

Abstract
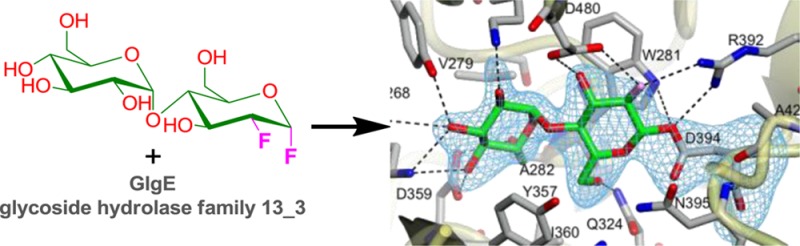
GlgE (EC 2.4.99.16) is an α-maltose 1-phosphate:(1→4)-α-d-glucan 4-α-d-maltosyltransferase of the CAZy glycoside hydrolase 13_3 family. It is the defining enzyme of a bacterial α-glucan biosynthetic pathway and is a genetically validated anti-tuberculosis target. It catalyzes the α-retaining transfer of maltosyl units from α-maltose 1-phosphate to maltooligosaccharides and is predicted to use a double-displacement mechanism. Evidence of this mechanism was obtained using a combination of site-directed mutagenesis of Streptomyces coelicolor GlgE isoform I, substrate analogues, protein crystallography, and mass spectrometry. The X-ray structures of α-maltose 1-phosphate bound to a D394A mutein and a β-2-deoxy-2-fluoromaltosyl-enzyme intermediate with a E423A mutein were determined. There are few examples of CAZy glycoside hydrolase family 13 members that have had their glycosyl-enzyme intermediate structures determined, and none before now have been obtained with a 2-deoxy-2-fluoro substrate analogue. The covalent modification of Asp394 was confirmed using mass spectrometry. A similar modification of wild-type GlgE proteins from S. coelicolor and Mycobacterium tuberculosis was also observed. Small-angle X-ray scattering of the M. tuberculosis enzyme revealed a homodimeric assembly similar to that of the S. coelicolor enzyme but with slightly differently oriented monomers. The deeper understanding of the structure–function relationships of S. coelicolor GlgE will aid the development of inhibitors of the M. tuberculosis enzyme.
GlgE (EC 2.4.99.16) is an α-maltose 1-phosphate:(1→4)-α-d-glucan 4-α-d-maltosyltransferase that is the defining enzyme of a recently discovered biosynthetic pathway in bacteria.1 This four-step pathway generates α-glucan from trehalose,2 whereby GlgE extends maltooligosaccharide acceptors with disaccharide units from the donor α-maltose 1-phosphate. Unusually, for a bacterial carbohydrate-active enzyme, GlgE is regulated by phosphorylation.3 The GlgE pathway genes are present in 14% of sequenced genomes of bacteria and archaea, making it half as common as the classical glycogen α-glucan pathway defined by GlgC and GlgA.4 One notable bacterium that possesses the GlgE pathway genes is Mycobacterium tuberculosis, the causative agent of tuberculosis that remains a significant global health problem.5 GlgE is itself a genetically validated anti-tuberculosis target with a novel mode of action,1 and the first inhibitor of GlgE has recently been reported.6 Thus, understanding structure–function relationships of GlgE would help in the development of inhibitors with therapeutic potential.
GlgE is the first member of CAZy (http://www.cazy.org) glycoside hydrolase subfamily GH13_37,8 to be characterized. Through comparison with other GH13 enzymes,9 GlgE is predicted to catalyze the transfer of maltosyl units with a double-displacement reaction mechanism giving overall retention of stereochemistry (Scheme 1). Thus, the attack of the donor substrate by a nucleophilic Asp residue is predicted to generate a β-maltosyl-enzyme intermediate that is subsequently liberated by an acceptor. An acid/base Glu residue is predicted to assist by protonating the phosphate leaving group and then deprotonating the incoming acceptor. Evidence in support of such a mechanism includes the retention of the α anomeric configuration of α-maltose 1-phosphate1 after its transfer to the nonreducing end10 of a maltooligosaccharide, giving an α-1,4-glycosidic linkage. As expected, no activity was detected with β-maltose 1-phosphate as the donor.1 In addition, the enzyme exhibited ping-pong kinetics, consistent with the inability of GlgE to bind the intact donor and acceptor simultaneously.1
Scheme 1. Proposed α-Retaining Double-Displacement Mechanism of GlgE.

This work presents evidence of the structures shown at the top left and top right.
The structure of Streptomyces coelicolor GlgE isoform I has been determined.10 Sequence and structural alignments with other GH13 enzymes11 suggest the nucleophile and acid/base catalytic residues of S. coelicolor GlgE isoform I are Asp394 and Glu423, respectively. A maltose-bound structure is consistent with this interpretation. However, attempts to obtain a structure with α-maltose 1-phosphate bound have thus far been unsuccessful.
Evidence of the existence of glycosyl-enzyme intermediates has been obtained with a number of glycosyl-transferring enzymes by using strategies to trap them kinetically. This has often involved the introduction of electron-withdrawing substituents around the sugar ring to destabilize the transition states of both catalytic steps, which are expected to have oxocarbenium ion character. Such mechanism-based inhibitors include 2-deoxy-2-fluoro,12,13 2-deoxy-2,2-difluoro, and 5-fluoro analogues.14 The disruption of potential hydrogen bonding interactions by substitution of a hydroxyl group with a fluorine further compromises these substrate analogues. To accelerate the first step, these analogues are often employed as their glycosyl fluorides, wherein the good fluoride leaving group facilitates the formation of the intermediate. This strategy has been used with great success with β-retaining enzymes, but there are far fewer examples with α-retaining enzymes. Most examples involve the use of 5-fluoro and 2-deoxy-2,2-difluoro analogues and the detection of intermediates using mass spectrometry (MS). There are a limited number of examples of α-d-retaining and related enzymes that have had their glycosyl-enzyme intermediate structurally characterized: a GH13 family cyclodextrin glycosyltransferase,15 a GH38 family α-mannosidase,16 a GH13 family amylosucrase,17 a GH31 family α-glycosidase,18 a GH13 family α-amylase,19 a GH27 α-galactosidase,20 a GH31 family α-xylosidase,21 a GH31 family α(1→4)-glucan disproportionating enzyme,22 and a GH31 α-(1,4)-glucan lyase.23 These were trapped using either fluorinated substrate analogues, substrate analogues that were unable to act as acceptors, or activated donor substrates with an enzyme lacking its acid/base residue.
We now report new evidence that supports the double-displacement mechanism in the reaction catalyzed by GlgE (Scheme 1). First, the crystal structure of the Michaelis complex between the S. coelicolor enzyme and α-maltose 1-phosphate was made possible by the substitution of the nucleophilic Asp394 with Ala. Second, a covalent catalytic intermediate was trapped using a 2-deoxy-2-fluoro-α-maltosyl fluoride substrate analogue. This was assisted by the substitution of the acid/base Glu423 residue of the S. coelicolor enzyme with Ala. The resulting maltosyl-enzyme intermediate was characterized by mass spectrometry (MS) and X-ray crystallography. There are few examples of GH13 family glycosyl-enzyme intermediate structures being determined, and this is the first using a 2-deoxy-2-fluoro analogue. Such an intermediate could also be identified by MS with the wild-type M. tuberculosis enzyme. Finally, high-resolution models of the S. coelicolor and M. tuberculosis enzymes were validated and compared in solution using small-angle X-ray scattering, providing the first glimpse of the structure of GlgE from a human pathogen.
Experimental Procedures
2-Deoxy-2-fluoro-α-maltosyl Fluoride
Thin layer chromatography was performed on precoated silica plates (Merck 60 F254, 0.25 mm). Compounds were visualized by being heated after being dipped in a solution of 5% (w/v) H2SO4 in ethanol. Flash chromatography was performed on silica gel columns (Biotage KP-Sil Silica, 60 Ǻ, 32–63 μm) fitted to a Biotage (Uppsala, Sweden) SP1 automated purification system. 19F spectra were recorded at 376 MHz on a Bruker Avance III 400 MHz spectrometer (Bruker Biospin Ltd.). Data were analyzed using a Topspin 2.1 software spectrometer (Bruker Biospin Ltd.), with J values in hertz.
3,6,2′,3′,4′,6′-Hexa-O-acetyl-2-deoxy-2-fluoro-α-d-maltosyl fluoride was prepared according to a published procedure24 with modifications (Scheme S1 of the Supporting Information). Boron trifluoride etherate (42 μL, 0.36 mmol) was added to a stirred solution of maltal per-O-acetate25 (0.5 g, 0.9 mmol) and XeF2 (0.17 g, 1 mmol) in dry dichloromethane (10 mL) at −70 °C. The reaction mixture was gradually allowed to warm to 0 °C, stirred at 0 °C for 2.5 h, and diluted with dichloromethane (50 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (50 mL) and water (50 mL), dried over MgSO4, filtered, and evaporated to yield the crude product (0.5 g) as an amorphous solid. Thin layer chromatography [1:2 (v/v) ethyl acetate:n-hexane] showed the presence of two products; the slower-running α isomer (Rf = 0.4) was purified by flash chromatography using an isocratic elution [1:2 (v/v) ethyl acetate:n-hexane] to give a syrup (100 mg, 21%): 19F NMR (CDCl3) δ −150.7 (d, JF1,F2 19.1, F-1), −205.0 (d, JF2,F1 19.1, F-2). 3,6,2′,3′,4′,6′-Hexa-O-acetyl-2-deoxy-2-fluoro-α-d-maltosyl fluoride was deacetylated using catalytic sodium methoxide according to published methods.24 The deprotected product was purified by flash chromatography using an isocratic elution [4:1 (v/v) ethyl acetate:methanol], yielding a colorless syrup.
Site-Directed Mutagenesis
Muteins were generated using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies). Conserved catalytic residues Asp394 and Glu423 were substituted with alanine residues to abolish their functions. The forward primers for mutating Asp394 and E423 were 5′-GCGCATCTTCCGCGTCGCCAACCCCCACACCAAAC-3′ and 5′-CTTCCTGGCCGCGGCCTTCACCCG-3′ (mutated bases underlined), respectively. Mutations within expression plasmids were confirmed by DNA sequencing.
Expression and Purification of GlgE Proteins
Expression and purification of S. coelicolor GlgE isoform I and M. tuberculosis GlgE were conducted as previously reported.1,10
Structure Determination and Refinement
Crystals of the two S. coelicolor GlgE muteins were obtained as previously described10 and soaked in a crystallization solution [15% (w/v) polyethylene glycol 3350, 0.2 M sodium citrate, and 15% (v/v) ethylene glycol] containing ligand prior to mounting. The α-maltose 1-phosphate10 complex of the D394A mutein was obtained by soaking a protein crystal for 1 h in a 5 mM solution of the ligand. The 2-deoxy-2-fluoro-α-maltosyl fluoride-derived complex of the E423A mutein was obtained by soaking a protein crystal for 210 s in a 5 mM solution of the ligand, and as a control, a similar crystal was soaked in 5 mM maltose for 180 s.
Crystals were flash-cooled in LithoLoops (Molecular Dimensions) by being plunged into liquid nitrogen and stored in Unipuck cassettes (MiTeGen) prior to being transported to the synchrotron. The crystals were subsequently transferred robotically to the goniostat on station I24 or I04-1 at the Diamond Light Source (Oxfordshire, U.K.) and maintained at 100 K with a Cryojet cryocooler (Oxford Instruments). Diffraction data were recorded using either a Pilatus 6M or 2M detector (Dectris). The resultant images were processed using the XIA2 expert system.26 X-ray data collection statistics are summarized in Table 1.
Table 1. X-ray Data Collection and Refinement Statistics.
| D394A mutein with α-maltose 1-phosphate | E423A mutein with maltose (control) | E423A mutein with a 2-deoxy-2-fluoro-β-maltosyl modification | |
|---|---|---|---|
| Data Collection | |||
| beamline | I24 | I04-1 | I04-1 |
| wavelength (Å) | 0.978 | 0.917 | 0.917 |
| detector | Pilatus 6M | Pilatus 2M | Pilatus 2M |
| resolution rangea (Å) | 104.53–2.55 (2.62–2.55) | 37.78–2.29 (2.35–2.29) | 50.54–2.40 (2.47–2.40) |
| space group | P41212 | P41212 | P41212 |
| cell parameters (Å) | a = b = 113.52, c = 313.60 | a = b = 114.18, c = 313.64 | a = b = 113.00, c = 313.86 |
| total no. of measured intensitiesa | 949841 (65056) | 1007908 (73110) | 582247 (40239) |
| no. of unique reflectionsa | 67726 (4748) | 93835 (6826) | 79517 (5527) |
| multiplicitya | 14.0 (13.7) | 10.7 (10.7) | 7.3 (7.3) |
| mean I/σ(I)a | 11.4 (3.0) | 15.4 (3.0) | 14.7 (2.0) |
| completenessa (%) | 99.7 (96.6) | 99.9 (99.9) | 99.5 (95.3) |
| Rmergea,b | 0.169 (0.659) | 0.088 (0.576) | 0.094 (0.991) |
| Rmeasa,c | 0.192 (0.564) | 0.097 (0.634) | 0.109 (1.150) |
| CC1/2a,d | 0.998 (0.804) | 0.999 (0.882) | 0.998 (0.657) |
| Wilson B value (Å2) | 54.1 | 51.1 | 43.7 |
| Refinement | |||
| resolution rangea (Å) | 91.95–2.55 (2.62–2.55) | 37.78–2.29 (2.35–2.29) | 50.54–2.40 (2.47–2.40) |
| no. of reflectionse | |||
| working | 64195 | 89028 | 75425 |
| free | 3423 | 4697 | 3985 |
| Rwork/Rfreea,f | 0.185/0.215 (0.275/0.328) | 0.195/0.220 (0.331/0.335) | 0.184/0.208 (0.335/0.355) |
| Ramachandrang (%) | |||
| favored | 97.9 | 97.9 | 98.0 |
| allowed | 1.8 | 2.1 | 1.8 |
| disallowed | 0.3 | 0.0 | 0.2 |
| root-mean-square deviation for bond distances (Å) | 0.011 | 0.011 | 0.011 |
| root-mean-square deviation for bond angles (deg) | 1.41 | 1.36 | 1.36 |
| no. of protein residues (ranges) (chain A, chain B) | 649 (15–663), 649 (15–663) | 649 (15–663), 649 (15–663) | 649 (15–663), 649 (15–663) |
| no. of water molecules | 437 | 480 | 418 |
| no. of ligand molecules | 2 | 2 | 2 |
| mean B factor (Å2) | |||
| protein | 45.3 | 52.4 | 53.4 |
| water | 37.5 | 43.9 | 43.2 |
| ligand | 54.1 | 45.6 | 47.2 |
| overall | 45.0 | 52.0 | 53.0 |
| Protein Data Bank entry | 4CN1 | 4CN6 | 4CN4 |
Figures in parentheses indicate values for the outer resolution shell.
Rmerge = ∑hkl∑i|Ii(hkl) – ⟨I(hkl)⟩|/∑hkl∑iIi(hkl).
Rmeas = ∑hkl[N/(N – 1)]1/2 × ∑i|Ii(hkl) – ⟨I(hkl)⟩|/∑hkl∑iIi(hkl), where Ii(hkl) is the ith observation of reflection hkl, ⟨I(hkl)⟩ is the weighted average intensity for all observations i of reflection hkl, and N is the number of observations of reflection hkl.
CC1/2 is the correlation coefficient between intensities taken from random halves of the data set.
The data set was split into “working” and “free” sets consisting of 95 and 5% of the data, respectively. The free set was not used for refinement.
Rwork and Rfree were calculated as follows: R = ∑(|Fobs – Fcalc|)/∑|Fobs| × 100, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
As calculated using MolProbity.51
The data sets were processed in space group P41212 with approximate cell parameters of a = b = 113 Å and c = 314 Å and, in contrast to the majority of previous data sets,10 did not suffer from severe twinning. The structures were determined by molecular replacement using one subunit from the apo-GlgE structure [Protein Data Bank (PDB) entry 3ZSS] as input to PHASER.27 In all cases, this was successful in placing two copies of the subunit in the asymmetric unit. Despite the apparent isomorphism with the only untwinned data set from the previous study [i.e., the complex with α-cyclodextrin (PDB entry 3ZST)],10 the resultant asymmetric unit corresponded to the biological dimer for all structures, rather than to halves of two separate biological dimers as seen previously. In each case, electron density maps, inspected using COOT,28 were consistent with the expected point mutations and provided clear evidence of ligand binding at the donor pocket. The structures were rebuilt and completed through several iterations of refinement (with local noncrystallographic symmetry restraints) in REFMAC5,29 and manual adjustment in COOT. In the latter stages, translation libration screw refinement was used with a total of eight translation libration screw domains, which were defined using the translation libration screw motion determination server (http://skuld.bmsc.washington.edu/∼tlsmd/).30 Refinement statistics are summarized in Table 1. All structural figures were prepared using CCP4MG.31
Enzymatic Assay
Unless otherwise specified, all enzyme assays were performed at 21 °C using an end-point assay involving the detection of inorganic phosphate with malachite green.1 Chemicals were purchased from Sigma-Aldrich. Reaction mixtures (25 μL) consisted of 100 mM Bis-Tris propane (pH 7.0), 50 mM NaCl, 1 mM maltohexaose, and 0.25 mM α-maltose 1-phosphate.10 Reactions were initiated by the addition of α-maltose 1-phosphate and mixtures assayed for 10 min before reactions were quenched with 175 μL of malachite green reagent [0.011% (w/v) in 1 M HCl containing 1% (w/v) ammonium molybdate and 0.037% (v/v) Triton N101],32 followed by incubation for 20 min at 21 °C. The absorbance at 630 nm was measured on a SpectraMax Plus microplate spectrophotometer using SoftMax Pro version 3.1.1. Initial reaction rates for S. coelicolor GlgE E423A were determined using 28 μM protein with assay mixtures terminated over the first 12 min. The activity of S. coelicolor GlgE D394A was measured at protein concentrations between 0.75 and 9.6 μM. Inhibition of S. coelicolor GlgE E423A by 1 mM 2-deoxy-2-fluoro-α-maltosyl fluoride was tested by preincubation of 28 μM GlgE E423A for 5–60 min. The reaction of wild-type S. coelicolor GlgE with 2-deoxy-2-fluoro-α-maltosyl fluoride as a donor was monitored by MALDI-TOF mass spectrometry. Reaction mixtures (20 μL) comprising 1 mM maltotetraose, 5 mM 2-deoxy-2-fluoro-α-maltosyl fluoride, and 18 μM protein were incubated for 20 min at 21 °C before being analyzed.1
MALDI-TOF, Orbitrap, and LC–MS
2-Deoxy-2-fluoro-α-maltosyl fluoride (2 mM) was added to either 100 μM wild-type or E423A S. coelicolor GlgE or wild-type M. tuberculosis GlgE in 20 mM Bis-Tris propane (pH 7.0) or 20 mM Tris (pH 8.5), respectively, and incubated for at least 5 min at 21 °C. An aliquot (1 μL, approximately 100 pmol) was diluted in 50 μL of 40 mM HCl. One microgram of pepsin (porcine, Princeton Separations, Adelphia, NJ) was added, and the sample was incubated at 37 °C for 16 h. An aliquot (approximately 1 pmol) was diluted into 20 μL of 0.1% trifluoroacetic acid and applied to a nanoAcquity (Waters, Manchester, U.K.) UPLC system running at a flow rate of 250 nL/min connected to an LTQ-Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA). Peptides were trapped using a precolumn (Symmetry, C18, 5 μm, 180 μm × 20 mm, Waters), which was then switched in line to an analytical column (BEH, C18, 1.7 μm, 75 μm × 250 mm, Waters) for separation. Peptides were eluted with a gradient of 3 to 40% acetonitrile in water, containing 0.1% formic acid, at a rate of 0.67%/min. The m/z (e.g., 853.9 with a z of +2) of the expected modified peptide was used on an inclusion list for the detection by the mass spectrometer.
Raw files were processed with MaxQuant version 1.3.0.533 (http://maxquant.org) to generate recalibrated peaklist files that were used for a database search using an in-house Mascot 2.4 Server (Matrix Science Limited, London, U.K.). Mascot searches were performed on the Uniprot sptrembl20121031 database with taxonomy set to S. coelicolor, a 6 ppm precursor tolerance, a 0.6 Da fragment tolerance, zero missed cleavages, and a mass of 326.1 as a variable modification. Mascot search results were imported and evaluated in Scaffold version 4.0.4 (proteomsoftware.com, Portland, OR). Additional LTQ-Orbitrap methods are described in the Supporting Information. The pepsin-digested sample was analyzed with an Ultraflex MALDI-TOF/TOF (Bruker, Coventry, U.K.). It was spotted onto a Prespotted AnchorChip (PAC) MALDI target plate (Bruker) and analyzed using methods optimized for peptides and for fragmentation using LIFT technology. The acquired spectra were processed in FlexAnalysis (Bruker).
The intact mass analysis was performed using LC–MS on a Synapt G2 HDMS mass spectrometer coupled to an Acquity UPLC system (Waters). The protein (around 200 pmol) was loaded onto a C4 reversed phase column (BEH300, C4, 1.7 μm, 1 mm × 50 mm, Waters) and eluted with a gradient from 10 to 80% acetonitrile in 0.1% formic acid (v/v) in 13 min. The mass spectrometer was run in MS/sensitivity/positive mode with standard settings. Spectra under the LC peak were combined and deconvoluted using the MaxEnt1 tool in MassLynx version 4.1 (Waters).
SAXS Data Collection
S. coelicolor and M. tuberculosis GlgE proteins were analyzed at several protein concentrations each in ranges of 0.6–11.0 and 0.5–7.9 mg/mL, respectively. The synchrotron radiation X-ray scattering data were collected on beamline X33 of the EMBL Hamburg on storage ring DORIS III (DESY, Hamburg, Germany)34 using a PILATUS 1M pixel array detector, a sample–detector distance of 2.7 m, and a wavelength (λ) of 1.5 Å. The range of momentum transfer (s) from 0.01 to 0.5 Å–1 was covered (s = 4π sin θ/λ, where 2θ is the scattering angle). To monitor the radiation damage, eight successive 15 s exposures of protein solutions were compared, and no significant changes were observed. The data were normalized to the intensity of the transmitted beam and radially averaged; the scattering of the buffer was subtracted, and the difference curves were scaled for protein concentration. The low-angle data measured at lower protein concentrations were extrapolated to infinite dilution and merged with the higher-concentration data to yield the final composite scattering curves.35 PRIMUS was used for data processing.36
Determination of the Radius of Gyration and Molecular Mass by SAXS
The forward scattering [I(0)] and the radius of gyration (Rg) were evaluated using the Guinier approximation37 assuming that at very small angles (s < 1.3/Rg) the intensity is represented as I(s) = I(0) exp[−(sRg)2/3]. The radii of gyration were also computed from the entire scattering patterns using the indirect transform package GNOM.38 The Fourier transform of the scattering profile also provides the pair distribution function of the particle [p(r)] and the maximal size (Dmax). The molecular mass of the solute was evaluated by comparison of the forward scattering with that of the reference solution of bovine serum albumin (66 kDa). The excluded volume of the hydrated particle was computed from the small-angle portion of the data (s < 0.25 Å–1) using the Porod invariant.39 For globular proteins, Porod volumes in cubic nanometers are ∼1.7 times the molecular masses in kilodaltons.40
Molecular Modeling Using SAXS Data
Theoretical scattering profiles of the crystallographic and predicted models of GlgE were calculated with CRYSOL.41 Given the atomic coordinates, the program minimizes the discrepancy in the fit to the experimental intensity by adjusting the excluded volume of the particle and the contrast of the hydration layer. The discrepancy (χ2) between the measured and calculated SAXS profiles is defined as
where N is the number of experimental points, c is a scaling factor, Icalc(sj) and Iexp(sj) are the calculated and experimental scattering intensities, respectively, and σ(sj) is the experimental error at momentum transfer sj.
The crystallographic and homology models of dimeric proteins were refined by a brute force global search program GENCRY developed in house. The program rotates and moves the monomer, constructs the dimer using a P2 symmetry axis, and calculates the fit by CRYSOL. With the initial model as a starting point, monomers were displaced up to ±0.2 nm along each axis with an increment of 0.1 nm and rotated around the center of mass by Euler angles α, γ (±0.1 rad), and 0 rad < β < 0.2 rad with an increment of 0.05 rad. CRYSOL fits from the 15625 resulting models to the experimental SAXS data were ranked based on the χ2 value to select the best fitting model.
Results
Binding of α-Maltose 1-Phosphate to GlgE
We have previously reported structures of S. coelicolor GlgE isoform I (PDB entry 3ZSS in Figure 1 and Figure S1A of the Supporting Information), including those with α-maltose bound (PDB entry 3ZT5 in Figure S1B of the Supporting Information).10 The putative nucleophile (Asp394) and general acid/base (Glu423) catalytic residues were close to the reducing end of the maltose, suggesting the glucose rings occupied subsites −1 and −242 of the donor substrate, α-maltose 1-phosphate. However, it was not possible to obtain a structure of GlgE with the donor bound, presumably because it was slowly but excessively hydrolyzed over the time scale of protein crystallization.
Figure 1.
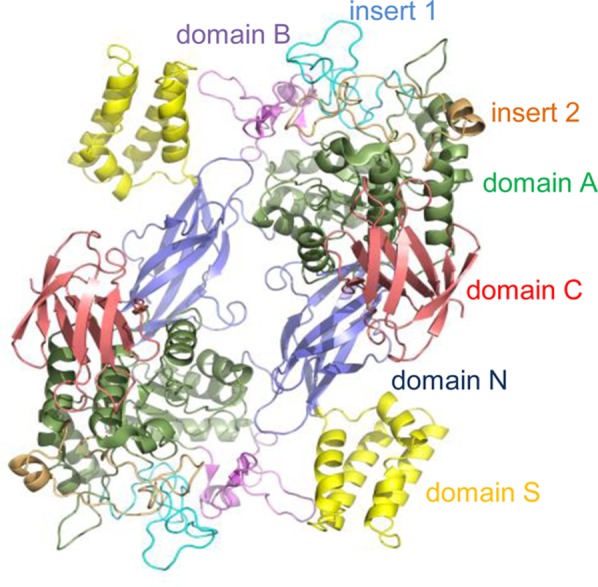
Domains within a dimer of S. coelicolor GlgE isoform I (PDB entry 3ZSS).10
To determine a structure with the intact donor substrate bound, the predicted nucleophilic residue, Asp394, was mutated to Ala. The maltosyl transferase activity of the D394A mutein was >4 orders of magnitude lower than that of the wild-type protein as expected. A crystal of the D394A mutein was soaked in 5 mM α-maltose 1-phosphate, and a ligand-bound structure was successfully determined to 2.55 Ǻ resolution (PDB entry 4CN1 in Figure 2A and Figure S1C of the Supporting Information). The ability to observe the intact substrate was consistent with a significantly weaker ability of the mutein to catalyze its hydrolytic side reaction as expected. The substitution of Asp394 with Ala was confirmed in the electron density map, but no other significant differences in the protein structure were observed (Figures S1 and S2 of the Supporting Information). The interactions between the protein and the sugar rings were very similar to those observed in the maltose-bound structures,10 and the overall conformations of the sugar rings were likewise similar (Figure 3). This supports the assignment of subsites −2 and −1 and is consistent with the structures of other GH13 enzymes.43 Therefore, the phosphate group of α-maltose 1-phosphate was located in what was predicted to be subsite +1.10 This group made hydrogen bonding interactions with the side chains of Asn352 and Tyr357 from domain B and Asn395 and Glu423 from domain A, as previously predicted10 (Figure 2A and Figures S1C and S2B of the Supporting Information). The carboxyl group of Glu423 formed an ∼3.0 Å hydrogen bond to one of the oxygen atoms of the phosphate group and was only ∼3.7 Å from the phosphate ester oxygen atom. The proximity of this conserved amino acid to the phosphate group is consistent with it being the general acid/base catalytic residue that protonates the leaving group. The side chain of Glu423 also interacted with the backbone NH group of Asn395, which could have a role in defining its pKa, a possibility that would require further investigation. Interestingly, the phosphate does not sit in the “tucked under” conformation seen for GT35 phosphorylases such as glycogen phosphorylase as well as many nucleotide sugar-dependent GTs.44,45 This difference likely reflects the quite different mechanisms followed: double-SN2 displacement versus internal nucleophilic substitution SNi.46 While it is tempting to speculate further about the existence and role of each hydrogen bonding interaction observed in this ligand-bound structure with a mutated enzyme, the hydrogen bonding network may be a little different in the true Michaelis complex.
Figure 2.
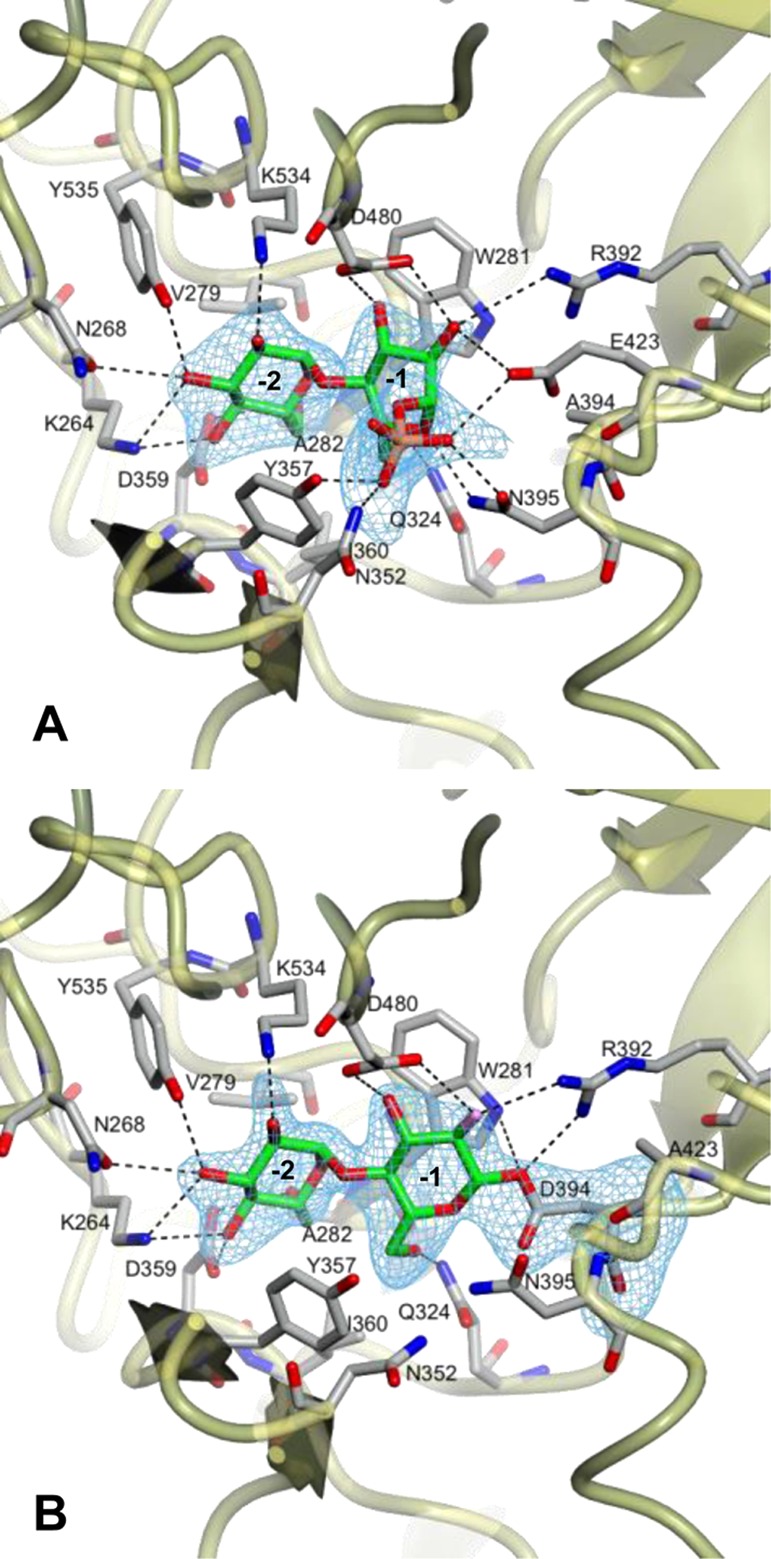
Structures of GlgE with (A) α-maltose 1-phosphate bound to the D394A mutein (PDB entry 4CN1) and (B) the covalent intermediate formed between the E423A mutein and 2-deoxy-2-fluoro-α-maltosyl fluoride (PDB entry 4CN4). Difference electron density “omit” maps were generated for bound ligands using phases from final models without ligand coordinates after application of small random shifts to the models and re-refining. The corresponding stereo images are shown in Figure S1 of the Supporting Information. Some amino acids interacting with the ligands have been omitted for the sake of clarity, but all are shown in Figure S2 of the Supporting Information. Subsites −1 and −2 are labeled.
Figure 3.
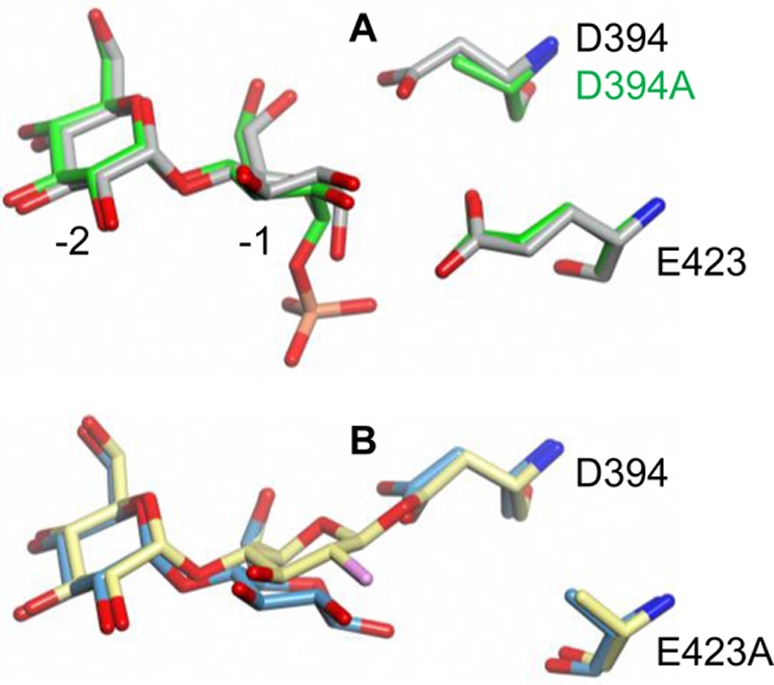
Superposed structures of (A) maltose (PDB entry 3ZT5, gray) and α-maltose 1-phosphate (PDB entry 4CN1, green) bound to wild-type and D394A GlgE, respectively, and (B) maltose (PDB entry 4CN6, blue) and the trapped 2-deoxy-2-fluoro-β-maltosyl intermediate (PDB entry 4CN4, yellow) bound to the E423A mutein. Comparison of the structures in panel B reveals that the anomeric carbon moves 1.6 Ǻ to allow the formation of a covalent bond between the disaccharide and D394. The glucose rings in each structure adopt the low-energy 4C1 conformation. The orientation shown is similar to that in Scheme 1 with subsites −1 and −2 labeled in panel A.
A Covalent Maltosyl-GlgE Intermediate
To capture a maltosyl-GlgE intermediate for structural studies, a donor substrate analogue was used together with a mutated form of GlgE. 2-Deoxy-2-fluoro-α-maltosyl fluoride was first synthesized (Scheme S1 of the Supporting Information) as an analogue for which both chemical steps (formation and hydrolysis of the intermediate) should be slow because of the destabilization of the oxocarbenium ion-like transition states by the fluorine at position 2.12,13 The further presence of the good anomeric fluoride leaving group should reaccelerate the first step, making the intermediate kinetically accessible (Scheme 1). Thus, the 2-fluoromaltosyl-enzyme intermediate would be expected to be much longer-lived than that formed during normal turnover.
The activity of the wild-type enzyme with 2-deoxy-2-fluoro-α-maltosyl fluoride was tested using maltotetraose as an acceptor. MS analysis of the product mixture clearly showed the successive extension of the acceptor by 326 Da consistent with the addition of 2-deoxy-2-fluoromaltosyl groups (Figure S3 of the Supporting Information). 2-Deoxy-2-fluoro-α-maltosyl fluoride inhibited turnover of the normal substrate: after preincubation for 5 min with 1 mM analogue, 70% of the normal activity with α-maltose 1-phosphate was lost, and longer preincubations did not result in any further loss of activity (Figure S4 of the Supporting Information). The residual 30% activity was therefore consistent with the analogue being a slow substrate rather than a dead-end inhibitor.14
The presence of any covalent modification of GlgE with the substrate analogue was assessed using MS of the intact wild-type protein. Incubation of the protein with the substrate analogue led to an increase in the mass of 326.4 Da (from 77496.5 to 77822.9 Da with a predicted change of 326.3 Da) for a small proportion of the protein, consistent with the modification of some of the GlgE with a 2-deoxy-2-fluoromaltosyl group.
To slow the turnover of the 2-fluoromaltosyl intermediate further, a mutation was introduced into GlgE. Having already obtained evidence for Glu423 being the general acid/base, we mutated this residue to Ala, giving an E423A mutein. This mutation would have little effect on the formation of the intermediate from a glycosyl fluoride for the reasons described above. However, the absence of this residue and its associated general base catalytic role would lower the rate of the intermediate’s decay. Indeed, the E423A mutein turned over the normal substrate, α-maltose 1-phosphate, ∼500 times slower than did the wild-type enzyme.
The modification of the E423A mutein with the substrate analogue was assessed using MS. An essentially quantitative increase in mass of 326.3 Da was observed (Figure S5 of the Supporting Information), consistent with the modification of the entire sample. The mass of the modified protein reverted fully to that of the unmodified protein after 24 h at 4 °C, showing that it was ultimately sensitive to hydrolysis given sufficient time. The location of the modification along the polypeptide chain was then determined using pepsin digestion at low pH, which would assist in preserving the integrity of the ester linkage of the glycosyl-enzyme intermediates, followed by MS. Only 19% sequence coverage was obtained, presumably reflecting the poor sequence specificity of pepsin. Nevertheless, a modification within a peptide fragment consistent with 392RVDNPHTKPVAF403 was identified, which gave a loss of 326.17 Da after LIFT fragmentation to give the expected unmodified peptide mass of 1380.83 Da (Figure S6 of the Supporting Information). Peptide sequencing using Orbitrap MS/MS analysis of b- and y-ion series from the modified peptide identified the Asp residue within 392RVDNPHTKPVAF403 as the site of modification (Table 2). This was consistent with Asp394 being the catalytic nucleophile as predicted.
Table 2. Orbitrap MS/MS Analysis of Modified Peptides Derived from GlgE after Exposure to 2-Deoxy-2-fluoro-α-maltosyl Fluoride.
| E423A S. coelicolor GlgE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R392a | V | D*b | N | P | H | T | K | P | V | A | F403 | |
| b-ions | 157.10 | 256.17 | 697.30 | 811.34 | 908.40 | 1045.45 | 1146.50 | 1274.60 | 1371.65 | 1470.72 | 1541.76 | – |
| y-ions | – | 1550.73 | 1451.67 | 1010.54 | 896.49 | 779.44 | 662.38 | 561.33 | 433.24 | 336.19 | 237.12 | 166.08 |
| M. tuberculosis GlgE | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F415a | R | V | D*b | N | P | H | T | K | P | P | N | F427 | |
| b-ions | 148.07 | 304.17 | 403.24 | 844.37 | 958.41 | 1055.46 | 1192.52 | 1293.57 | 1421.67 | 1518.72 | 1615.77 | 1729.81 | – |
| y-ions | – | 1747.82 | 1591.72 | 1492.66 | 1051.53 | 937.48 | 840.43 | 703.37 | 602.32 | 474.23 | 377.18 | 280.12 | 166.08 |
The peptide was sequenced by assignment of the m/z of singly charged b-ions and y-ions.
D* denotes the modified residue with an observed mass change within the b-ion series of 441.13 Da in both cases that was consistent with the predicted value (115.03 Da for the Asp plus 326.10 Da for the 2-deoxy-2-fluoromaltosyl group, giving 441.16 Da).
The structural integrity of the E423A mutein was then confirmed by determining its structure to 2.3 Ǻ resolution after soaking a crystal with maltose (PDB entry 4CN6 in Table 1 and Figure S1D of the Supporting Information). The mutation of Glu423 to Ala was confirmed, and the presence of the β anomer rather than the α anomer of maltose was observed. It would therefore appear that the loss of the Glu423 side chain in this mutein allowed the energetically more favorable β anomer to be accommodated within its active site. Apart from these minor differences, the structure of the mutein resembled that of the wild-type protein.10
Finally, a crystal of the E423A mutein was soaked with 5 mM 2-deoxy-2-fluoro-α-maltosyl fluoride and flash-frozen after 210 s. Its structure was determined to 2.4 Ǻ resolution (PDB entry 4CN4 in Table 1, Figure 2B, and Figure S1E of the Supporting Information). There were no significant changes in the overall protein structure compared with that of the maltose-bound form, but there was clear continuous electron density consistent with Asp394 being modified with a 2-deoxy-2-fluoro-β-maltosyl group. In the process of forming the covalent bond, the reducing end sugar was tilted within subsite −1 toward the Asp394 residue such that its anomeric carbon moved 1.6 Å compared with that in the corresponding maltose-bound structure (Figure 3). By contrast, the nonreducing end glucose ring within subsite −2 took up a position similar to those of all other structures with this subsite occupied. Indeed, the nonreducing end sugar ring was consistently associated with better resolved electron density and lower B factors (although this was less pronounced for the maltose-bound form of the E423A mutein). The hydrogen bonding interactions between the protein and the sugar rings were broadly similar to those of other structures. Small differences included an additional interaction between Arg392 and the C1 oxygen of the newly formed covalent bond (Figure 2B and Figures S1E and S2C of the Supporting Information).
Relevance of the S. coelicolor GlgE to M. tuberculosis GlgE
The enzymes from S. coelicolor and M. tuberculosis have a lot in common; they have 51% identical sequences with essentially complete conservation within the donor site, share similar net secondary structures according to circular dichroism spectroscopy, and have very similar kinetic properties, including indistinguishable Km values for α-maltose 1-phosphate.3,10 To further explore their commonalities, the M. tuberculosis enzyme was exposed to 2-deoxy-2-fluoro-α-maltosyl fluoride. MALDI-TOF MS identified an increase in mass of the intact protein of 324 Da, from 80316 Da (80316 Da, expected) to 80640 Da, consistent with formation of a covalent intermediate despite it being the wild-type protein rather than an active site mutein. Pepsin digestion and Orbitrap MS/MS analysis gave 44% sequence coverage and identified 415FRVDNPHTKPPNF427 as a modified peptide with D418 forming the covalent glycosidic linkage (Table 2). This Asp residue was indeed predicted to be the nucleophilic residue based on sequence alignments.10
Although we are developing a good understanding of the structure of the S. coelicolor enzyme, a high-resolution structure of M. tuberculosis GlgE has proven to be elusive. It was already known that both enzymes formed dimers in solution according to analytical ultracentrifugation.1,10 To explore their structural similarities further, they were both subjected to SAXS analysis at several protein concentrations (Figure 4A and Figure S7 and Table S1 of the Supporting Information). The proteins had similar radii of gyration (Rg = 40 ± 1 Å) consistent with dimeric assemblies as expected. Further, the maximal sizes of the particles (Dmax) derived from the p(r) function analysis (Figure S7 of the Supporting Information) were in good agreement with the maximal distance between surface amino acids of ∼130 Å observed in the crystallographic homodimer of S. coelicolor GlgE.
Figure 4.
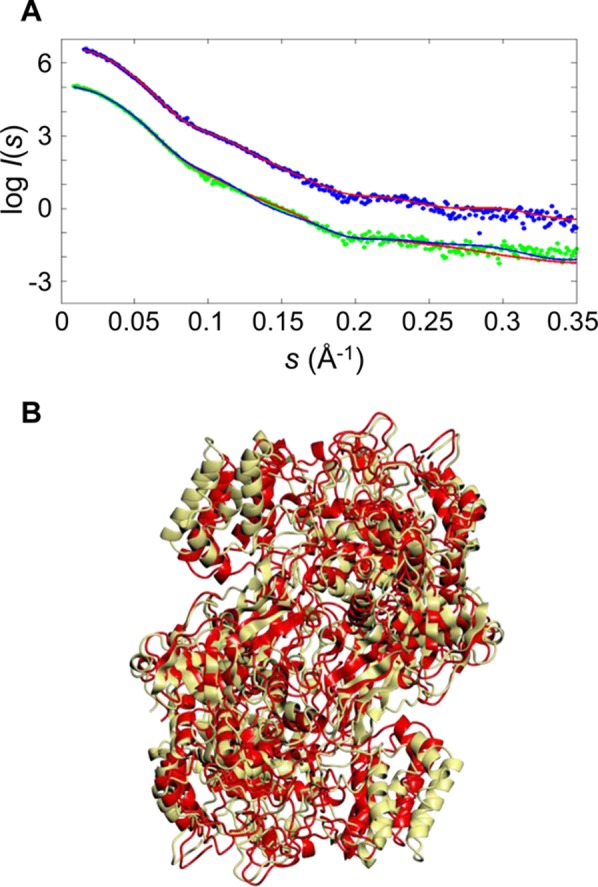
SAXS analysis of GlgE proteins. The upper curves (A) show the SAXS data and fit for the S. coelicolor GlgE protein (χ = 1.03; experimental data colored blue and the theoretical profile colored red on the basis of its X-ray crystal structure). The lower curves show the M. tuberculosis GlgE SAXS data and fit of the theoretical profile of the initial homology model based on the S. coelicolor GlgE crystal structure (χ = 1.34; experimental data colored green and fit colored blue) and after the GENCRY rigid body refinement giving a significantly better fit particularly in the range of 0.1–0.2 Å–1 (χ = 1.09; theoretical fit colored red), consistent with a better relative orientation of the monomers. The SAXS profiles are displaced along the logarithmic axis for the sake of clarity. The homology model of the M. tuberculosis GlgE dimer (B) based on the S. coelicolor GlgE structure before (yellow) and after (red) rigid body refinement gave a root-mean-square deviation of 6.8 Å. The overall orientation of the dimer is similar to that in Figure 1.
Despite the similar radii of gyration and homodimeric states of both proteins in solution, the SAXS patterns displayed noticeable differences in the momentum transfer range from 0.1 to 0.2 Å–1, i.e., in the resolution range from 60 to 30 Å, corresponding to quaternary structure (Figure 4A and Figure S7 of the Supporting Information). The difference in the overall structure was further corroborated by a shift of the main maximum of the M. tuberculosis GlgE pair distribution function p(r) compared to that of S. coelicolor GlgE (Figure S7 of the Supporting Information).
The theoretical scattering profile of the crystal structure of the S. coelicolor GlgE dimer computed by CRYSOL agreed well with the experimental data recorded, with a discrepancy χ of 1.03 (Figure 4A), indicating that the homodimer observed in the crystal structure is probably preserved in solution. The S. coelicolor GlgE model gave a poorer fit to the experimental scattering from M. tuberculosis GlgE with a χ of 1.34. To verify whether the misfit was caused by the difference in species, a homology model of the dimeric M. tuberculosis GlgE was generated using the SWISSMODEL server,47 with the crystal structure of S. coelicolor GlgE serving as a structural template. The homology model still yielded a poor fit to the data from M. tuberculosis GlgE with a χ of 1.38, and most of the systematic deviations in the fit remained in the range of 0.1–0.2 Å–1 (Figure 4A). This observation indicated that the most likely cause of the misfit was differences in the orientation of the two monomers in the dimeric assemblies of the two enzymes.
To refine the M. tuberculosis dimer model, an exhaustive rigid body search in which one monomer of the M. tuberculosis homology model was rotated and moved around its position within the context of a dimeric assembly was employed. The best fitting model with a χ of 1.09 yielded a good fit to the experimental data (Figure 4A). The refined model had a root-mean-square deviation of 6.8 Å from the original homology model of M. tuberculosis GlgE (Figure 4B).
Discussion
The structure of the GlgE D394A mutein with α-maltose 1-phosphate bound provides supporting evidence for the assignment of subsites −2, −1, and +1. Although there are no significant changes to the structure of the protein once this donor substrate has bound to the enzyme, it is likely that domain B acts like a lid to allow entry and exit to the otherwise enclosed donor site. Subsite +1 is expected to be able to bind the nonreducing sugar ring of an acceptor molecule once the phosphate group has departed, but no structures with wild-type or mutated proteins in which this is the case have thus far been determined. It is likely that other more distant subsites, such as those responsible for binding cyclodextrins near the donor site,10 are primarily responsible for the affinity of the enzyme for acceptor substrates. This is supported by the shortest acceptor being maltotetraose.1,10
The trapping of a maltosyl-Asp394 intermediate unequivocally confirms the identity of the nucleophile. In addition, superposition of all three structures presently described shows that the oxygen atom of Asp394 that forms the glycosyl intermediate is aligned along the trajectory of the C1–O bond of α-maltose 1-phosphate and is ∼3.5 Å from C1 (Figure 3A). This arrangement is consistent with the first step of the proposed mechanism (Scheme 1). The proximity of the Glu423 carboxyl group to the phosphate group of the donor substrate (Figure 2A and Figure S1C of the Supporting Information), together with the slowing of GlgE activity in the D423A mutein, supports the role of this amino acid as the acid/base catalytic residue. Importantly, this evidence strongly supports the proposed double-displacement mechanism (Scheme 1).
The structure of the maltosyl-enzyme intermediate shows few changes in the protein compared with the corresponding maltose-bound structures, except for of course the presence of electron density consistent with the formation of a covalent bond between the substrate and the enzyme. For this bond to form, the Asp394 nucleophilic side chain did not move significantly, but the reducing end sugar ring of the donor molecule in subsite −1 tilted toward it, giving an ∼1.6 Å shift in the position of C1. Similar ∼1 Å movements in C1 of the sugar ring have been observed in other enzymes such as a α-retaining GH13 family cyclodextrin glycosyltransferase.15
The ability of the sugar ring to tilt in subsite −1 is potentially relevant to the conformational requirements of an α-retaining mechanism. The low-energy 4C1 conformation is appropriate for the loss of the axial phosphate group for stereoelectronic reasons.48 Such a conformation is indeed observed in the crystal structure with the donor substrate bound (Figure 2A and Figure S1C of the Supporting Information). It is expected that the first transition state with oxocarbenium character would adopt a 4H3 half-chair conformation (Scheme 1). It has been argued that a glucosyl-enzyme intermediate in an α-retaining enzyme that catalyzes transglycosylation reactions would adopt a 4C1 conformation to prolong its lifetime, thus allowing the catalytic cycle to be completed before any hydrolytic side reaction could take place.15 Such a low-energy conformation of the intermediate is observed with GlgE (Figure 2B and Figure S1E of the Supporting Information). Indeed, the C2–C1–O5–C5 torsion angle of this sugar ring is within 9° of the angle of −65° observed in crystalline α-maltose49 in all of the reported ligand-bound structures of GlgE. However, for this conformation to be adopted without moving the Asp394 side chain, the sugar ring had to tilt instead. The deglycosylation step would be expected to involve a 1S3 skew boat conformation, allowing the glucose–Asp394 bond to adopt a pseudoaxial position. This is feasible without moving the Asp394 side chain because all of the ring except for C1 could tilt back toward its original position (Scheme 1). To minimize the chance of hydrolysis, it is tempting to speculate that such a conformation is adopted only when the acceptor is also bound. The second transition state, analogous to the first, would then lead to the formation of the product.
GlgE is efficient in catalyzing reversible phosphorolysis and transglycosidation reactions with little observable hydrolytic activity.1,10 The suppression of hydrolytic reactions could be due in part to the binding of the acceptor affecting the conformation of the intermediate, as discussed above. In addition, inspection of the electron density near the intermediate revealed the presence of water molecules within the active site hydrogen-bonded to the 2′-OH group of the maltosyl group and Tyr357 (Figure 5). Importantly, none of these water molecules were positioned appropriately to attack C1 of the intermediate, with the closest water molecule being 5.0 Å away, and at an angle of ∼30° to the expected trajectory. This water molecule would have to move for the structure to accommodate an appropriately positioned water molecule for nucleophilic attack. Therefore, the structure is consistent with the possibility of GlgE utilizing this strategy to avoid hydrolysis, as has been suggested for other enzymes such as a GH31 family α-transglucosylase.22
Figure 5.
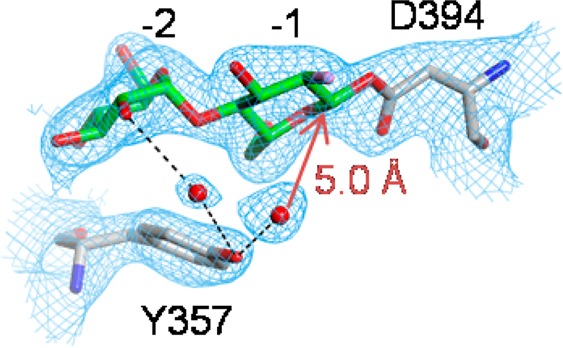
Hydrogen-bonded water molecules near the 2-deoxy-2-fluoro-β-maltosyl intermediate of the E423A mutein of GlgE (PDB entry 4CN4). The orientation shown is similar to that in Scheme 1 with subsites −1 and −2 labeled. Superimposed on the coordinates is the final 2mFobs – DFcalc, αcalc electron density map calculated at 2.4 Å resolution.
The structures of only three glycosyl-enzyme intermediates in the GH13 family have been determined previously using either 5-fluoro substrate analogues, substrate analogues that were unable to act as acceptors, or activated donor substrates with an enzyme lacking its acid/base residue.15,17,19 GlgE is the first example of a GH13 family enzyme to have its intermediate structurally characterized using a 2-deoxy-2-fluoro analogue, showing that it is not necessary to use the synthetically more challenging 5-fluoro or 2-deoxy-2,2-difluoro strategies in this case. Although it was necessary to introduce a mutation into GlgE to give the intermediate sufficient longevity to allow its crystal structure to be determined, the intermediate could also be observed with wild-type proteins by MS. The lack of success in such trapping with α-glycosidases previously has been suggested to be due to less development of positive charge at the neighboring C1 in the transition states of these α-retaining enzymes than in β-retaining enzymes.50 This could be due in part to the alignment of the oxygen atom of the Asp carboxyl near the oxygen atom of the sugar ring in GH13 family enzymes.19 However, this alignment was also observed in the intermediate of GlgE (Figure 2B and Figure S1E of the Supporting Information). Thus, it remains unclear why the strategy worked better with GlgE than with other GH13 family enzymes.
GlgE is an interesting enzyme in its own right, given that it is the defining enzyme of the GH13_3 subfamily and a recently discovered bacterial α-glucan biosynthetic pathway. However, its potential as a new target for therapies against tuberculosis has also attracted interest.6 The S. coelicolor and M. tuberculosis enzymes share a great deal in common, allowing the former to be potentially used as a model for the latter. Nevertheless, a few differences have been identified, including a 23-fold lower affinity of the M. tuberculosis enzyme for its acceptor substrates.10 It has been suggested that this could be due to a longer loop on the enzyme’s surface that could have an impact on the acceptor binding site. SAXS analysis has confirmed that both enzymes assemble into similar homodimers but that the monomers within the M. tuberculosis dimer are oriented somewhat differently. Whether this could have an impact on enzyme activity is hard to ascertain. Furthermore, it has not been possible to establish whether the longer loop could have an impact on the acceptor site given the limited resolution afforded by this technique. Nevertheless, the successful trapping of a 2-fluoromaltosyl-M. tuberculosis enzyme intermediate gives us confidence that the S. coelicolor enzyme is a good model for the development of inhibitors of the M. tuberculosis enzyme that target the donor site.
Acknowledgments
We acknowledge Diamond Light Source for time on beamlines I24 and I04-1 under proposal MX1219.
Glossary
Abbreviations
- LC
liquid chromatography
- MALDI-TOF
matrix-assisted laser desorption ionization time-of-flight
- MS
mass spectrometry
- SAXS
small-angle X-ray scattering.
Supporting Information Available
Detailed Orbitrap methods, Figures S1–7, Scheme S1, and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (entries 4CN1, 4CN4, and 4CN6).
This work was supported by the United Kingdom Biotechnology and Biological Sciences Research Council (Grants BB/I012850/1 and BB/J004561/1), the John Innes Foundation, and, for the SAXS experiments, the European Commission Seventh Framework Programme (FP7/2007-2013) under Grant 226716.
The authors declare no competing financial interest.
Supplementary Material
References
- Kalscheuer R.; Syson K.; Veeraraghavan U.; Weinrick B.; Biermann K. E.; Lui Z.; Sacchettini J. C.; Besra G.; Bornemann S.; Jacobs W. R. (2010) Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an α-glucan pathway. Nat. Chem. Biol. 6, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miah F.; Koliwer-Brandl H.; Rejzek M.; Field R. A.; Kalscheuer R.; Bornemann S. (2013) Flux through trehalose synthase flows from trehalose to the α anomer of maltose in mycobacteria. Chem. Biol. 20, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiba J.; Syson K.; Baronian G.; Zanella-Cleon I.; Kalscheuer R.; Kremer L.; Bornemann S.; Molle V. (2013) Mycobacterium tuberculosis maltosyltransferase GlgE, a genetically validated anti-tuberculosis target, is negatively regulated by Ser/Thr phosphorylation. J. Biol. Chem. 288, 16546–16556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G.; Chater K. F.; Bornemann S. (2011) Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology 157, 1565–1572. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. (2011) The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Veleti S. K.; Lindenberger J. J.; Ronning D. R.; Sucheck S. J. (2014) Synthesis of a C-phosphonate mimic of maltose-1-phosphate and inhibition studies on Mycobacterium tuberculosis GlgE. Bioorg. Med. Chem. 22, 1404–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam M. R.; Danchin E. G. J.; Rancurel C.; Coutinho P. M.; Henrissat B. (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of α-amylase-related proteins. Protein Eng., Des. Sel. 19, 555–562. [DOI] [PubMed] [Google Scholar]
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B.; Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644. [DOI] [PubMed] [Google Scholar]
- Syson K.; Stevenson C. E. M.; Rejzek M.; Fairhurst S. A.; Nair A.; Bruton C. J.; Field R. A.; Chater K. F.; Lawson D. M.; Bornemann S. (2011) Structure of a Streptomyces maltosyltransferase GlgE: A homologue of a genetically validated anti-tuberculosis target. J. Biol. Chem. 286, 38298–38310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter J. D.; Withers S. G. (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 4, 885–892. [DOI] [PubMed] [Google Scholar]
- Withers S. G.; Street I. P.; Bird P.; Dolphin D. H. (1987) 2-Deoxy-2-fluoroglucosides: A novel class of mechanism-based glucosidase inhibitors. J. Am. Chem. Soc. 109, 7530–7531. [Google Scholar]
- Withers S. G.; Rupitz K.; Street I. P. (1988) 2-Deoxy-2-fluoro-d-glycosyl fluorides. A new class of specific mechanism-based glycosidase inhibitors. J. Biol. Chem. 263, 7929–7932. [PubMed] [Google Scholar]
- Rempel B. P.; Withers S. G. (2008) Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology 18, 570–586. [DOI] [PubMed] [Google Scholar]
- Uitdehaag J. C. M.; Mosi R.; Kalk K. H.; van der Veen B. A.; Dijkhuizen L.; Withers S. G.; Dijkstra B. W. (1999) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat. Struct. Biol. 6, 432–436. [DOI] [PubMed] [Google Scholar]
- Numao S.; Kuntz D. A.; Withers S. G.; Rose D. R. (2003) Insights into the mechanism of Drosophila melanogaster Golgi α-mannosidase II through the structural analysis of covalent reaction intermediates. J. Biol. Chem. 278, 48074–48083. [DOI] [PubMed] [Google Scholar]
- Jensen M. H.; Mirza O.; Albenne C.; Remaud-Simeon M.; Monsan P.; Gajhede M.; Skov L. K. (2004) Crystal structure of the covalent intermediate of amylosucrase from Neisseria polysaccharea. Biochemistry 43, 3104–3110. [DOI] [PubMed] [Google Scholar]
- Lovering A. L.; Lee S. S.; Kim Y. W.; Withers S. G.; Strynadka N. C. J. (2005) Mechanistic and structural analysis of a family 31 α-glycosidase and its glycosyl-enzyme intermediate. J. Biol. Chem. 280, 2105–2115. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Li C. M.; Williams L. K.; Rempel B. P.; Brayer G. D.; Withers S. G. (2009) Directed “in situ” inhibitor elongation as a strategy to structurally characterize the covalent glycosyl-enzyme intermediate of human pancreatic α-amylase. Biochemistry 48, 10752–10764. [DOI] [PubMed] [Google Scholar]
- Guce A. I.; Clark N. E.; Salgado E. N.; Ivanen D. R.; Kulminskaya A. A.; Brumer H.; Garman S. C. (2010) Catalytic mechanism of human α-galactosidase. J. Biol. Chem. 285, 3625–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J.; Izumi A.; Ibatullin F. M.; Nakhai A.; Gilbert H. J.; Davies G. J.; Brumer H. (2011) Structural and enzymatic characterization of a glycoside hydrolase family 31 α-xylosidase from Cellvibrio japonicus involved in xyloglucan saccharification. Biochem. J. 436, 567–580. [DOI] [PubMed] [Google Scholar]
- Larsbrink J.; Izumi A.; Hemsworth G. R.; Davies G. J.; Brumer H. (2012) Structural enzymology of Cellvibrio japonicus Agd31B protein reveals α-transglucosylase activity in glycoside hydrolase family 31. J. Biol. Chem. 287, 43288–43299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozeboom H. J.; Yu S.; Madrid S.; Kalk K. H.; Zhang R.; Dijkstra B. W. (2013) Crystal structure of α-1,4-glucan lyase, a unique glycoside hydrolase family member with a novel catalytic mechanism. J. Biol. Chem. 288, 26764–26774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter J. D.; Adam M. J.; Braun C.; Namchuk M.; Tull D.; Withers S. G. (1993) Synthesis of 2-deoxy-2-fluoro monosaccharides and oligosaccharide glycosides from glycals and evaluation as glycosidase inhibitors. Carbohydr. Res. 249, 77–90. [DOI] [PubMed] [Google Scholar]
- Hehre E. J.; Kitahata S.; Brewer C. F. (1986) Catalytic flexibility of glycosylases: The hydration of maltal by β-amylase to form 2-deoxymaltose. J. Biol. Chem. 261, 2147–2153. [PubMed] [Google Scholar]
- Winter G. (2010) xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190. [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111. [Google Scholar]
- McNicholas S.; Potterton E.; Wilson K. S.; Noble M. E. M. (2011) Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D67, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetta P. A.; Alvarez L. J.; Remack P. S.; Candia O. A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Blanchet C. E.; Zozulya A. V.; Kikhney A. G.; Franke D.; Konarev P. V.; Shang W. F.; Klaering R.; Robrahn B.; Hermes C.; Cipriani F.; Svergun D. I.; Roessle M. (2012) Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Crystallogr. 45, 489–495. [Google Scholar]
- Franke D.; Kikhney A. G.; Svergun D. I. (2012) Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods Phys. Res., Sect. A 689, 52–59. [Google Scholar]
- Konarev P. V.; Volkov V. V.; Sokolova A. V.; Koch M. H. J.; Svergun D. I. (2003) PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. [Google Scholar]
- Guinier A. (1939) La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann. Phys. 12, 161–237. [Google Scholar]
- Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503. [Google Scholar]
- Porod G. (1982) General theory. In Small angle X-ray scattering (Glatter O., and Kratky O., Eds.) pp 17–51, Academic Press, London. [Google Scholar]
- Petoukhov M. V.; Franke D.; Shkumatov A. V.; Tria G.; Kikhney A. G.; Gajda M.; Gorba C.; Mertens H. D. T.; Konarev P. V.; Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D.; Barberato C.; Koch M. H. J. (1995) CRYSOL: A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773. [Google Scholar]
- Davies G. J.; Wilson K. S.; Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor E. A.; Janeček S.; Svensson B. (2001) Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta 1546, 1–20. [DOI] [PubMed] [Google Scholar]
- Martin J. L.; Johnson L. N.; Withers S. G. (1990) Comparison of the binding of glucose and glucose 1-phosphate derivatives to T-state glycogen phosphorylase-B. Biochemistry 29, 10745–10757. [DOI] [PubMed] [Google Scholar]
- Persson K.; Ly H. D.; Dieckelmann M.; Wakarchuk W. W.; Withers S. G.; Strynadka N. C. J. (2001) Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8, 166–175. [DOI] [PubMed] [Google Scholar]
- Breton C.; Fournel-Gigleux S.; Palcic M. M. (2012) Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 22, 540–549. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Vasella A.; Davies G. J.; Böhm M. (2002) Glycosidase mechanisms. Curr. Opin. Chem. Biol. 6, 619–629. [DOI] [PubMed] [Google Scholar]
- Takusagawa F.; Jacobson R. A. (1978) Crystal and molecular structure of α-maltose. Acta Crystallogr. B34, 213–218. [Google Scholar]
- Zechel D. L.; Withers S. G. (2000) Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. (2007) MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.