandem with Kpn21L; Enterobacteria phage P4 |
| Kpn49R | 1296810–1345944 | 49135 | Yes | Alth | tRNA-Arg | Phage, gpII intron | Islander, PHAST | Cronobacter phage ENT47670 |
| Kpn38RybB | 1823069–1785570 | 37500 | Yes | Yesi | RybB | Phage, gpII intron | PHAST | Enterobacteria phage Fels 2 |
| Kpn29S | 1966140–1994683 | 28544 | Yes | No | tRNA-Ser | Phage | Islander | |
| Kpn23SapB | 2286456–2309756 | 23301 | Yes | No | No | Phage, integron, gpII intron | ||
| Phast.2 | 2342484–2388101 | 45618 | No | Yesj | No | Phage, gpII intron | PHAST | Salmonella phage Fels 1 |
| Kpn40GuaA | 3969748–4010194 | 40447 | Yes | No | No | Phage, gpII intron | PHAST | Salmonella phage SPN1S |
| Kpn37X | 4129454–4166020 | 36567 | Yes | No | tmRNA | Phage | Islander, PHAST | Salmonella phage RE-2010; also called tmGI_Kp35 (JF764793) |
| Kpn55F | 4603724–4658623 | 54900 | Yes | Yesk | tRNA-Phe | T4SS, ParAB | Islander | Damage |
| Kpn16Fis | 4919120–4935114 | 15995 | Yes | Yesl | No | Phage | PHAST | Salmonella phage ST64B |
Islands integrated into RNA genes are assigned the orientation of the target gene; remaining islands are arbitrarily assigned the genomic orientation.
Putative att sites identified at each flank.
Status in the closely related K. pneumoniae HS11286 chromosome; No/Yes, absent/present in HS11286; Alt, alternative island in HS11286 at the Kpn2146 island site.
The PHAST islands are strongly phage-like; we note where other islands have proteins suggestive of phage function, and other features of interest.
All islands had phyloblocks support.
Damage denotes loss of an acceptor stem nucleotide in the tRNA gene fragment [31]; phage names are the closest relatives determined by PHAST.
CP003200.1/581777-594029; with IS903 inserted at position 4245 with 9-bp DR; directly at tRNA-Leu att site with no intervening Kpn21L.
50403-bp alternative island at CP003200.1/1288317-1338720 with same initial 1989 bp as Kpn49R.
CP003200.1/1778319-1808718; 2 mismatches; with 7201 bp deleted from peg.320 to 3′ end of gpII intron.
CP003200.1/2277469-2325549; with ISEc22 at position 15471 with 8-bp DR.
CP003200.1/4502805-4558770; with IS903B insert at position 9777 with 9-bp DR.
CP003200.1/4819193-4835187; no mismatches.
Figure 5. Learned phyloblocks identify a new island and the highly variable capsular polysaccharide and lipopolysaccharide synthesis gene cluster (cps-lps).
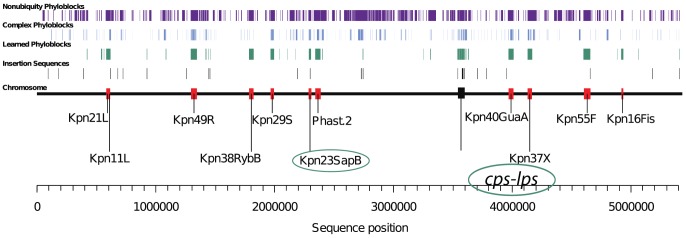
Nonubiquity phyloblocks: those missing in at least one of the 11 reference chromosomes. Complex phyloblocks: those requiring more than one gain/loss event to reconcile the phylotype with the genome tree of Fig. 1. As a percentage of their combined 411 kbp, the learned phyloblocks mapped either to the training islands (81.9%), the two newly indicated regions (12.0%), insertion sequences (2.1%), or to small scattered regions that did not show hallmarks of islands (4.0%). Red segments: the 11 final islands (including a tandem array of Kpn21L and Kpn11L). Circles, the two newly indicated regions.
To summarize, the 11 islands identified here (Table 3) amount to 365 kbp. Ten islands were precisely determined, having found an integrase gene and both attL and attR sites. Two islands had damage in the attR tRNA fragment, as has been previously observed [27]. Only five of these islands were found in the closely related strain K. pneumoniae HS11286.
The island Kpn23SapB has an In127 integron fragment containing an aadA2 cassette (Fig. S2 in File S1). An upstream IS26 insertion has displaced the integron Pc promoter, yet generated a new plausible promoter with the −35 TTGCA from IS26, a 17 bp spacer, and a −10 TTTCAT from the integron. This aadA2 is the only island-borne resistance determinant identified here. However, some mobile genes with currently unknown function may eventually prove to be new virulence or resistance genes; the islands are enriched in hypothetical genes (Table S3 in File S1). Considering non-hypothetical genes, nine islands primarily possess phage genes, while Kpn55F encodes plasmid-like ParAB and some type IV secretion system functions indicative of an integrative conjugative element (ICE). Islands contain five of the six chromosomal group II intron copies.
Operon fusion and translocation at the cps-lps polysaccharide synthesis locus
Learned phyloblocks indicated, in addition to a new island, the genomic locus of capsular polysaccharide (cps) and lipopolysaccharide (lps) synthesis genes (Fig. 6). This region is not an integrase-mobilized genomic island, yet the cps cluster is known to be so highly varied as to suggest horizontal transfer of genes within the array [62]. The capsule is the outermost cell surface, a key Klebsiella pathogenicity determinant subject to immune surveillance. In other Enterobacteriaceae, the large cps and lps gene clusters are typically separate, but in Klebsiella, lps is found immediately downstream of cps. Nevertheless there normally appears to be transcriptional separation between Klebsiella cps and lps; cps terminates with the reverse-oriented gene uge, and an lps promoter has been found in the large intergenic space between uge and lps (Fig. 6A) [62]. The Kpn2146 cps-lps region has undergone a major rearrangement with gene-regulatory consequences (Fig. 6B). The terminal cps P3 transcription unit is deleted from its usual site, fusing the lps operon to the main cps operon. Morever this cps P3 unit has translocated to a nearby location, within a complex array of insertion sequences. In this new location the P3 unit is transcriptionally isolated, whereas at the usual location transcription could be supplemented by the upstream P2. Deletion of a polysaccharide synthesis gene cluster by homologous recombination between repeated manCB units has been noted before [63], but in our case the translocation has preserved the deleted cps subcluster.
Figure 6. Operon translocation and fusion at the cps-lps polysaccharide synthesis locus.

The cps P1, P2 and P3 promoters are taken from [68], while a promoter (Plps) has been mapped in K. pneumoniae MGH 78578 to the intergenic space between uge and the first lps gene [69]. A) The cps-lps region of K. pneumoniae 342, which is typical of Klebsiella. Genes of cps are in yellow (common in most strains) or blue (varying in gene identity, count, and order); genes of lps are in red. The manCB unit (orange arrows) is occasionally found in cps, and occasionally in lps, and here unusually in both. The diamond represents the JUMPstart DNA/RNA motif at whose ops sequence RfaH is loaded onto the elongating RNA polymerase in place of NusG, preventing Rho-based termination for the small number of long transcription units that are controlled by ops-RfaH, and physically coupling the elongating RNA polymerase to the trailing ribosome [70]. B) Kpn2146 cps-lps. The boxed cps P3 unit has been deleted from its usual site, and moreover translocated to a nearby position, apparently by transposition and/or homologous recombination mechanisms; note the complex pattern of surrounding IS insertions and the directly repeated flanking sequence copies (gray arrows).ΔIS, incomplete IS copy; dotted lines, gene or IS interrupted by ISs; GT, glucosyl transferase, Hyp, hypothetical.
Circular transposition intermediates of ISKpn21
Above we demonstrated transposition of bla CTX-M-15 from a resident plasmid to the chromosome by sequence comparison. Another way to assess the potential of a transposon to disseminate antibiotic resistance genes is to identify active transposition intermediates. Such intermediates have previously been found in vivo as free molecules unintegrated into chromosomes or plasmids, in circular, linear or tandem repeat linear forms [64], in the two-step transposition mechanism used by elements of the IS3, IS30, IS21 and IS256 families. We present here a novel approach for detecting circular transposition intermediates, through high-throughput sequencing. Examining the termini of ISKpn21, we found MiSeq reads where ISKpn21 ends were linked, and separated by 5-bp direct repeat from one of the two integrated copies (Table S3 and Fig. S4 in File S1). Possible explanations for these sequences are i) that what we had assembled as single copies were instead tandem genomic repeats, or ii) that these are from circular molecules free from the genome. We tested the integrated ISKpn21 copies by PCR and found each to be present as a single unit, not as a tandem (Fig. S4 in File S1). We also tested for a genome-free circle (or possibly genome-free tandem) and observed the indicated PCR product. The copy number of each circle and each end of its integrated parent ISKpn21 copy was measured, yielding an average circle:parent ratio of 3.72%±0.84%, presuming no sequencing bias. The pKpn2146c copy of ISKpn21 has different direct repeat sequences at its two flanks, perhaps due to recombination between different ancestral copies. Finding only the left end direct repeat in its circle sequence suggests, without achieving statistical significance, that the left end of ISKpn21 preferentially attacks the right end during circularization. We propose that ISKpn21 and perhaps the entire ISNCY family use the two-step transposition mechanism of the IS3 family.
Using PacBio reads to detect homologously recombinant subpopulations
Above we used sequence comparison to demonstrate homologous recombination at high copy repeats as a mechanism for reassorting resistance determinants. Here we present a new method for measuring recombinant subpopulations in a bacterial culture. Small numbers of PacBio reads disagreed with the preponderant assembly pattern across the 8 copies of the rRNA operon and the 8 copies of a group II intron (Fig. S5 in File S1). To the extent that the PCR-free PacBio method is not expected or known to produce in vitro homologous recombination artifacts, our data indicated that approximately ∼4% of this bacterial culture was recombinant across these repeats.
Klebsiella phylogeny revises taxonomy
We expanded the phylogenetic analysis used in our learned phyloblocks analysis, to produce a robust genome-based phylogenetic analysis of Klebsiella (Fig. 1). This reveals a clade with Kpn2146 and fellow members of multi-locus sequence type (ST) 11, K. pneumoniae HS11286 and K. pneumoniae JM45, from which sprang a tight clade of heavily sequenced K. pneumoniae ST258 and ST512 hospital strains; Kpn2146 is the only bla NDM-1-containing member of this clade (or indeed our entire tree). The surrounding and subtending of Enterobacter aerogenes and Raoultella with Klebsiella taxa of long standing, with 100% bootstrap support, suggests that all should be subsumed under Klebsiella and that the genus Raoultella, defined based on analysis of only two genes [62], should be abandoned.
Conclusions
A single relatively small Illumina read set, combined with a PacBio set of longer but less accurate reads, was sufficient to assemble the genome despite the numerous repeat and high-GC regions, with no need for gap closure by PCR. Moreover we demonstrated direct detection of an active transposable element by high-throughput sequencing. Our novel read-visualization tools (http://bioinformatics.sandia.gov/software/index.html) were useful for working through problematic areas, and this software was developed into a greedy contig assembler.
The known extensive antibiotic-resistance profile of Klebsiella pneumoniae ATCC BAA-2146 (Kpn2146) was explained and additional resistances, which remain to be tested experimentally, were suggested by the genome sequence. Several mechanisms were identified for the mobility of resistance genes: i) acquisition of plasmids and genomic islands, ii) integron cassette swapping (whole or partial integrons account for eight antibiotic-resistance genes), iii) transposition events from chromosome to plasmid leading to greater disseminability of resistance, and vice versa leading to greater stability in the genome, and iv) homologous recombination at high copy repeats. Gaining more insight into such key evolutionary mechanisms, beyond simply identifying them, often comes through technological advances. Here we have made novel use of high-throughput sequencing technologies to inform both transposition and homologous recombination.
Numerous mobile genetic elements were identified. The eleven genomic islands were identified by three different methods that were based on the preference of islands for tRNA gene integration sites [31], clustering of phage genes [32], and a novel phylogenomic approach introducing phyloblocks, DNA segments with shared phylogenetic profiles that may be applicable in more general studies of horizontal gene transfer (Fig. 6). A recent study of the closely related ST258 K. pneumoniae, published while our manuscript was under review, also found numerous islands and indicated the cps locus as the major non-island chromosomal site of variation among strains [65].
The Kpn2146 genome illustrates the massive arsenal of antibiotic-resistance genes, and agile repertoire of mobile genetic elements, that the emerging CRE bacteria have at their disposal for adapting to new challenges. Homologous recombination at multicopy sequences [66], site-specific recombination by resolvases [67], switching of integron cassettes, and transpositions have shaped Klebsiella plasmid mosaicism.
Supporting Information
Supplementary materials: additional genomic features and supplementary tables, figures, and references.
(DOCX)
Acknowledgments
The authors thank Guilin Wang at Yale Genome Sequencing Center for assistance, Thomas Jové (U. Limoges) for supplying names for intact integrons, and Chris Whitfield (U. Guelph) for helping identify cps-lps genes.
Funding Statement
This work was funded by Sandia National Laboratories' Grand Challenge LDRD (Laboratory-Directed Research and Development, grant number 165683) program. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin company, for the U.S. Department of Energy's National Nuclear Security Administration under Contract DE-AC04-94AL85000. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gupta N, Limbago BM, Patel JB, Kallen AJ (2011) Carbapenem-resistant Enterobacteriaceae: epidemiology and prevention. Clin Infect Dis 53: 60–67. [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention (2013) Vital signs: carbapenem-resistant Enterobacteriaceae . MMWR Morb Mortal Wkly Rep 62: 165–170. [PMC free article] [PubMed] [Google Scholar]
- 3. Shon AS, Bajwa RP, Russo TA (2013) Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence 4: 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL (2012) Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 25: 682–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, et al. (2009) Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 53: 5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Z, Qlu S, Wang Y, Liu S, Wang Z, et al. (2011) Coexistence of bla NDM-1 with the prevalent bla OXA23 and bla IMP in pan-drug resistant Acinetobacter baumannii isolates in China. Clin Infect Dis 52: 692–693. [DOI] [PubMed] [Google Scholar]
- 7. Huang TW, Chen TL, Chen YT, Lauderdale TL, Liao TL, et al. (2013) Copy number change of the NDM-1 sequence in a multidrug-resistant Klebsiella pneumoniae clinical isolate. PLoS One 8: e62774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castanheira M, Deshpande LM, Mathai D, Bell JM, Jones RN, et al. (2011) Early dissemination of NDM-1- and OXA-181-producing Enterobacteriaceae in Indian hospitals: report from the SENTRY Antimicrobial Surveillance Program, 2006–2007. Antimicrob Agents Chemother 55: 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moellering R (2010) NDM-1—a cause for worldwide concern. N England J Med 363: 2377–2379. [DOI] [PubMed] [Google Scholar]
- 10. Pfeifer Y, Cullik A, Witte W (2010) Resistance to cephalosporins and carbapenems in Gram-negative bacterial pathogens. Int J Med Microbiol 300: 371–379. [DOI] [PubMed] [Google Scholar]
- 11. Walsh TR, Weeks J, Livermore DM, Toleman MA (2011) Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11: 355–362. [DOI] [PubMed] [Google Scholar]
- 12. Hernández-Allés S, Albertí S, Alvarez D, Doménech-Sánchez A, Martínez-Martínez L, et al. (1999) Porin expression in clinical isolates of Klebsiella pneumoniae . Microbiology 145 (Pt 3): 673–679. [DOI] [PubMed] [Google Scholar]
- 13. Warner DM, Yang Q, Duval V, Chen M, Xu Y, et al. (2013) Involvement of MarR and YedS in carbapenem resistance in a clinical isolate of Escherichia coli from China. Antimicrob Agents Chemother 57: 1935–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Centers for Disease Control and Prevention (2010) Detection of Enterobacteriaceae isolates carrying metallo-β-lactamase - United States, 2010. MMWR Morb Mortal Wkly Rep 59: 750. [PubMed] [Google Scholar]
- 15. Leski T, Vora GJ, Taitt CR (2012) Multidrug resistance determinants from NDM-1-producing Klebsiella pneumoniae in the USA. Int J Antimicrob Agents 40: 282–284. [DOI] [PubMed] [Google Scholar]
- 16. Broberg CA, Palacios M, Miller VL (2013) Whole-genome draft sequences of three multidrug-resistant Klebsiella pneumoniae strains available from the American Type Culture Collection. Genome Announc 1: e00312–00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim H, Jebrail MJ, Sinha A, Bent ZW, Solberg OD, et al. (2013) A microfluidic DNA library preparation platform for next-generation sequencing. PLoS One 8: e68988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, et al. (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41: D226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu B, Pop M (2009) ARDB—antibiotic resistance genes database. Nucleic Acids Res 37: D443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, et al. (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67: 2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M (2006) ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34: D32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moura A, Soares M, Pereira C, Leitão N, Henriques I, et al. (2009) INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25: 1096–1098. [DOI] [PubMed] [Google Scholar]
- 24. Cleary JM, Smith DW, Harding NE, Zyskind JW (1982) Primary structure of the chromosomal origins (oriC) of Enterobacter aerogenes and Klebsiella pneumoniae: comparisons and evolutionary relationships. J Bacteriol 150: 1467–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carattoli A (2013) Plasmids and the spread of resistance. Int J Med Microbiol 303: 298–304. [DOI] [PubMed] [Google Scholar]
- 26. Carattoli A, Bertini A, Villa L, Falbo V, Hopkins KL, et al. (2005) Identification of plasmids by PCR-based replicon typing. J Microbiol Methods 63: 219–228. [DOI] [PubMed] [Google Scholar]
- 27. Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, et al. (2011) PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun 79: 4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Angiuoli SV, Salzberg SL (2011) Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27: 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56: 564–577. [DOI] [PubMed] [Google Scholar]
- 30. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- 31. Mantri Y, Williams KP (2004) Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res 32: D55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39: W347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beiko RG, Harlow TJ, Ragan MA (2005) Highways of gene sharing in prokaryotes. Proc Natl Acad Sci USA 102: 14332–14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doyon J-P, Scornavacca C, Gorbunov KY, Szöllősi GJ, Ranwez V, et al.. (2011) An efficient algorithm for gene/species trees parsimonious reconciliation with losses, duplications and transfers. Comparative genomics: Springer. pp. 93–108.
- 35. Duvernay C, Coulange L, Dutilh B, Dubois V, Quentin C, et al. (2011) Duplication of the chromosomal bla SHV-11 gene in a clinical hypermutable strain of Klebsiella pneumoniae . Microbiology 157: 496–503. [DOI] [PubMed] [Google Scholar]
- 36. Ramirez MS, Tolmasky ME (2010) Aminoglycoside modifying enzymes. Drug Resist Updat 13: 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramirez MS, Nikolaidis N, Tolmasky ME (2013) Rise and dissemination of aminoglycoside resistance: the aac(6′)-Ib paradigm. Front Microbiol 4: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brisse S, Milatovic D, Fluit AC, Verhoef J, Martin N, et al. (1999) Comparative in vitro activities of ciprofloxacin, clinafloxacin, gatifloxacin, levofloxacin, moxifloxacin, and trovafloxacin against Klebsiella pneumoniae, Klebsiella oxytoca, Enterobacter cloacae, and Enterobacter aerogenes clinical isolates with alterations in GyrA and ParC proteins. Antimicrob Agents Chemother 43: 2051–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim HB, Park CH, Kim CJ, Kim E-C, Jacoby GA, et al. (2009) Prevalence of plasmid-mediated quinolone resistance determinants over a 9-year period. Antimicrob Agents Chemother 53: 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martínez-Martínez L, Pascual A, Jacoby GA (1998) Quinolone resistance from a transferable plasmid. Lancet 351: 797–799. [DOI] [PubMed] [Google Scholar]
- 41. Rodríguez-Martínez JM, Velasco C, García I, Cano ME, Martínez-Martínez L, et al. (2007) Mutant prevention concentrations of fluoroquinolones for Enterobacteriaceae expressing the plasmid-carried quinolone resistance determinant qnrA1 . Antimicrob Agents Chemother 51: 2236–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang H, Duan G, Zhu J, Zhang W, Xi Y, et al. (2013) Prevalence and characterisation of plasmid-mediated quinolone resistance and mutations in the gyrase and topoisomerase IV genes among Shigella isolates from Henan, China, between 2001 and 2008. Int J Antimicrob Agents 42: 173–177. [DOI] [PubMed] [Google Scholar]
- 43. Whiteway J, Koziarz P, Veall J, Sandhu N, Kumar P, et al. (1998) Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli . J Bacteriol 180: 5529–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Villa L, Feudi C, Fortini D, Garcia-Fernandez A, Carattoli A (2014) Genomics of KPC-producing Klebsiella pneumoniae sequence type 512 clone highlights the role of RamR and ribosomal S10 protein mutations in conferring tigecycline resistance. Antimicrob Agents Chemother 58: 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hentschke M, Wolters M, Sobottka I, Rohde H, Aepfelbacher M (2010) ramR mutations in clinical isolates of Klebsiella pneumoniae with reduced susceptibility to tigecycline. Antimicrob Agents Chemother 54: 2720–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Albertí S, Rodríquez-Quiñones F, Schirmer T, Rummel G, Tomás JM, et al. (1995) A porin from Klebsiella pneumoniae: sequence homology, three-dimensional model, and complement binding. Infect Immun 63: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcillán-Barcia MP, Francia MV, de la Cruz F (2009) The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol Rev 33: 657–687. [DOI] [PubMed] [Google Scholar]
- 48. Sabate M, Navarro F, Miro E, Campoy S, Mirelis B, et al. (2002) Novel complex sul1-type integron in Escherichia coli carrying bla CTX-M-9 . Antimicrob Agents Chemother 46: 2656–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carattoli A, Villa L, Poirel L, Bonnin RA, Nordmann P (2012) Evolution of IncA/C bla CMY-2-carrying plasmids by acquisition of the bla NDM-1 carbapenemase gene. Antimicrob Agents Chemother 56: 783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Partridge SR, Brown HJ, Hall RM (2002) Characterization and movement of the class 1 integron known as Tn2521 and Tn1405. Antimicrob Agents Chemother 46: 1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Partridge SR, Iredell JR (2012) Genetic contexts of blaNDM-1. Antimicrob Agents Chemother 56: : 6065–6067; author reply 6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poirel L, Bonnin RA, Boulanger A, Schrenzel J, Kaase M, et al. (2012) Tn125-related acquisition of bla NDM-like genes in Acinetobacter baumannii . Antimicrob Agents Chemother 56: 1087–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Toleman MA, Spencer J, Jones L, Walsh TR (2012) bla NDM-1 is a chimera likely constructed in Acinetobacter baumannii . Antimicrob Agents Chemother 56: 2773–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pasquali F, Kehrenberg C, Manfreda G, Schwarz S (2005) Physical linkage of Tn3 and part of Tn1721 in a tetracycline and ampicillin resistance plasmid from Salmonella Typhimurium. J Antimicrob Chemother 55: 562–565. [DOI] [PubMed] [Google Scholar]
- 55. Osborn AM, Bruce KD, Strike P, Ritchie DA (1997) Distribution, diversity and evolution of the bacterial mercury resistance (mer) operon. FEMS Microbiol Rev 19: 239–262. [DOI] [PubMed] [Google Scholar]
- 56. Ford PJ, Avison MB (2004) Evolutionary mapping of the SHV β-lactamase and evidence for two separate IS26-dependent bla SHV mobilization events from the Klebsiella pneumoniae chromosome. J Antimicrob Chemother 54: 69–75. [DOI] [PubMed] [Google Scholar]
- 57. Jacoby GA (2009) AmpC β-lactamases. Clinical microbiology reviews 22: 161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bailey JK, Pinyon JL, Anantham S, Hall RM (2011) Distribution of the bla TEM gene and bla TEM-containing transposons in commensal Escherichia coli . J Antimicrob Chemother 66: 745–751. [DOI] [PubMed] [Google Scholar]
- 59. Tian SF, Chu YZ, yi Chen B, Nian H, Shang H (2011) ISEcp1 element in association with bla CTX-M genes of E. coli that produce extended-spectrum-lactamase among the elderly in community settings. Enferm Infecc Microbiol Clin 29: 731–734. [DOI] [PubMed] [Google Scholar]
- 60. Coelho A, González-López JJ, Miró E, Alonso-Tarrés C, Mirelis B, et al. (2010) Characterisation of the CTX-M-15-encoding gene in Klebsiella pneumoniae strains from the Barcelona metropolitan area: plasmid diversity and chromosomal integration. Int J Antimicrob Agents 36: 73–78. [DOI] [PubMed] [Google Scholar]
- 61. Tolmasky ME, Crosa JH (1993) Genetic organization of antibiotic resistance genes (aac(6′)-Ib, aadA, and oxa9) in the multiresistance transposon Tn1331 . Plasmid 29: 31–40. [DOI] [PubMed] [Google Scholar]
- 62. Shu HY, Fung CP, Liu YM, Wu KM, Chen YT, et al. (2009) Genetic diversity of capsular polysaccharide biosynthesis in Klebsiella pneumoniae clinical isolates. Microbiology 155: 4170–4183. [DOI] [PubMed] [Google Scholar]
- 63. Jensen SO, Reeves PR (2004) Deletion of the Escherichia coli O14:K7 O antigen gene cluster. Can J Microbiol 50: 299–302. [DOI] [PubMed] [Google Scholar]
- 64. Ton-Hoang B, Polard P, Haren L, Turlan C, Chandler M (1999) IS911 transposon circles give rise to linear forms that can undergo integration in vitro. Mol Microbiol 32: 617–627. [DOI] [PubMed] [Google Scholar]
- 65. DeLeo FR, Chen L, Porcella SF, Martens CA, Kobayashi SD, et al. (2014) Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae . Proceedings of the National Academy of Sciences 111: 4988–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Partridge SR, Zong Z, Iredell JR (2011) Recombination in IS26 and Tn2 in the evolution of multiresistance regions carrying bla CTX-M-15 on conjugative IncF plasmids from Escherichia coli . Antimicrob Agents Chemother 55: 4971–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sampei G, Furuya N, Tachibana K, Saitou Y, Suzuki T, et al. (2010) Complete genome sequence of the incompatibility group I1 plasmid R64. Plasmid 64: 92–103. [DOI] [PubMed] [Google Scholar]
- 68. Arakawa Y, Wacharotayankun R, Nagatsuka T, Ito H, Kato N, et al. (1995) Genomic organization of the Klebsiella pneumoniae cps region responsible for serotype K2 capsular polysaccharide synthesis in the virulent strain Chedid. J Bacteriol 177: 1788–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Seo J-H, Hong JS-J, Kim D, Cho B-K, Huang T-W, et al. (2012) Multiple-omic data analysis of Klebsiella pneumoniae MGH 78578 reveals its transcriptional architecture and regulatory features. BMC Genomics 13: 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Artsimovitch I, Landick R (2002) The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell 109: 193–203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials: additional genomic features and supplementary tables, figures, and references.
(DOCX)