

Abstract
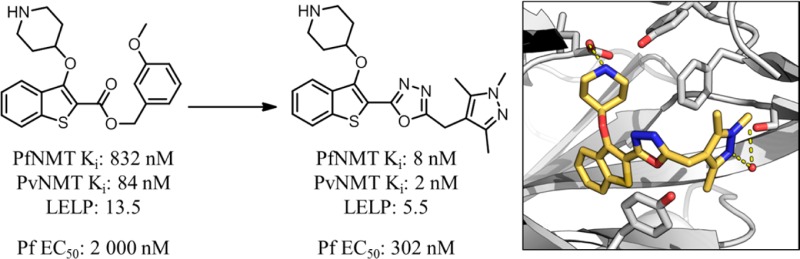
N-Myristoyltransferase (NMT) is an essential eukaryotic enzyme and an attractive drug target in parasitic infections such as malaria. We have previously reported that 2-(3-(piperidin-4-yloxy)benzo[b]thiophen-2-yl)-5-((1,3,5-trimethyl-1H-pyrazol-4-yl)methyl)-1,3,4-oxadiazole (34c) is a high affinity inhibitor of both Plasmodium falciparum and P. vivax NMT and displays activity in vivo against a rodent malaria model. Here we describe the discovery of 34c through optimization of a previously described series. Development, guided by targeting a ligand efficiency dependent lipophilicity (LELP) score of less than 10, yielded a 100-fold increase in enzyme affinity and a 100-fold drop in lipophilicity with the addition of only two heavy atoms. 34c was found to be equipotent on chloroquine-sensitive and -resistant cell lines and on both blood and liver stage forms of the parasite. These data further validate NMT as an exciting drug target in malaria and support 34c as an attractive tool for further optimization.
Introduction
Malaria is an infectious disease caused by parasites of the genus Plasmodium and is a world health crisis of paramount urgency. Malaria was responsible for over 200 million cases and 1 million deaths in 2010 alone,1 primarily affecting developing countries and children under the age of 5.2 Although five species of Plasmodium parasite are known to infect humans,2,3 two species are responsible for the majority of morbidity and mortality: Plasmodium falciparum (Pf) and Plasmodium vivax (Pv). These species are the focus of the work described in this paper.
The current treatment for malaria is combination therapy, typically comprising artemisinin derivatives and a companion drug such as lumefantrine, mefloquine, or amodiaquine. These drugs (and the majority of antimalarials)4,5 target only the symptomatic blood-stage forms of the parasite; drugs that target additional life stages (such as asymptomatic liver stage parasites) are in high demand.6 Furthermore, resistance to chloroquine is long established7 and signs of artemisinin resistance have been detected along the eastern8 and western borders9 of Thailand, compounding the urgent requirement for additional therapeutic agents targeting Plasmodium parasites.
Although there has been a great deal of funding and expertise directed toward antimalarial drug discovery over the past decade, the majority of therapeutics in clinical development are either elaborations of existing pharmacophores, reformulations/combinations of existing drugs, or novel molecules that function by unknown mechanisms of action.10 In order to combat resistance and achieve the goal of malaria eradication, a range of therapies targeting a variety of biological mechanisms and parasite life stages are required.
N-Myristoylation is the covalent attachment of myristate, a saturated 14-carbon fatty acid, to the N-terminal glycine of target proteins from the acyl source myristoyl-coenzyme A (CoA). This transformation is catalyzed by N-myristoyltransferase (NMT), a monomeric enzyme found ubiquitously in eukaryotes. Myristoylation is a widespread modification that occurs co- and post-translationally and has hundreds of putative substrates in vivo.11 Furthermore, myristoylation of proteins is known to modulate a variety of properties (such as protein localization12 and stability13) and is implicated in a variety of critical biological pathways.14,15
In the specific context of malaria, several important parasitic proteins have been shown to require myristoylation in order to localize correctly and to carry out their biological functions,16−18 and the genetic essentiality of NMT in P. berghei (Pb), the infectious species in the murine model of malaria, has been confirmed by conditional knockdown experiments.19 Furthermore, we recently reported the validation of NMT as an essential and chemically tractable drug target in P. falciparum. Using an integrated chemical biology approach, we identified the NMT substrate proteins in the blood stage of the parasite and demonstrated with small molecule tools that on-target inhibition of myristoylation disrupts the function of multiple specific downstream pathways, resulting in rapid cell death.20 Herein, we describe the development of a previously described series of benzo[b]thiophene inhibitors into 34c, a potent and selective parasite NMT inhibitor instrumental to the validation of this drug target. We also report further investigations into the utility of NMT inhibitors in drug-resistant cell lines and liver stage parasites.
Results and Discussion
Previous work reported discovery of a series of benzo[b]thiophene-based inhibitors of P. falciparum (Pf)NMT and P. vivax (Pv)NMT, exemplified by 1 (Figure 1).211 represents a promising starting point for hit to lead development but has only moderate enzyme affinity and high lipophilicity and contains a potentially metabolically labile ester group. Further development therefore focused on removal of this high-risk functionality combined with a 100-fold improvement in enzyme affinity, reduced lipophilicity, and controlled molecular weight. Little is currently known of the potential for toxicity resulting from mammalian NMT inhibition, and previous data have shown that a potent Homo sapiens (Hs)NMT inhibitor is not toxic to mice at high doses.22 Although selectivity over HsNMT is desirable, selectivity at the cellular level was considered the more critical determinant for progression.
Figure 1.
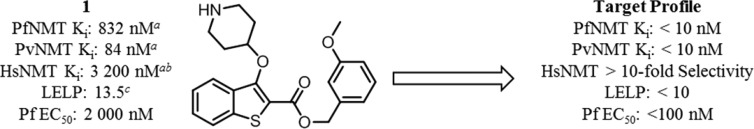
2,3-Substituted benzo[b]thiophene PfNMT and PvNMT inhibitor 1 and the target profile for the development of this series. Footnote a: Ki values are quoted in place of IC50 values as a means of expressing the inhibitor affinity while correcting for differing Michaelis constants (Km) between enzymes. Enzyme Ki values are calculated from the IC50 values using the Cheng–Prusoff equation, the definition of which is given in the Experimental Section.23 IC50 values are the mean value of two or more determinations, and standard deviation is within 20% of the IC50. Footnote b: No significant difference in inhibition between HsNMT1 and HsNMT2 isoforms has been observed in this series; therefore, the HsNMT affinities reported in this work refer to HsNMT1. Footnote c: LELP = cLogP/LE. LE = [−log(Ki)](1.374)/(no. of heavy atoms), with cLogP determined with ChemAxon, which can be obtained from http://www.chemaxon.com/products/calculator-plugins/logp/.
The metric ligand efficiency (LE) has been used extensively in early stage drug development, and its utility has been well documented.24 The main criticism of LE is the omission of any reference to the lipophilicity of the compound in question. Lipophilicity has been implicated as one of the most important properties of a potential drug molecule; it is critical to many related properties including solubility, permeability, and distribution.25 In vivo, highly lipophilic compounds will be more likely to partition from plasma to organic components such as membranes and proteins, increasing biological promiscuity (and in turn toxicity).26 Furthermore lipophilicity tends to inflate throughout drug development.27 Indeed, a recent review on metrics in drug discovery has stated “optimizing LE and LLE (ligand lipophilic efficiency, pIC50 – log P) in concert is an important success factor for hit and lead optimization in drug discovery projects”.28
Ligand efficiency dependent lipophilicity (cLogP/LE, LELP) has been proposed as a way to incorporate affinity, lipophilicity, and molecular size into a single metric for the triage of molecules29 and has been shown to be a strong predictor of druglikeness.30 It is proposed that a low LELP will reduce the chance of later stage failure due to nonspecific toxicity; marketed drugs have an average LELP of approximately 6, and desirable leads (LE > 0.3, cLogP < 3) display LELP < 10.29 In order to control the lipophilicity and molecular weight of the current series, LELP was used to determine the druglikeness or physicochemical quality of the inhibitors tested. For clarity, all LELP values quoted in this work refer to PfNMT LE. Consideration of the LELP of 1 (13.5, Figure 1) emphasizes the requirement for significant improvement in the physicochemical profile of this series.
Previous work showed that an appropriately placed 3-methoxyphenyl substituent can form π–π and hydrogen bonding interactions with Phe105 and Ser319 of PvNMT, respectively.31 Attempts to replicate these interactions within the benzothiophene series yielded only a small affinity enhancement, but previously reported crystallographic evidence indicated that extension of the linker between the ester and methoxyphenyl moieties might result in improved interactions.21 A series of phenethyl esters and amides were therefore synthesized and assayed against a panel of NMTs (Scheme 1, Table 1).
Scheme 1. Synthesis of Phenethyl Esters and Amides.

Reagents and conditions: (a) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, hydroxybenzotriazole, N,N-diisopropylethylamine, MeCN, rt, 18 h, 36–57%; (b) benzotriazo-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate, N,N-diisopropylethylamine, DCM, rt, 18 h, 69–93%; (c) 10% TFA in DCM (v/v), rt, 2 h, 42–97%. 2 was prepared as described previously.21
Table 1. Enzyme Affinity and Plasmodium falciparum LELP for Phenethyl Esters and Amides.
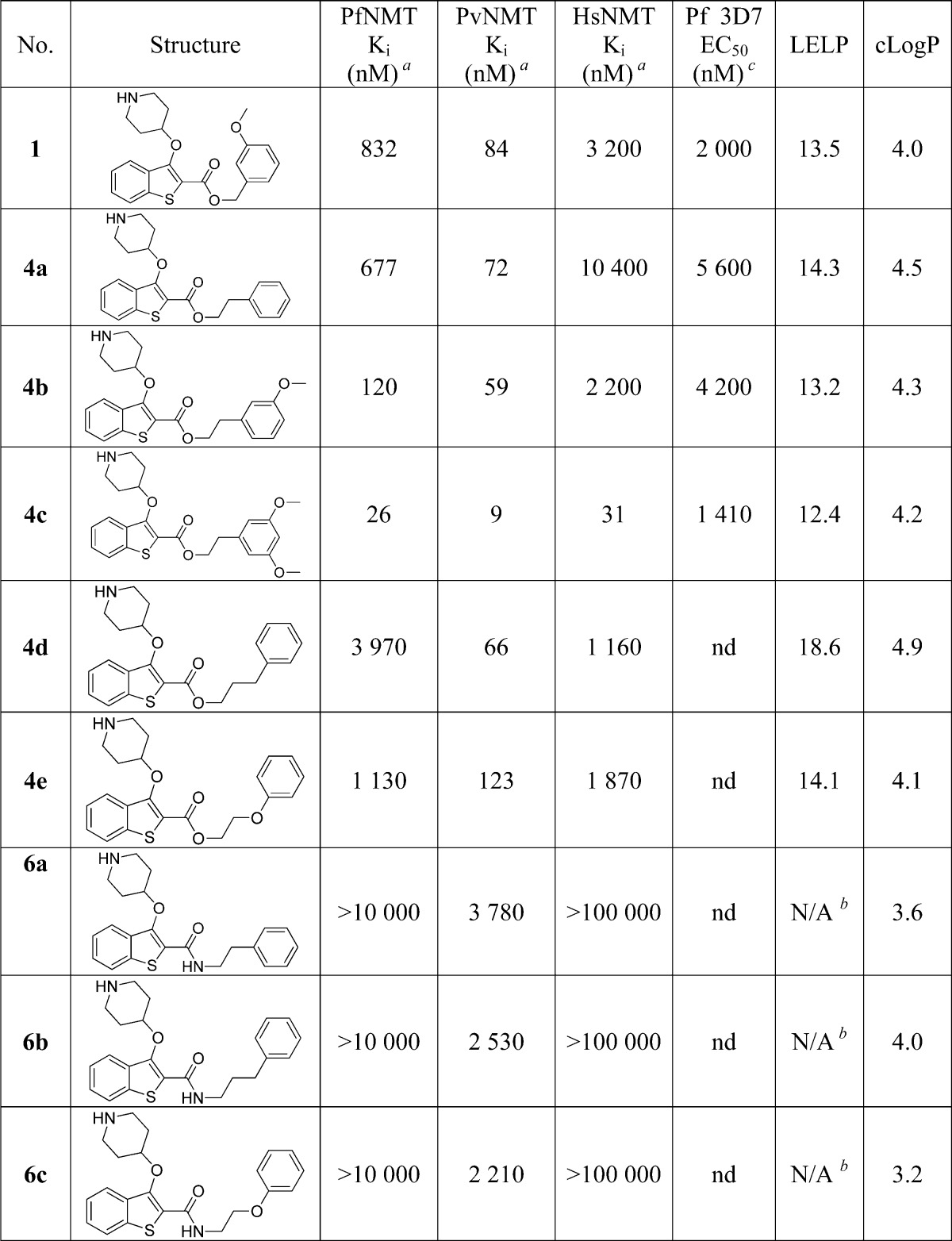
Ki values are the mean value of two or more determinations, and standard deviation is within 20% of the Ki.
LELP not calculated as enzyme affinity above measurable range.
3D7 EC50 values are determined using a Sybr Green fluorometric assay.
This hypothesis was successfully validated, in that moving from 1 to 4b produced a 6-fold improvement in PfNMT affinity, retained the high PvNMT affinity, and pleasingly had little effect on HsNMT affinity. However, despite this improvement, the accompanying lipophilic methylene unit resulted in only a marginally improved LELP. Furthermore, the antiparasitic activity of 4b was reduced compared to 1.
Further SAR mimicked that described previously for benzyl esters:31 adding a second hydrogen bond acceptor (4c) significantly improved affinity but removed all selectivity, resulting in a compound with high affinity against all three enzymes. Esters are strongly preferred over amides (4a vs 6a, 4d vs 6b, 4e vs 6c), and the active site for PvNMT appears to be more promiscuous than that of PfNMT and HsNMT. Intriguingly, increasing the linker length by a further methylene (4a vs 4d) reduced affinity for PfNMT, maintained affinity for PvNMT, and increased affinity for HsNMT. This reinforces the requirement for the ethyl spacer and indicates that selectivity is achievable through interactions with residues in this area of the enzyme. It is notable that the antiparasitic activity of these compounds is slightly reduced, with only the high affinity 4c displaying improved activity compared to 1. It may be that the increased lipophilicity of this series has reduced the free fraction available for binding. 4c also displays the best LELP score of any analogues tested; further development shall continue to focus on targeting a LELP of <10.
Ester Bioisosteric Replacement
The increased linker length between the phenyl moiety and the ester presented an excellent opportunity to investigate replacing the ester moiety with a heterocycle. In earlier analogues this modification was not explored, since it would have resulted in four contiguous aromatic rings, with likely deleterious effects on the solubility and toxicity profile. However, the incorporation of an extra methylene unit adds an extra degree of freedom and interrupts the π-system, eliminating this potential issue. Crystallography suggested that the interactions of the ester with the enzyme were confined to long-distance π-interactions with Tyr211 and Phe105. In this scenario oxadiazoles are common bioisosteric replacements for methylene esters,32 displaying increased biological stability but with similar size and shape and increased potential for π-interactions. Both 1,2,4-regioisomers were prepared using complementary routes (Scheme 2).
Scheme 2. Synthesis of Two Regioisomers of 1,2,4-Oxadiazole Bioisosteres.

Reagents and conditions: (a) NH2OH·H2O, EtOH, rt, 5 h, 98–99%; (b) 2, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, hydroxybenzotriazole, DMF, 140 °C, 3 h, 10–31%; (c) 10% TFA in DCM (v/v), rt, 2 h, 13–99%; (d) bromoacetonitrile, t-BuOK, THF, 0 °C to rt, 15 min, 88%; (e) tert-butyl 4-hydroxypiperidine-1-carboxylate, diisopropyl azodicarboxylate, PPh3, THF, rt 1.5 h, 78%; (f) RCH2CO2H, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, hydroxybenzotriazole, MeCN, rt, 18 h; (g) 4 Å molecular sieves, toluene, 18 h, 110 °C, 54–68% over two steps.
Replacement of the ester linker with a 1,2,4-oxadiazole produced an increase in affinity for all three enzymes, presumably due to π-interactions within the active site and/or increased rigidification of the linker with respect to 4b. A single methylene between the oxadiazole linker and the phenyl moiety was preferred (9a vs 9b). Introduction of a m-methoxy substituent improved affinity, although only by a small degree (9a vs 9c, 14a vs 14b), and replacement of the methoxy group with a weakly hydrogen bonding and lipophilic chlorine reduced affinity for all three enzymes (9c vs 9d). Interestingly, in 4c addition of the second methoxy substituent produced an increase in affinity against PfNMT and HsNMT, whereas in 9e selectivity is improved with respect to 9c. Although this result was quite promising, the solubility of 9e is very low (data not shown), limiting the potential of this substitution pattern in further development. As with the ester extension, this optimization is accompanied by an increase in heavy atom count resulting in only a small increase in LELP, still above the target value of 10. Nonetheless, both isomers of 1,2,4-oxadiazoles appear to be promising bioisosteres for the potentially problematic ester functionality.
Improvements in LELP: Capitalizing on Matched Molecular Pairs Analysis
The developments detailed above improved enzyme affinity and removed a potentially labile ester group, making the compound series significantly more druglike. A consideration of LELP across the series highlights that the increase in enzyme affinity from 1 to 9c has been achieved at the expense of increased lipophilicity and molecular weight. This results in only a small improvement in LELP from 13.5 to 12.5, still substantially above the desired value of 10 for a promising lead. The optimization of biological potency at the expense of molecular properties is a common pitfall in drug development that has been associated with clinical attrition.29 The next stage in development therefore focused on improving the LELP of the series by improving affinity while reducing lipophilicity and molecular weight.
Several strategies for lowering lipophilicity have been described in the recent literature; one of the most successful is matched molecular pairs analysis,33 the concept of replacing a moiety with a “matched pair”, a structural analogue that displays favorable physicochemical properties. One of the most studied (and striking) examples is the replacement of 1,2,4-oxadiazoles with the symmetrical 1,3,4-oxadiazole isomer.34,35 Because of the contrasting dipole moment of each heterocycle, 1,3,4-oxadiazoles display on average 10-fold lower lipophilicity and more favorable profiles with respect to metabolic stability, hERG inhibition, and aqueous solubility.35 Switching to this matched pair thus represented an attractive opportunity to lower lipophilicity and improve physicochemical properties by making a minimal structural change to the series, crucially without adding molecular weight. The mixed hydrazide intermediate 17a/b was synthesized readily from ethyl ester 15, and this was converted into the 1,3,4-oxadiazole analogue 20a/b, and additionally the 1,2,4-triazole analogue 18 from 17a (Scheme 3). Although less well studied than 1,3,4-oxadiazoles, 1,2,4-triazoles are known to have a lower log D than 1,2,4-oxadiazoles,36 as well as a higher aromatic stabilization energy.37
Scheme 3. Synthesis of 1,3,4-Oxadiazole and 1,2,4-Triazole Linker Bioisosteres.

Reagents and conditions: (a) NH2NH2·H2O, EtOH, 78 °C, 24 h, 75%; (b) RCH2C(O)Cl, N,N-diisopropylethylamine, DCM, rt, 15 min, 75–91%; (c) POCl3, 100 °C, 1 h; (d) ammonium acetate, acetic acid, 140 °C, 1.5 h, 6% over two steps; (e) TsCl, 1,2,2,6,6 pentamethylpiperidine, DCM, rt, 3 h, 48–65%; (f) TFA, DCM, rt, 2 h, 55–98%.
Triazole 18 displayed a disappointing affinity profile, with no change in affinity for HsNMT but a decrease in affinity against the NMTs of the two parasite species (9a, 14a vs 18, Table 2). Nevertheless, an accompanying decrease in cLogP resulted in a significant improvement in the LELP for 18, 9.5 compared to 12.4 and 12.8 for 9a and 14a, respectively. This scaffold is therefore likely to have improved aqueous solubility and druglike properties compared to the 1,2,4-oxadiazoles, which will need to be considered in the future development of this series.
Table 2. Enzyme Affinity and LELP of Bioisosteric Heterocycle-Linked Inhibitors.
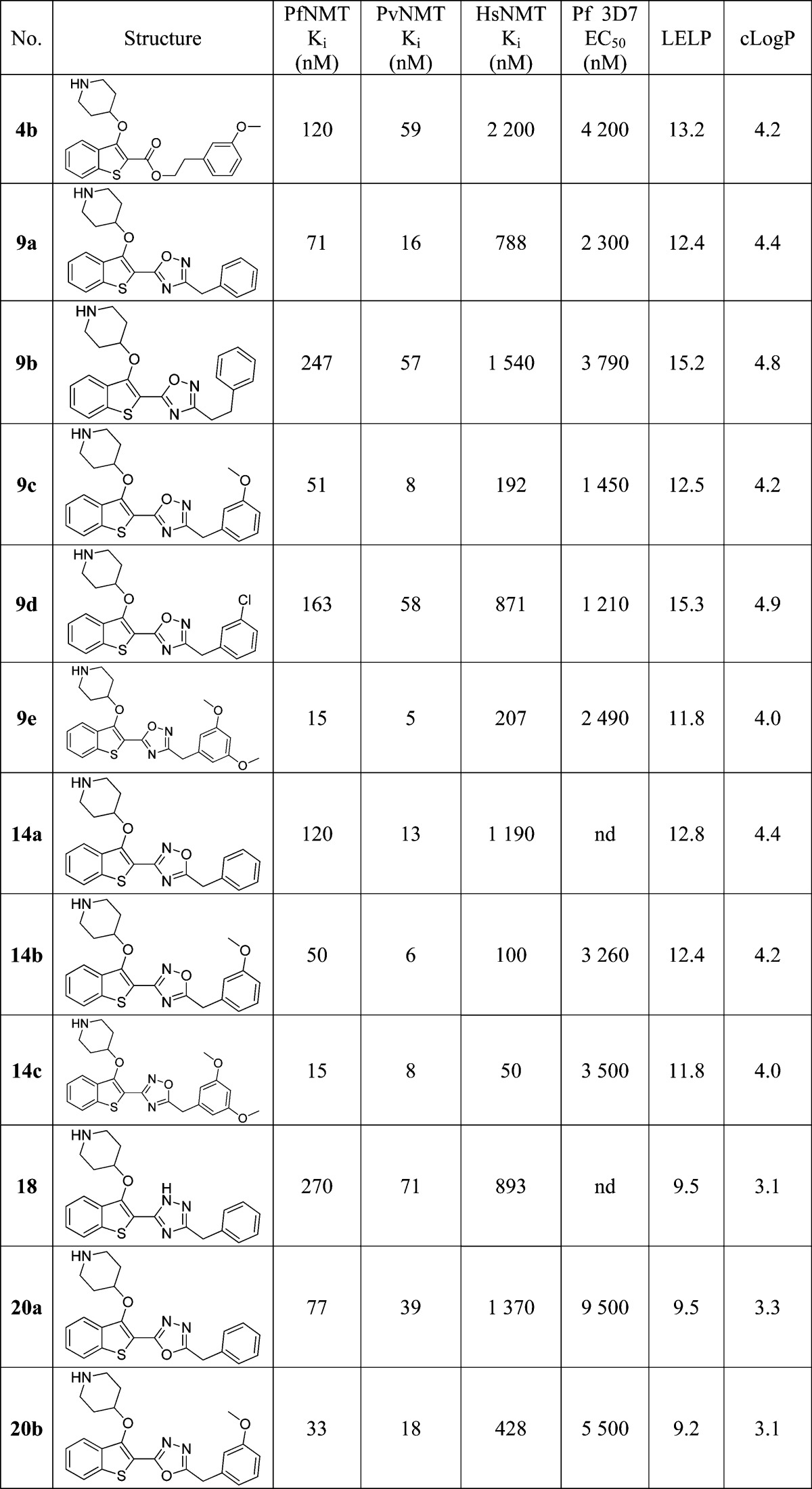
As might be expected from the small difference in affinity between the 1,2,4-oxadiazole regioisomers (for example, 9c vs 14b), moving to the 1,3,4-oxadiazole 20a/b had only a small effect on Pf/Pv NMT affinity (Table 2). However, this modification unexpectedly decreased HsNMT affinity (20b vs 9c, 14b), resulting in a 13-fold selectivity window between PfNMT and HsNMT, 24-fold between PvNMT and HsNMT. In addition to the improved selectivity profile, 20a/b has a significantly lower cLogP than 9a/c and 14a/b, resulting in a greatly improved LELP. Indeed, 18, 20a, and 20b all display LELP within the target range for promising lead compounds (LELP < 10).
Crystallography confirmed the binding mode of 20b in the peptide substrate pocket of PvNMT (Figure 2A, PDB entry 4CAE), showing that the methoxyphenyl moiety makes hydrophobic contacts with Phe105 and a polar contact with Ser319, in addition to the interactions observed previously for 1.21 Furthermore, the oxadiazole is “sandwiched” between the aromatic residues Phe105 and Tyr211, potentially explaining the affinity improvement observed when the ester in 4b is replaced by an oxadiazole in 9c/14b (Figure 2B).
Figure 2.
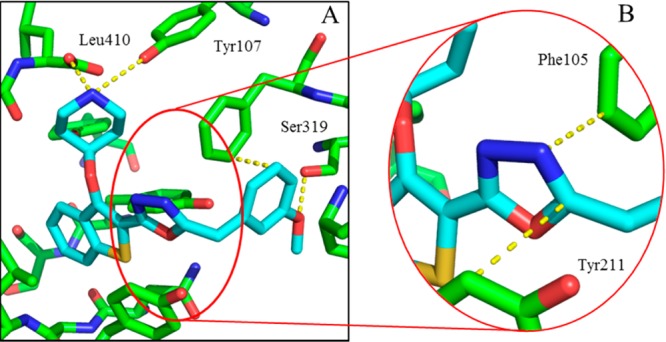
X-ray crystal structure of 20b (blue) bound to PvNMT (green). (A) 20b bound to PvNMT. The 3-methoxyphenyl substituent forms the intended interactions with Ser319 and Phe105, in addition to the deeply buried hydrophobic scaffold and salt bridge interaction observed in 1. (B) The oxadiazole linker is sandwiched between two aromatic residues, rationalizing the affinity enhancement in moving to an aromatic heterocycle from the ester linker in 4b. Dashed lines are drawn to highlight key interactions between the enzyme and the ligand.
Improvements in LELP: Investigations of the Solvated Pocket
The crystal structure of 20b bound to PvNMT indicated that there was a voluminous solvent-filled pocket surrounding the methylene group α to the methoxyphenyl, inviting the introduction of functionality that could either stabilize the water molecules in this pocket or displace water to the bulk solvent (Figure 3).38,39 In addition, this position is a potential target for oxidative metabolism and blocking with alternative substituents may provide benefits during subsequent lead development.
Figure 3.
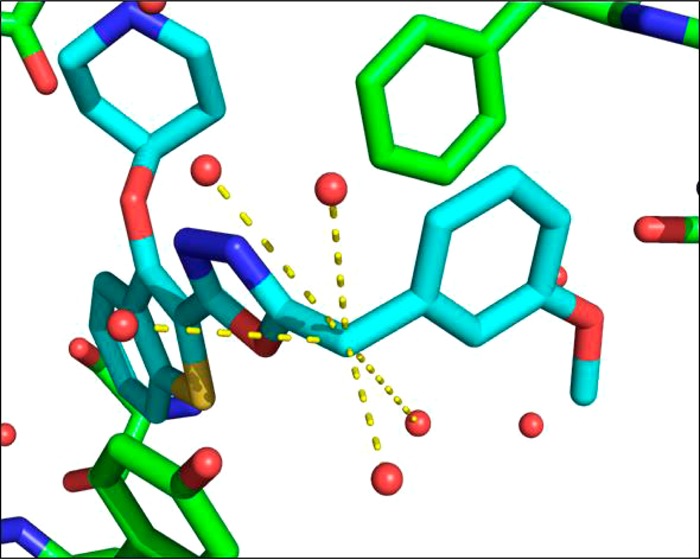
X-ray crystal structure of 20b (blue) bound to PvNMT (green). Further inspection of the water molecules within the active site shows that the benzylic CH2 occupies a heavily solvated pocket, indicating that substitution may result in more favorable energetics within the enzyme active site. Dashed lines indicate water molecules within 5 Å of the benzylic position.
The abundance of readily available α-substituted phenylacetic acids meant this strategy could be explored extensively, using previously developed chemistry: a variety of substitutions were introduced as shown in Scheme 4 and Table 3.
Scheme 4. Synthesis of α-Substituted Phenyl 1,3,4-Oxadiazole Inhibitors.

Reagents and conditions: (a) Ph-CR1R2-CO2H, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, hydroxybenzotriazole, THF, DMF, rt, 18 h, 48–99%; (b) TsCl, 1,2,2,6,6pentamethylpiperidine, DCM, rt, 18 h; (c) 10% TFA in DCM (v/v), rt, 2 h, 4–40% over two steps.
Table 3. Enzyme Affinity and LELP of α-Substituted Phenyl 1,3,4-Oxadiazole Inhibitors.
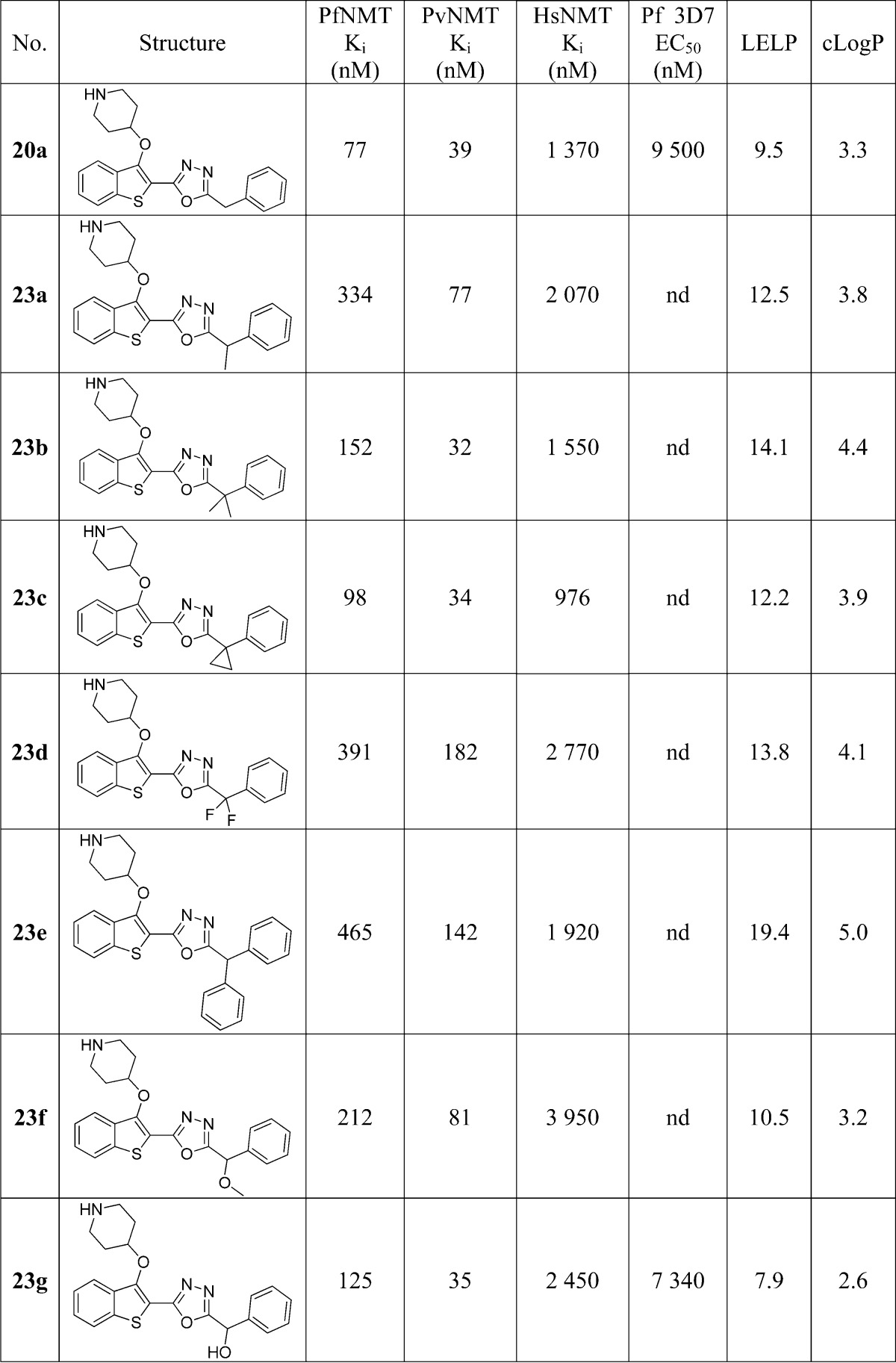
The results show that a wide variety of substitutions at this position produce only small changes in enzyme affinity, reinforcing the notion that there is flexibility in the binding pocket. Of the hydrophobic substituents, cyclopropyl results in the highest affinity: thus, 23c retains the activity of 20a but with a concomitant increase in LELP. The most significant result is 23g, which is approximately equipotent to 23a with the addition of a polar hydroxyl group. This increases the hydrophilicity and decreases the LELP but increases the molecular complexity (with the addition of a stereocenter) and introduces a further hydrogen bond donor (which may adversely affect membrane permeability). Weighing these detriments against the moderate improvements these modifications provided, the decision was made to progress development with the unsubstituted template as in 20a/b with the knowledge that functionality could be reintroduced during future lead development should a solubilizing/metabolism blocking group be required.
Improvements in LELP: Optimization of Polar Contacts
The next area of interest was modification of the methoxyphenyl portion of the molecule. The rationale for this change was twofold: (i) the methoxyphenyl portion contributes significantly to the lipophilicity of these molecules and, as such, presents a target for improvement of LELP;40 (ii) aryl ethers are notoriously poor H-bond acceptors,41 and so replacement may result in a stronger hydrogen bond with Ser319 and an improvement in affinity.
Brand et al. previously described the development of a series of highly potent Trypanosoma brucei NMT inhibitors42 that form a hydrogen bond to the conserved Ser319 (Ser330 in Leishmania major NMT) residue, for example, via a 1,3,5-trimethylpyrazole moiety (PDB entry 2WSA). Building on this observation, we selected three distinct heterocycles to replace the methoxyphenyl substituent (Scheme 5) on the basis that nitrogen atoms conjugated within π-systems are typically excellent hydrogen bond acceptors,41 and these moieties would have reduced lipophilicity relative to the methoxyphenyl parent compound (Table 4). The binding mode in PvNMT (Figure 2) indicated that the heterocycles in molecules 34a–c (bearing a methylene linker) would not make direct contacts to the desired residues. For this reason, compounds 35a–c with an extended two-carbon linker were also synthesized in the expectation that this would place the heterocycle directly adjacent to Ser319, albeit with the entropic and lipophilic penalty associated with a longer alkyl chain.
Scheme 5. Synthesis of Five-Membered Heterocyclic Methoxyphenyl Replacements.

Reagents and conditions: (a) NaH, ethyl bromoacetate, THF, 0 °C, 18 h, 78%; (b) methyl 3-bromopropionate, K2CO3, DMF, 55 °C, 18 h, 30%; (c) NH2NH2·H2O, MeOH, rt, 3 h, 83–99%; (d) n = 1, NH2OH·HCl, K2CO3, EtOH, 78 °C, 3 h, 12%; n = 2, NH2OH·HCl, H2O, MeOH, 60 °C, 18 h, 89%; (e) MeNHNH2, AcOH, 3 h, rt, 73–95%; (f) LiOH·H2O, MeOH, rt, 18 h, 51–95%; (g) 16, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, hydroxybenzotriazole, THF, DMF, rt, 18 h, 48–99%; (h) TsCl, 1,2,2,6,6-pentamethylpiperidine, DCM, rt, 18 h; (i) 10% TFA in DCM (v/v), rt, 2 h, 3–26% over two steps.
Table 4. Enzyme Affinity and LELP of Heterocyclic Methoxyphenyl Replacements.
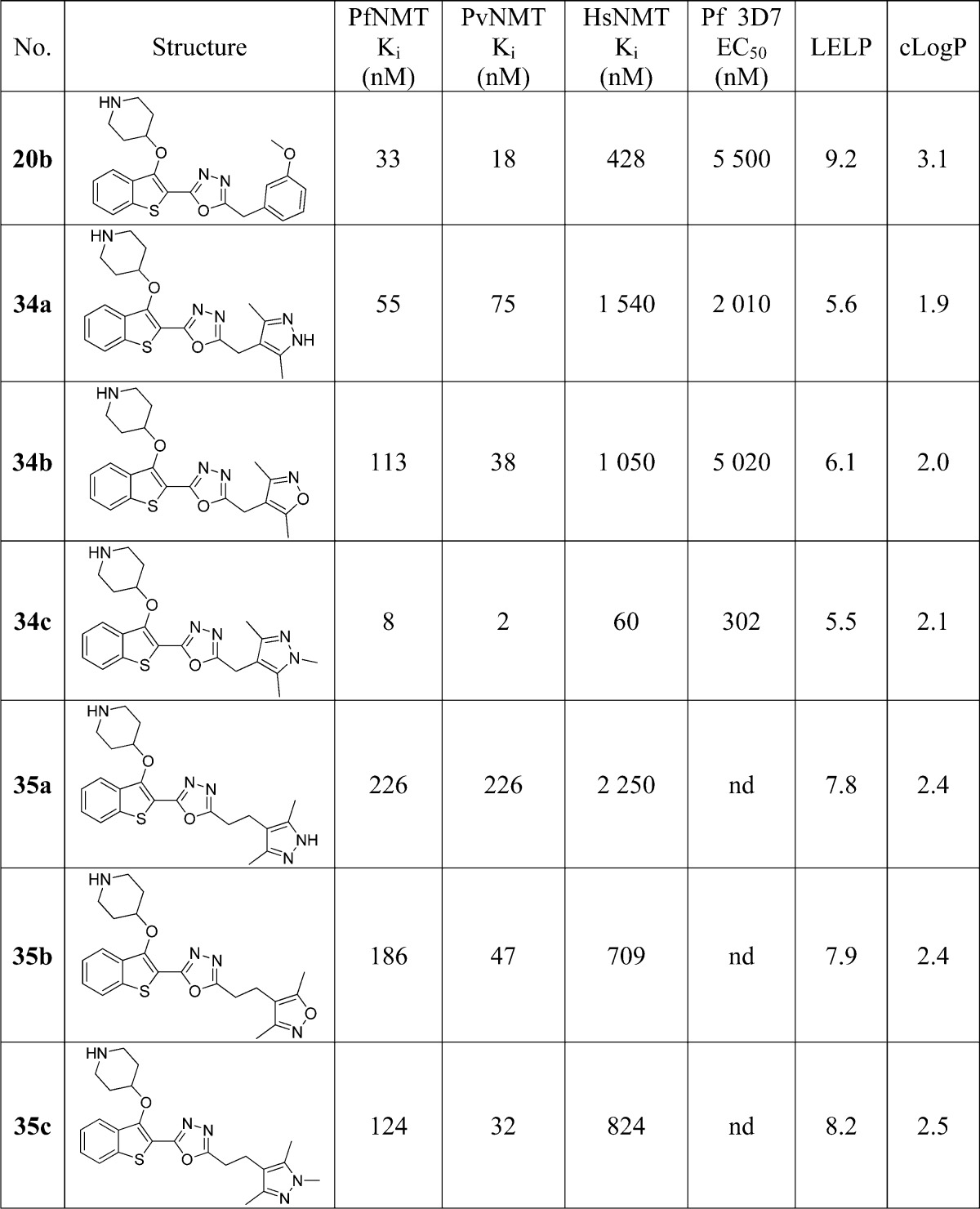
The SAR obtained across these analogues was unexpected: in all cases PvNMT and PfNMT had higher affinity for the methylene linked 34 series, although this was not always the case for HsNMT (34b vs 35b). Of the three heterocycles, isoxazoles (34b and 35b) and 1H-pyrazoles (34a and 35a) had decreased affinity compared to the methoxyphenyl parent 20b. Interestingly, 34a displays 30-fold selectivity between PfNMT and HsNMT, a significant improvement over the 8- to 9-fold observed in 34b/34c.
Addition of the N-methyl group in 34c produces a marked increase in affinity against all enzymes, resulting in the highest affinity Plasmodium spp. NMT inhibitor reported to date with single-digit nanomolar Ki against both enzymes. Furthermore, this compound displays nanomolar efficacy against the parasite, allowing further biological evaluation of this drug target (vide infra). The affinity enhancement imparted by the methylpyrazole is accompanied by a reduction in cLogP (relative to methoxyphenyl 20b) resulting in an excellent LELP of 5.5, close to the reported average LELP of marketed drugs.29 Consequently, this compound has excellent druglike properties; we have previously reported the excellent aqueous solubility (>8 mM) and in vitro pharmacokinetic properties of this molecule.20 Although the selectivity between PfNMT and HsNMT is 8-fold (30-fold vs PvNMT), comparison to 34a shows that significant changes in the selectivity profile may potentially be achieved by altering the interactions with this binding pocket.
The binding mode of 34c was elucidated by crystallography, as briefly described previously20 (Figure 4, PDB entry 2YNE). As predicted during the compound design process, the pyrazole of 34c does not directly interact with the target Ser319, indicating a subtler basis for the affinity improvement. The pyrazole N-2 interacts indirectly with Ser319 via a water molecule, with the methyl groups occupying hydrophobic regions of the pocket (Figure 4B). The improved enzyme affinity is likely a result of the strong pyrazole–water hydrogen bond combined with fit complementarity within the binding site. In addition, the compound makes all the interactions observed with previous members of the series, including the salt bridge interaction with Leu410/Tyr107, a sandwiched oxadiazole linker, and deeply buried benzothiophene scaffold (Figure 4A).
Figure 4.
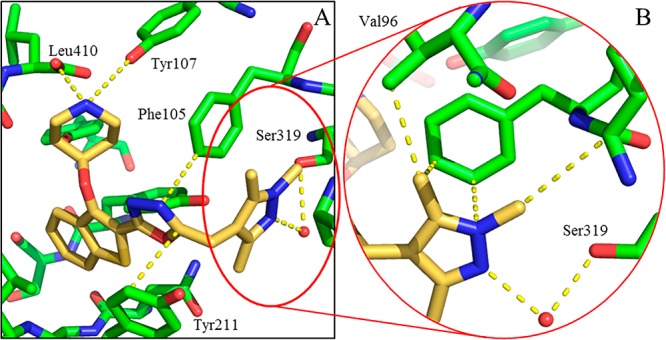
Binding mode of 34c (gold) bound to PvNMT. (A) 34c (gold) bound to PvNMT (green), showing piperidine–Leu410 salt bridge interaction, deeply buried benzothiophene scaffold, and 1,3,5-trimethylpyrazole heterocycle bound within the Ser319 hydrophobic pocket. (B) Enlarged view of the 1,3,5-trimethylpyrazole of 34c (gold) with PvNMT (green). This shows the water-bridged interaction between the pyrazole and Ser319, as well as multiple hydrophobic contacts between the heterocycle and the binding pocket.
The binding mode of 34a in PvNMT was also determined by crystallography (Figure 5A, PDB entry 4CAF). This compound displays a ∼40-fold lower binding affinity to PvNMT than 34c; however, the binding modes of these two compounds are extremely similar (Figure 5B). The only point of differentiation is the pyrazole heterocycle which forms a direct polar interaction with Ser319 as opposed to the water-bridged interaction observed in Figure 4B. Indeed, this water molecule is excluded from the pocket occupied by 34a, perhaps indicating that it is involved in stabilizing Ser319 and therefore that its expulsion to the bulk solvent is energetically unfavorable.
Figure 5.
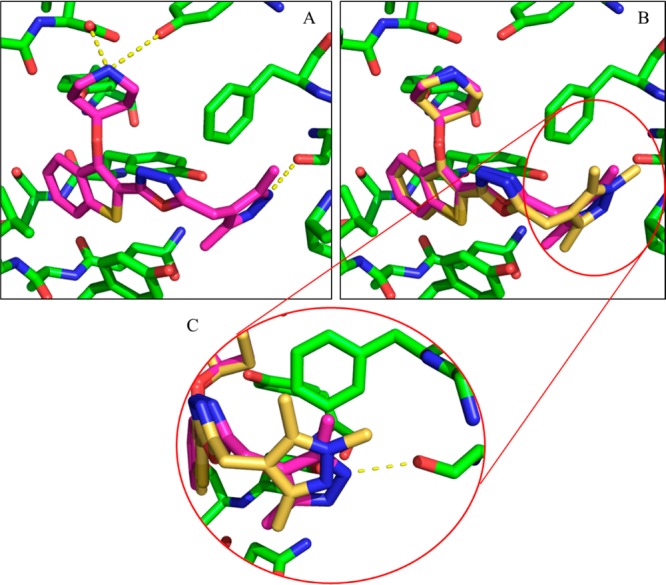
Binding mode of 34a (pink) bound to PvNMT. (A) 34a (pink) forms all previously observed interactions with the enzyme. (B) Comparison of the binding modes of 34a and 34c reaffirms this similarity, showing that the only point of differentiation is in the Phe105/Ser319 binding site occupied by the pyrazole. (C) Enlarged view of the 1,3,5-trimethylpyrazole of 34c (gold) and 3,5-dimethylpyrazole of 34a (pink) with PvNMT (green). The pyrazole of 34a forms a direct interaction with Ser319 (3.2 Å), and the water involved in the bridged interaction in Figure 4B has been excluded from the pocket.
Blood and Liver Stage in Vitro Cellular Activity
34c provides excellent enzyme affinity and LELP, and the cellular efficacy enabled further biological evaluation of this inhibitor. 34c was tested against a variety of parasites in vitro, including both drug-resistant cell lines and liver stage parasites, and for mammalian cell toxicity against human hepatocellular carcinoma cell line HepG2. Activity was directly compared to chloroquine (36) and atovaquone (37) where relevant.
3D7 and NF54 are chloroquine-sensitive strains of the parasite, whereas K1 and Dd2 are chloroquine-resistant strains, as evidenced by the greatly reduced potency of this drug against these strains (Table 5). 34c was previously shown to act on-target in blood stage drug-sensitive (3D7) parasites.20 Pleasingly, 34c is active against an additional drug-sensitive line (NF54) and there is very little loss of potency for drug-resistant cell lines (less than 4-fold between NF54 and Dd2), indicating that NMT inhibition may be an effective mechanism to target drug-resistant parasites in a clinical context.
Table 5. In Vitro Cellular Assay Data for 34c against Both Parasitic and Human Cells, Compared to Chloroquine (36) and Atovaquone (37).
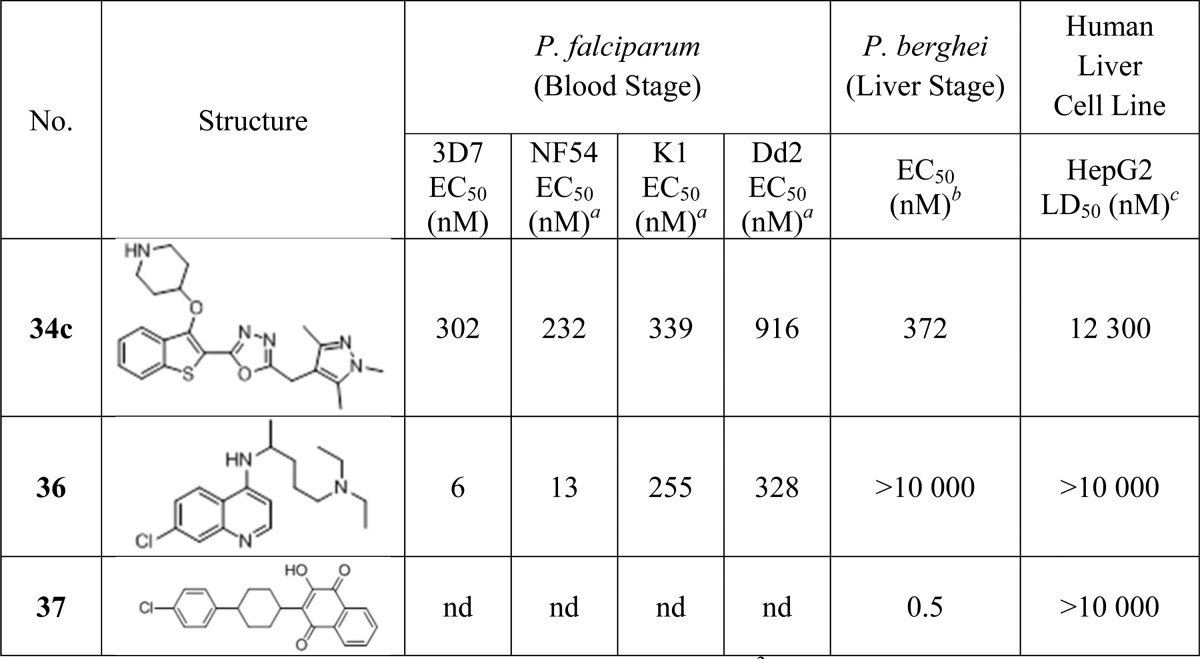
NF54, K1, and Dd2 EC50 values are determined using a [3H]hypoxanthine incorporation assay (performed by Dr. Sergio Wittlin and Dr. Christian Scheurer, Swiss Tropical and Public Health Institute).
P. berghei liver stage EC50 values determined using a luciferase bioluminescence assay. EC50 value is the mean of two determinations. Standard deviation is within 50% of the EC50.
LD50 values determined using an MTS cellular viability assay. LD50 value is the mean of six determinations. Standard deviation is within 20% of the LD50.
In addition, 34c was tested against a Plasmodium berghei (Pb) liver stage model to determine whether NMT is likely to be a viable drug target for clearance of liver stage parasites. The PbNMT enzyme affinity of 34c was determined at 14 nM, validating an approximate comparison with the Pf blood stage models. 34c had a liver stage EC50 of 372 nM, which, although significantly lower than the atovaquone standard, is in excellent agreement with the blood stage results. This provides evidence that NMT inhibition is also a promising mechanism for targeting liver stage parasites and may therefore be an effective pathway for the disruption of multiple parasite life stages.
Lastly, 34c displays up to ∼40-fold selectivity between parasitic and HepG2 cell lines (14-fold in the case of Dd2). This represents an encouraging window for a compound with 10-fold enzyme selectivity: if HepG2 cytotoxicity is due to NMT inhibition, then a more selective compound will have a larger window, and if the lethality is nonspecific, then the cell potency and toxicity appear sufficiently decoupled to be optimized independently. The Medicines for Malaria Venture dictates that validated hit compounds should display cellular selectivity of greater than 10-fold, whereas preclinical candidates should be >100-fold selective.43 With this target in mind, 34c displays reasonable selectivity for a lead compound: a further 3-fold improvement in efficacy without accompanying toxicity will achieve the required window for a candidate molecule. Note that the moderate selectivity is a functional of low efficiacy rather than high toxicity; the LD50 for 34c is approximately the top concentration tested (10 μM) for the antimalarial standards.
Effects of LELP on Cellular Activity
The metric LELP was used to guide the development of this compound series, based on the hypothesis that compounds with a low LELP (ideally <10) will have fewer off-target effects and as a result are more druglike.29 In an attempt to assess if LELP is a useful metric in this regard, the cellular EC50 against the 3D7 parasite line was plotted against NMT affinity for all tested members of this series. Analysis of the EC50/Ki correlation is shown in Figure 6.
Figure 6.
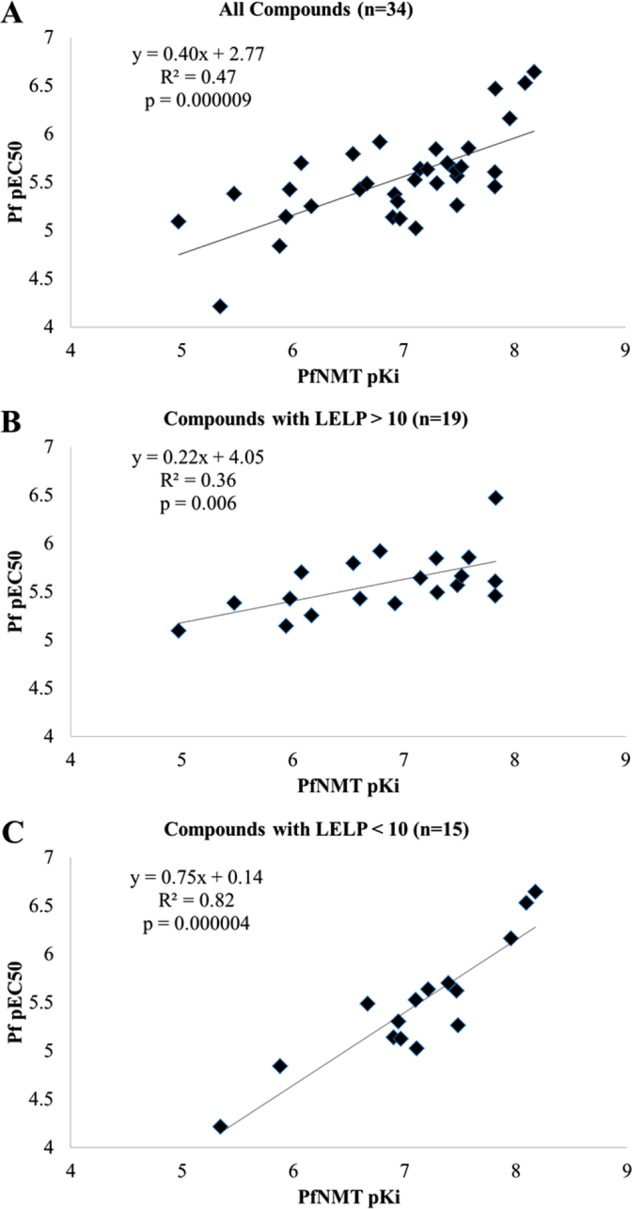
(A) Plot of cellular potency vs enzyme affinity for all members of this series tested in both assays. (B) Plot of cellular potency vs enzyme affinity for all compounds with a LELP of >10. (C) Plot of cellular potency vs enzyme affinity for all compounds with a LELP of <10.
An initial assessment of cellular potency vs enzyme affinity showed only a weak correlation (R2 = 0.47) between these two parameters (Figure 6A) and a relatively flat gradient (0.40). However, when the compounds were separated into those with a leadlike LELP score (LELP < 10) and those without (LELP > 10), a qualitative assessment showed an improved correlation for those compounds with a low LELP value (Figure 6C vs Figure 6B). Figure 6C shows that for those compounds with LELP < 10 there is a strong correlation between cellular potency and enzyme affinity (R2 = 0.82) and a gradient significantly closer to 1 than in the full data set (0.75 vs 0.40). In contrast, for compounds with a LELP of >10 there is a very weak correlation between EC50 and Ki (R2 = 0.36) and a flat gradient (0.23), indicating that antiparasitic activity from these compounds derives predominantly from a mechanism independent of NMT inhibition.
This observation is consistent with the hypothesis that high-LELP compounds (with relatively high lipophilicity and low LE) are more promiscuous than low-LELP analogues29 and therefore may exhibit cytotoxic activity by a variety of nonspecific mechanisms. Low-LELP analogues are predicted to bind more specifically to the target enzyme, and this is supported by the improved correlation between NMT affinity and antiparasitic activity in this data set; although wary of “correlation inflation”,44 this analysis appears to provide a robust predictor of cell efficacy when applied to these models. It would be unwise to invoke LELP < 10 as a rule rather than a guideline, as this is unlikely to be a binary boundary, with some compounds in either subset behaving not as predicted. Nevertheless, it provides encouraging evidence that LELP is a useful metric for guiding the hit-to-lead optimization process. Furthermore, this analysis highlights the danger of assuming that weakly active (LE < 0.3), lipophilic compounds (cLogP > 3) function in cellular systems by the presumed mechanism of action, without further validation.
Conclusion
Previously reported work described the discovery of 1 by a lead-hopping approach, as a ligand efficient inhibitor of plasmodial NMT.21 This work details LELP-guided development of this series, optimizing enzyme affinity while retaining selectivity over HsNMT to yield 34c, a high affinity druglike plasmodial NMT inhibitor. Development from 1 to 34c achieved a 100-fold improvement in enzyme affinity coupled with a 100-fold reduction in lipophilicity, at the expense of only two additional heavy atoms. This further demonstrates the utility of medicinal chemistry strategies such as matched molecular pairs analysis, bioisosterism, and metric-guided optimization in hit-to-lead development.
34c has antiparasitic activity in vitro, exhibiting similar potency over four parasite strains including two drug-resistant ones. In addition, this compound is equipotent against blood and liver stage parasites and displays selectivity over human liver host cells. It has been previously shown that this molecule exerts its antiparasitic activity via inhibition of NMT and moreover that it can reduce parasitemia in vivo.20 Taken together, this biological characterization shows that NMT is a highly promising target for the development of a new generation of antimalarial drugs. Further work will focus on optimization of cellular potency and in vivo pharmacokinetics, with the aim of producing a candidate series for the treatment of malaria.
Experimental Section
General
All chemicals were purchased from Sigma-Aldrich Ltd. (Gillingham, UK), Acros Organics (Geel, Belgium) and Alfa Aesar (Heysham, UK) and used without further purification. Moisture sensitive reactions were performed under nitrogen atmosphere using dried glassware, anhydrous solvents, and standard syringe/septa techniques.
Silica gel normal phase column chromatography was performed on an Isolera (Biotage, U.K.) automated apparatus with SNAP silica cartridges (Biotage, U.K.). Mobile phase consisted of n-hexane (solvent A) and ethyl acetate (solvent B), and standard gradient consisted of x% solvent B for 1 column volumes, x% to y% B for 10 column volumes, and then y% B for 2 column volumes. x and y are defined in the characterization section of the compound of interest.
Final compounds were purified by one of two methods: either HPLC or LC–MS. Postchromatography, organic solvents were removed by partial evaporation under reduced pressure and then compounds were dried by lyophilization overnight.
HPLC involved the following: Gilson semipreparative reverse phase HPLC system equipped with a HICHROM C18 column (250 mm × 21.2 mm), no. 306 pumps, and a Gilson UV/vis detector, detecting at 220 nm. The mobile phase consisted of H2O + 0.1% formic acid (solvent A) and MeOH + 0.1% formic acid (solvent B), with an elution method of 0–2 min 50% B, 2–30 min 50–98% B, 30–32 min 98%, 32–32.5 min 2% B at a flow rate of 12 mL/min.
LC–MS involved the following: RP-HPLC/MS on a Waters 2767 system equipped with a photodiode array and an ESI mass spectrometer using a XBridge Prep C18 (5 μm, 19 mm × 100 mm) column, equipped with an XBridge Prep C18 guard column (5 μm, 19 mm × 10 mm). The following elution method was used: gradient of solvent A and solvent B (as above) of 0–10 min 50–98% B, 10–12 min 98% B, 12–13 min 98–50% B, 13–17 min 50% B. Flow rate was 20 mL/min.
The purity of the title compounds was verified by reverse phase LC–MS on a Waters 2767 system equipped with a photodiode array and an ESI mass spectrometer using a XBridge C18 (5 μm, 4.6 mm × 100 mm) column, equipped with an XBridge C18 guard column (5 μm, 4.6 mm × 20 mm). The following elution method was used: gradient of solvent A and solvent B (as above) of 0–10 min 5–98% B, 10–12 min 98% B, 12–13 min 98–5% B, 13–17 min 5% B. Flow rate was 1.2 mL/min. Purity of tested compounds was ≥95% unless otherwise specified.
1H and 13C NMR spectra were respectively recorded on 400 and 101 MHz Bruker AV instruments at room temperature unless specified otherwise. In these cases 1H and 13C NMR spectra were respectively recorded on 500 and 126 MHz Bruker AV instruments at room temperature and were referenced to residual solvent signals. Data are presented as follows: chemical shift in ppm, integration, multiplicity (br = broad, app = apparent, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), and coupling constants in Hz.
Mass spectra were obtained from the Mass Spectrometry Service of Department of Chemistry, Imperial College London.
Prototypical Procedure for Preparation of Ester Family 4
1 and 2 were prepared as described previously.21
tert-Butyl 4-((2-(((3-Methoxybenzyl)oxy)carbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (3b)
To a solution of 2 (50 mg, 0.13 mmol) in dry acetonitrile (2 mL) were added hydroxybenzotriazole (27 mg, 0.20 mmol), N,N-diisopropylethylamine (26 μL, 0.16 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (30 mg, 0.16 mmol). The reaction mixture was stirred at room temperature for 15 min. 2-(3-Methoxyphenyl)ethanol (22 μL, 0.15 mmol) was then added and reaction mixture allowed to stir at room temperature for 18 h. The reaction mixture was concentrated under reduced pressure, dissolved in 10 mL of saturated ammonium chloride solution, and 3b was extracted with 3 × 10 mL ethyl acetate. Combined organic layers were then washed with brine (10 mL), dried over magnesium sulfate, concentrated under reduced pressure and crude product was purified by flash chromatography (10g SNAP cartridge, 6–50% B, Rf = 5.1 column volumes) to give 3b as a colorless oil (24 mg, 36%). 1H NMR (CDCl3, δ, ppm) 7.86 (1H, d, J = 8.0), 7.75 (1H, d, J = 8.2), 7.51–7.46 (1H, m), 7.43–7.37 (1H, m), 7.32 (1H, dd, J = 8.0, 7.8), 7.04 (1H, d, J = 7.8), 7.02–7.00 (1H, m), 6.90 (1H, dd, J = 8.0, 2.3), 5.35 (2H, s), 4.74–4.66 (1H, m), 3.94–3.86 (2H, m), 3.84 (3H, s), 3.07–2.98 (2H, m), 1.98–1.88 (2H, m), 1.85–1.73 (2H, m), 1.48 (9H, s).
3-Methoxyphenethyl 3-(Piperidin-4-yloxy)benzo[b]thiophene-2-carboxylate (4b)
To a solution of 3b (23.0 mg, 0.05 mmol) in dichloromethane (1 mL) was added trifluoroacetic acid (100 μL), and the solution was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure and purified by HPLC, yielding 4b as a colorless oil (5 mg, 18%). tR = 12.3 min; 1H NMR (CDCl3, δ, ppm) 7.82 (1H, d, J = 8.0), 7.76 (1H, d, J = 8.1), 7.56–7.48 (1H, m), 7.47–7.40 (1H, m), 7.33 (1H, app t, J = 7.9), 7.04 (1H, d, J = 7.4), 7.01–6.98 (1H, m), 6.91 (1H, dd, J = 8.2, 2.4), 5.34 (2H, s), 4.88–4.81 (1H, m), 3.84 (3H, s), 3.54–3.44 (2H, m), 3.11–3.01 (2H, m), 2.22–2.11 (4H, m); 13C NMR (CDCl3, δ, ppm) 161.48, 159.84, 153.90, 138.23, 136.98, 134.06, 129.82, 128.39, 125.07, 123.14, 122.61, 120.34, 115.87, 113.81, 113.79, 76.10, 66.82, 55.29, 41.01, 28.07; ESI HRMS, found 398.1425 (C22H24NO4S, [M + H]+, requires 398.1426).
Prototypical Procedure for Preparation of 1,2,4-Oxadiazole (9/14b)
tert-Butyl 4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (8a)
To a solution of 2 (40 mg, 0.11 mmol) in N,N-dimethylformamide (1 mL) were added hydroxybenzotriazole (16 mg, 0.12 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (22 mg, 0.12 mmol), and the reaction mixture was stirred at room temperature for 30 min. N′-Hydroxy-2-phenylacetimidamide was added (17.5 mg, 0.12 mmol), and the mixture was stirred at 140 °C for 3 h. The reaction mixture was concentrated under reduced pressure, dissolved in 20 mL of saturated ammonium chloride solution, and 8a was extracted with 3 × 20 mL of ethyl acetate. The combined organic layers were washed with saturated potassium carbonate solution (20 mL), brine (20 mL), dried over magnesium sulfate, concentrated under reduced pressure and crude product was purified by flash chromatography (10 g SNAP cartridge, 6–50% B, Rf = 7.5 column volumes) to give 8a as a clear yellow oil (16 mg, 31%). 1H NMR (CDCl3, δ, ppm) 7.87 (1H, d, J = 7.8 Hz), 7.80 (1H, d, J = 8.0 Hz), 7.50 (1H, ddd, J = 8.0, 7.1, 1.2 Hz), 7.46–7.28 (6H, m), 4.63–4.54 (1H, m), 4.16 (2H, s), 3.98–3.87 (2H, m), 3.00–2.91 (2H, m), 2.01–1.93 (2H, m), 1.90–1.78 (2H, m), 1.49 (9H, s).
3-Benzyl-5-(3-(piperidin-4-yloxy)benzo[b]thiophen-2-yl)-1,2,4-oxadiazole (9a)
9a was prepared as in 4b replacing 3b with 8a (16 mg, 0.03 mmol) and purified by LC–MS, yielding 9a as a white solid (12 mg, 94%). tR = 4.22 min; 1H NMR (CDCl3, δ, ppm) 7.82 (2H, d, J = 8.1 Hz), 7.52 (1H, dd, J = 8.1, 7.1 Hz), 7.46 (1H, dd, J = 8.1, 7.1 Hz), 7.40–7.28 (5H, m), 4.80–4.72 (1H, m), 4.16 (2H, s), 3.56–3.45 (2H, m), 3.04–2.95 (2H, m), 2.22–2.15 (4H, m); 13C NMR (CDCl3, δ, ppm) 170.28, 169.61, 152.34, 137.32, 135.57, 133.82, 129.29, 128.92, 128.37, 127.44, 125.57, 123.45, 122.72, 109.93, 76.97, 40.73, 32.49, 28.4l; ESI HRMS, found 392.1425 (C22H22N3O2S, [M + H]+, requires 392.1433).
3-Hydroxybenzo[b]thiophene-2-carbonitrile (10)
To a solution of methyl-2-mercaptobenzoate (1.64 mL, 11.9 mmol) and 2-bromoacetonitrile (0.92 mL, 13.1 mmol) in dry tetrahydrofuran (100 mL) at 0 °C was added potassium tert-butoxide (5.14 g, 71.3 mmol) gradually over 2 min. The reaction mixture was stirred and allowed to warm to room temperature over 15 min, quenched with 2 M hydrochloric acid to pH 2, and diluted with 75 mL of water. 10 was immediately extracted with 3 × 75 mL portions of ethyl acetate. The organic layers were combined, washed with 75 mL of brine, dried over magnesium sulfate, and concentrated under reduced pressure to give desired product 10 as a dark brown solid (2.03 g, 88%). 1H NMR (CDCl3, δ, ppm) 8.54 (1H, brs), 7.91 (1H, d, J = 8.1 Hz), 7.73 (1H, d, J = 8.2 Hz), 7.62–7.50 (1H, m), 7.51–7.40 (1H, m).
tert-Butyl 4-((2-Cyanobenzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (11)
To a solution of 10 (500 mg, 2.85 mmol) in tetrahydrofuran (7.5 mL) was added tert-butyl 4-hydroxypiperidine-1-carboxylate (1.15 g, 5.71 mmol) and triphenylphosphine (1.50 g, 5.71 mmol). The reaction mixture was stirred under nitrogen for 20 min and cooled to 0 °C, and diisopropyl azodicarboxylate (1.12 mL, 5.71 mmol) in tetrahydrofuran (10 mL) was added dropwise over 5 min. Reaction mixture was allowed to warm to room temperature and stirred for 1.5 h, then concentrated under reduced pressure and the crude product purified by flash chromatography (100 g SNAP cartridge, 2–18% B, Rf = 8.5 column volumes) to give 11 as a white solid (800 mg, 78%). 1H NMR (CDCl3, δ, ppm) 7.85 (1H, dd, J = 8.2, 0.9 Hz), 7.72 (1H, d, J = 8.2 Hz), 7.55 (1H, ddd, J = 8.2, 7.1, 1.1 Hz), 7.44 (1H, ddd, J = 8.2, 7.2, 0.9 Hz), 5.25 (1H, tt, J = 7.3, 3.6 Hz), 3.85–3.74 (2H, m), 3.46–3.36 (2H, m), 2.18–2.07 (2H, m), 1.98–1.87 (2H, m), 1.49 (9H, s).
(Z)-tert-Butyl 4-((2-(N′-Hydroxycarbamimidoyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (12)
To a solution of 11 (100 mg, 0.28 mmol) in ethanol (2 mL) was added hydroxylamine as 50% aqueous solution (170 μL, 2.79 mmol), and reaction mixture was stirred under refluxing conditions for 4 h. Reaction mixture was concentrated under reduced pressure to give 12 as a white solid (109 mg, 99%). 1H NMR (CDCl3, δ, ppm) 7.77–7.73 (1H, m), 7.72–7.67 (1H, m), 7.41–7.37 (2H, m), 6.25 (1H, brs), 5.55 (2H, brs), 4.50–4.41 (1H, m), 4.17–3.99 (2H, m), 2.87 (2H, ddd, J = 14.2, 11.7, 2.6 Hz), 2.12–2.00 (2H, m), 1.87–1.74 (2H, m), 1.48 (9H, s).
tert-Butyl 4-((2-(5-(3-Methoxybenzyl)-1,2,4-oxadiazol-3-yl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (13b)
To a solution of 3-methoxyphenylacetic acid (31 mg, 0.19 mmol) in acetonitrile (1 mL) were added hydroxybenzotriazole (27.5 mg, 0.21 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (39 mg, 0.21 mmol), and reaction mixture was stirred at room temperature for 15 min. 12 was added (80 mg, 0.21 mmol) and the mixture stirred at room temperature for 18 h. The reaction mixture was diluted with 20 mL of 0.5 M NaOH(aq) solution, and 13b was extracted with 3 × 20 mL of ethyl acetate. The combined organic layers were washed with saturated potassium carbonate solution (20 mL), brine (20 mL), dried over sodium sulfate, and concentrated under reduced pressure. Crude product (120 mg) and 4 Å molecular sieves (200 mg) were dissolved in toluene (3 mL), and reaction mixture was stirred at 110 °C for 18 h. The reaction mixture was filtered, concentrated under reduced pressure and crude product purified by flash chromatography (10g SNAP cartridge, 5–40% B, Rf = 8.0 column volumes) to give 13b as a clear orange oil (66 mg, 68%). 1H NMR (CDCl3, δ, ppm) 7.84 (1H, dd, J = 7.1, 1.2 Hz), 7.79 (1H, dd, J = 7.1, 1.2 Hz), 7.43 (2H, dd, J = 7.1, 1.2 Hz), 7.33–7.28 (1H, m), 6.97 (1H, d, J = 8.0 Hz), 6.95–6.92 (1H, m), 6.87 (1H, dd, J = 8.0, 2.4 Hz), 4.60–4.50 (1H, m), 4.28 (2H, s), 4.00–3.89 (2H, m), 3.83 (3H, s), 3.02–2.93 (2H, m), 2.01–1.92 (2H, m), 1.89–1.77 (2H, m), 1.48 (9H, s).
5-(3-Methoxybenzyl)-3-(3-(piperidin-4-yloxy)benzo[b]thiophen-2-yl)-1,2,4-oxadiazole (14b)
14b was prepared as in 4b replacing 3b with 13b (66 mg, 0.13 mmol) and purified by HPLC, yielding 14b as a white solid (21 mg, 40%). tR = 9.7 min; 1H NMR (CDCl3, δ, ppm) 8.03 (1H, dd, J = 7.0, 1.3 Hz), 7.92–7.84 (1H, m), 7.58–7.47 (2H, m), 7.31 (1H, app t, J = 7.9 Hz), 7.04–7.00 (1H, m), 6.99–6.94 (1H, m), 6.94–6.87 (1H, m), 4.66 (1H, tt, J = 7.7, 3.6 Hz), 4.43 (2H, s), 3.76 (3H, s), 3.44–3.27 (2H, m), 3.03–2.90 (2H, m), 2.13–2.03 (2H, m), 2.01–1.89 (2H, m); 13C NMR (CDCl3, δ, ppm) 178.57, 162.84, 159.46, 149.67, 136.80, 135.25, 133.88, 129.86, 127.34, 125.28, 123.55, 122.15, 121.40, 115.13, 112.72, 112.35, 76.88, 55.09, 40.82, 31.85, 28.20; ESI HRMS, found 422.1534 (C23H24N3O3S, [M + H]+, requires 422.1538).
Prototypical Procedure for Preparation of 1,3,4-Oxadiazole (23a)
tert-Butyl 4-((2-(Hydrazinecarbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (16)
To a solution of 15 (300 mg, 0.74 mmol) in ethanol (1 mL) was added hydrazine monohydrate (145 μL, 2.96 mmol). The reaction mixture was heated under refluxing conditions for 24 h and then concentrated under reduced pressure, yielding 16 as a yellow oil (217 mg, 75%). 1H NMR (CDCl3, δ, ppm) 7.79 (1H, d, J = 7.6 Hz), 7.74 (1H, d, J = 7.2 Hz), 7.48–7.37 (2H, m), 4.58 (1H, tt, J = 9.8, 4.1 Hz), 4.13–4.04 (2H, m), 2.95–2.82 (2H, m), 2.14–2.05 (2H, m), 1.92–1.78 (2H, m), 1.48 (9H, s).
tert-Butyl 4-((2-(2-(2-Phenylpropanoyl)hydrazinecarbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (21a)
To a solution of 16 (48 mg, 0.12 mmol) in tetrahydrofuran/N,N-dimethylformamide (4:1 v/v, 0.6 mL) were added hydroxybenzotriazole (8 mg, 0.06 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (28 mg, 0.15 mmol) and 2-phenylpropanoic acid (20 μL, 0.15 mmol). Reaction mixture was stirred at room temperature for 18 h and then diluted with 1.0 M NaOH(aq) (4 mL). 21a was extracted with EtOAc (2 × 5 mL). Combined organic layers were washed with brine (5 mL), dried over sodium sulfate, and concentrated under reduced pressure, yielding 21a as a colorless oil (63 mg, 98%). 1H NMR (CDCl3, δ, ppm) 7.82–7.76 (2H, m), 7.49–7.20 (7H, m), 4.68 (1H, tt, J = 10.0, 4.1 Hz), 4.19–4.06 (2H, m), 3.85 (1H, q, J = 7.2 Hz), 2.88–2.79 (2H, m), 2.16–2.05 (2H, m), 2.02–1.88 (2H, m), 1.60 (3H, d, J = 7.2 Hz), 1.48 (9H, s).
2-(1-Phenylethyl)-5-(3-(piperidin-4-yloxy)benzo[b]thiophen-2-yl)-1,3,4-oxadiazole (23a)
To a solution of 21a (63 mg, 0.12 mmol) and 1,2,2,6,6-pentamethylpiperidine (47 μL, 0.26 mmol) in dichloromethane (1 mL) was added m-toluenesulfonyl chloride (25 mg, 0.13 mmol), and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was then diluted with a further 2 mL of dichloromethane, washed with water (2 mL), washed with 1.0 M NaOH(aq) (2 mL), washed with brine (2 mL), dried over magnesium sulfate, and concentrated under reduced pressure. Crude reaction mixture was Boc-deprotected without further purification as in 4b, replacing 3b with 22a (10 mg, 0.02 mmol), and purified by HPLC yielding 23a as a yellow oil (7 mg, 14%). tR = 11.0 min; 1H NMR (CDCl3, δ, ppm) 8.44 (1H, brs), 7.85–7.73 (2H, m), 7.54–7.30 (7H, m), 4.65 (1H, brs), 4.44 (1H, q, J = 6.6 Hz), 3.53–3.35 (2H, m), 3.06–2.86 (2H, m), 2.20–2.01 (4H, m), 1.84 (3H, d, J = 6.6 Hz); ESI HRMS, found 406.1587 (C23H24N3O2S, [M + H]+, requires 406.1589).
Procedure for Preparation of 34c
Ethyl 3-Acetyl-4-oxopentanoate (24)
To a solution of sodium hydride (576 mg, 24.0 mmol) in anhydrous tetrahydrofuran (30 mL) cooled to 0 °C was added pentane-2,4-dione (2.05 mL, 20.0 mmol) in anhydrous tetrahydrofuran (40 mL), and the mixture was stirred for 1 h. Ethyl bromoacetate (2.66 mL, 24.0 mmol) in anhydrous tetrahydrofuran (30 mL) was then added, and the reaction mixture was stirred for 18 h. The reaction mixture was then washed with saturated NH4Cl(aq) (100 mL), and aqueous layer was back-extracted with EtOAc (100 mL). Combined organic layers were washed with brine (100 mL), dried over magnesium sulfate, and concentrated under reduced pressure, yielding 24 as a yellow oil (2.90 g, 78%). Mixture of diketone/enol tautomers 2:1 was observed by NMR in CDCl3 at room temperature. Diketone 1H NMR (CDCl3, δ, ppm) 4.20–4.12 (3H, m), 2.90 (2H, d, J = 7.3 Hz), 2.29 (6H, s), 1.31–1.25 (3H, m); enol 1H NMR (CDCl3, δ, ppm) 4.22–4.08 (2H, m), 3.25 (2H, s), 2.17 (6H, s), 1.32–1.23 (3H, m).
Ethyl 2-(1,3,5-Trimethyl-1H-pyrazol-4-yl)acetate (26c)
To a solution of 24 (400 mg, 2.15 mmol) in acetic acid (3 mL) was added methylhydrazine (125 μL, 2.37 mmol) dropwise, and reaction mixture was stirred at room temperature for 3 h. Reaction mixture was concentrated under reduced pressure, yielding 26c as a colorless oil (349 mg, 73%). 1H NMR (CDCl3, δ, ppm) 4.15 (2H, q, J = 7.1 Hz), 3.74 (3H, s), 3.35 (2H, s), 2.22 (6H, s), 1.28 (3H, t, J = 7.1 Hz).
2-(1,3,5-Trimethyl-1H-pyrazol-4-yl)acetic Acid (28c)
To a solution of 26c (300 mg, 1.53 mmol) in methanol (3 mL) was added lithium hydroxide monohydrate (642 mg, 15.3 mmol), and the mixture was stirred at room temperature for 18 h. Reaction mixture was diluted with water (20 mL) and acidifed with 2.0 M HCl(aq) to pH 4. Then 28c was extracted with EtOAc (3 × 20 mL). Combined organic layers were then dried over sodium sulfate and concentrated under reduced pressure, yielding 28c as a pink crystalline solid (130 mg, 51%). 1H NMR (CDCl3, δ, ppm) 3.80 (3H, s), 3.41 (2H, s), 2.24 (3H, s), 2.23 (3H, s).
tert-Butyl 4-((2-(2-(2-(1,3,5-Trimethyl-1H-pyrazol-4-yl)acetyl)hydrazinecarbonyl)benzo[b]thiophen-3-yl)oxy)piperidine-1-carboxylate (30c)
30c was prepared as in 21a replacing 2-phenylpropanoic acid with 28c (25 mg, 0.15 mmol), yielding 30c as an orange oil (43 mg, 66%). 1H NMR (CDCl3, δ, ppm) 9.75 (1H, d, J = 6.2 Hz), 7.89–7.72 (3H, m), 7.53–7.37 (2H, m), 4.75–4.65 (1H, m), 4.12–4.06 (2H, m), 3.76 (3H, s), 3.46 (2H, s), 2.86–2.82 (2H, m), 2.26 (6H, s), 2.14–2.09 (2H, m), 1.99–1.87 (2H, m), 1.48 (9H, s).
2-(3-(Piperidin-4-yloxy)benzo[b]thiophen-2-yl)-5-((1,3,5-trimethyl-1H-pyrazol-4-yl)methyl)-1,3,4-oxadiazole (34c)
34c was prepared as in 23a replacing 21a with 30c (66 mg, 0.12 mmol) and purified by LC–MS, yielding 34c as a yellow solid (9 mg, 17%). tR = 7.1 min; 1H NMR (CD3OD, δ, ppm) 8.47 (1H, brs), 7.99–7.88 (2H, m), 7.61–7.45 (2H, m), 4.81 (1H, tt, J = 7.2, 4.1 Hz), 4.12 (2H, s), 3.72 (3H, s), 3.56 (2H, ddd, J = 12.1, 7.5, 4.1 Hz), 3.11 (2H, ddd, J = 12.5, 7.5, 4.1 Hz), 2.29 (3H, s), 2.22 (3H, s), 2.21–2.04 (4H, m); 13C NMR (CD3OD, δ, ppm) 170.17, 160.26, 147.99, 134.99, 139.01, 134.93, 134.27, 128.98, 126.59, 124.49, 123.45, 120.38, 109.98, 77.73, 45.42, 42.37, 36.01, 29.56, 20.79, 9.52; ESI HRMS, found 424.1801 (C22H26N5O2S, [M + H]+, requires 424.1807).
Enzyme Inhibition Assay
All IC50 determinations were carried out using a 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) fluorescence assay, as described previously for HsNMT1,45 PvNMT,46 and PfNMT.21
IC50 values are the mean value of two or more determinations, and standard deviation is within 20% of the IC50 unless otherwise specified. Data were elaborated using Microsoft Office Excel 2010, and IC50 values were determined using GraFit 7.0 (Erithacus Software Ltd., U.K.) by nonlinear regression fitting, which were then quoted as Ki as defined below.
Ki Calculations
Ki values quoted are the Ki calculated from the experimentally determined IC50 values, the substrate concentration ([S]), and the Michaelis–Menten constant (Km) as described by the Cheng–Prusoff equation:23
| 1 |
For example, 34c had an experimentally determined PfNMT IC50 of 0.017 ± 0.002 μM. The Michaelis constant (Km) was 3.64 μM and the substrate concentration was 4.00 μM, resulting in a Ki of 0.008 μM. Km values were calculated as described previously.45
Plasmodium falciparum Sybr Green Viability Assay
Plasmodium falciparum Culture
Synchronous Plasmodium falciparum 3D7 late stage trophozoites at 33–36 h were used. Final parasitemia and hematocrit were 0.1–0.2% and 2%, respectively. Red blood cells used for the assay were centrifuged to remove the buffy coat and washed twice in Roswell Park Memorial Institute (RPMI) medium 1640 so that no white blood cells were present. The culture medium contained RPMI 1640 with 5 g/L Albumax, 0.025 g/L gentamycin, and 0.292 g/L l-glutamine.
SYBR Green Assay
Sterile 96-well black tissue culture plates (Costar) were used routinely for every assay. Drugs were diluted in culture medium and used in duplicate wells for each dilution of 10.0, 3.333, 1.111, 0.370, 0.123, 0.041, and 0.014 μM in a final volume of 100 μL per well. Chloroquine was used as a standard with 10 times reduced concentration range as above. Two sets of control were used in duplicate wells, one set with no added drugs (positive control) and one with uninfected red blood cells (negative control).
The plates were incubated at 37 °C for 48 h in a gas chamber flushed with 5% CO2, 5% O2, and 90% N2. After 48 h supernatants were taken from each well and replaced with fresh drug and incubated for a further 48 h in the same manner. At the end of the 96 h incubation, 25 μL of SYBR Green I dye (SYBR Green I nucleic acid gel stain 10000×, in DMSO from Invitrogen) in lysis buffer (1 μL dye to 1 mL of lysis buffer) was added to each well and stored overnight at −20 °C. The lysis buffer contained Tris-HCl (20 mM, pH 8.0), EDTA (2 mM), saponin (0.16%), and Triton X-100 (1.6% v/v).
Plates were warmed to room temperature, and fluorescence intensity was measured with a FLUOstar Omega microplate fluorescence reader (BMG Labtech). Values were expressed in relative fluorescence units. Binding of SYBR Green is specific for parasite DNA, as mature erythrocytes lack DNA and RNA. Fluorescence intensity unit was converted to percentage (%) of growth as follows:
Plasmodium falciparum [3H]Hypoxanthine Assay
This assay was performed by Dr. Sergio Wittlin and Dr. Christian Scheurer at the Swiss Tropical and Public Health Institute and is a modified version of the original hypoxanthine assay published by Desjardins et al.:47
Serial drug dilutions were prepared with a multichannel pipet, transferring 100 μL in a 2-fold serial dilution. Wells of rows A served as controls without drug. An amount of 100 μL of infected blood (parasitemia of 0.3%, 2.5% hematocrit) was added to all wells with a multipipette. The control wells (A9–A12) received uninfected blood of 2.5% hematocrit. The plates were incubated in an incubation chamber at 37 °C in an atmosphere containing 93% N2, 4% CO2, 3% O2. After 48 h, an amount of 50 μL of [3H]hypoxanthine (=0.5 μCi) solution was added to each well of the plate. The plates were incubated for another 24 h. The plates were then harvested with a Betaplate cell harvester (Wallac, Zurich, Switzerland). The dried filters were inserted into a plastic foil with 10 mL of scintillation fluid and counted in a Betaplate liquid scintillation counter (Wallac, Zurich, Switzerland). The results were recorded as counts per minute (cpm) per well at each drug concentration.
Data were analyzed using a graphic program (e.g., Excel) and expressed as percentage of the untreated controls. The 50% inhibitory concentration (IC50) value was evaluated by logit regression analysis.
Plasmodium berghei Liver Stage Assay
This assay is a slightly modified version of the assay previously described:5
HepG2-A16-CD81EGFP cells stably transformed to express a GFP-CD81 fusion protein were cultured at 37 °C in 5% CO2 in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FCS, 0.29 mg/mL glutamine, 100 units of penicillin, and 100 μg/mL streptomycin. The cells were seeded 24 h prior to infection into 1536-well plates at 3000 cells/well. The cells were pretreated for 12 h with the drug in a 12-point dilution series, and the cells were then infected with freshly dissected P. berghei sporozoites expressing luciferase (1000 sporozoites/well). After 48 h of incubation, the viability of P. berghei exoerythrocytic forms (EEF) was measured by bioluminescence. IC50 values were obtained using the measured bioluminescence intensity and a nonlinear variable slope four-parameter regression curve fitting model in Prism 6 (GraphPad Software Inc.).
HepG2 Toxicity Assay
Measurement of the ability of 34c to kill human cells (HepG2, human hepatocellular carcinoma) was performed using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS), cell viability assay, modified from the Promega Corporation Technical Bulletin No. TB169. The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS) plus 1% penicillin/streptomycin, incubated at 37 °C in humidified atmosphere with 5% CO2.
Then 200 μL of medium was added to wells 2–7 of a 12-well reservoir per replicate, along with 598 μL of medium in well 1. Compound was dissolved in DMSO at a top concentration of 50 mM, and 1.8 μL of this stock was added to column 1. Well 1 was mixed thoroughly with a 1 mL pipet, and 200 μL was transferred to well 2 and mixed thoroughly. This was repeated until well 7. Furthermore, a puromycin control was prepared by diluting 3 μL of puromycin stock (1 mg/mL) in 1497 μL of medium.
Cells were added to a 96-well plate at a concentration of 5000 cells per well (50 μL), excluding the exterior wells. To wells B2–G2 and B11–G11 is added 100 μL of 0.2% DMSO/medium (positive control wells). To wells B3–G3 was added 100 μL of puromycin/medium (negative control wells). To wells B4–B10, C4–C10, and D4–D10 was added 100 μL of each of the seven drug dilutions. This was repeated with a second compound if required in wells E4–E10, F4–F10, and G4–G10. The plate was placed in an incubator at 37 °C and cell growth/morphology inspected at 24, 48, and 72 h.
At 72 h, 20 μL of MTS/PMS solution (1.9 mg/mL MTS, 43.8 μg/mL PMS) was added to each assay well, and the plate returned to the incubator for 4 h. Fluorescence was then read at 490 nm on a SpectraMax M2e microplate reader from Molecular Devices. Data were elaborated using Microsoft Office Excel 2011, and LD50 values were determined using GraFit 7.0 (Erithacus Software Ltd., U.K.) by nonlinear regression fitting.
Crystallography
Crystals of ternary complexes of PvNMT, a nonhydrolyzable myristoyl-CoA analogue (NHM),48 and the inhibitors 20b and 34a were prepared as described previously.21
X-ray diffraction data were collected on synchrotron beamlines at the Diamond Light Source, Harwell, U.K., and processed using XDS49 and SCALA50 implemented within xia2.51
Model refinement was by maximum likelihood methods implemented in REFMAC52 using the protein chains of 4A95.pdb46 as a starting model interspersed with cycles of model building and adjustment using COOT.53 A summary of data collection and refinement statistics is in Supporting Information.
Acknowledgments
The authors are grateful to Andrew Bell, Jennie Hutton, and Zhiyong Yu for valuable discussions. We thank Sergio Wittlin and Christian Scheurer for performing assays on NF54, K1, and Dd2 Pf cell lines, and Elizabeth A. Winzeler and colleagues for enabling access to the liver stage assay. We also thank the Medicines for Malaria Venture (MMV) for funding Elizabeth A. Winzeler to support liver assays at University of California San Diego, and Sergio Wittlin to support cell-line testing at Swiss TPH. We thank William Heal and Zhiyong Yu for performing the HepG2 toxicity assay. We thank Munira Grainger for providing the parasites used in the in vitro assay, and Barbara Clough for performing the assay. We acknowledge the Diamond Light Source, Harwell, U.K., for the provision of data collection facilities and Shirley Roberts for expert crystal handling. This work was supported by the Engineering and Physical Sciences Research Council (Doctoral Prize Fellowship) and Medical Research Council (MRC Grants 0900278 and U117532067).
Glossary
Abbreviations Used
- nd
not determined
- Pb
Plasmodium berghei
- Pv
Plasmodium vivax
- Pf
Plasmodium falciparum
- Ld
Leishmania donovani
- NMT
N-myristoyltransferase
- Hs
Homo sapiens
- LE
ligand efficiency
- LELP
cLogP/LE
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- DMEM
Dulbecco’s modified Eagle medium
- FBS
fetal bovine serum
- PMS
phenazine methosulfate
Supporting Information Available
Experimental procedures and characterization of all intermediates and target compounds, full biological results, and X-ray crystallographic collection data and refinement statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ R.J.L.: Liverpool John Moores University, Egerton Court, 2 Rodney Street, Liverpool L1 2UA, U.K.
The authors declare no competing financial interest.
Accession Codes
The coordinates and structure factor files have been deposited in the Protein Data Bank under the accession codes 4CAE (PvNMT-NHM-20b) and 4CAF (PvNMT-NHM-34a).
Supplementary Material
References
- Murray C. J. L.; Rosenfeld L. C.; Lim S. S.; Andrews K. G.; Foreman K. J.; Haring D.; Fullman N.; Naghavi M.; Lozano R.; Lopez A. D. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 2012, 379, 413–431. [DOI] [PubMed] [Google Scholar]
- World Malaria Report 2011. http://www.who.int/malaria/world_malaria_report_2011/en/ (accessed Jan 18, 2012).
- Singh B.; Sung L. K.; Matusop A.; Radhakrishnan A.; Shamsul S. S. G.; Cox-Singh J.; Thomas A.; Conway D. J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004, 363, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Delves M.; Plouffe D.; Scheurer C.; Meister S.; Wittlin S.; Winzeler E. A.; Sinden R. E.; Leroy D. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 2012, 9, e1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M. C.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C. S.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T.-g.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E. R.; Mota M. M.; Clardy J. The next opportunity in anti-malaria drug discovery: the liver stage. PLoS Pathog. 2011, 7, e1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock D. A.; Nomura T.; Talley A. K.; Cooper R. A.; Dzekunov S. M.; Ferdig M. T.; Ursos L. M. B.; bir Singh Sidhu A.; Naudé B.; Deitsch K. W.; Su X.-z.; Wootton J. C.; Roepe P. D.; Wellems T. E. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 2000, 6, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phyo A. P.; Nkhoma S.; Stepniewska K.; Ashley E. A.; Nair S.; McGready R.; ler Moo C.; Al-Saai S.; Dondorp A. M.; Lwin K. M.; Singhasivanon P.; Day N. P.; White N. J.; Anderson T. J.; Nosten F. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 2012, 379, 1960–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines for Malaria Venture. Interactive R&D Portfolio. http://www.mmv.org/research-development/rd-portfolio (accessed Dec 8, 2013).
- Wright M. H.; Heal W. P.; Mann D. J.; Tate E. W. Protein myristoylation in health and disease. J. Chem. Biol. 2010, 3, 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tull D.; Heng J.; Gooley P. R.; Naderer T.; McConville M. J. Acylation-dependent and independent membrane targeting and distinct functions of small myristoylated proteins (SMPs) in Leishmania major. Int. J. Parasitol. 2012, 42, 239–247. [DOI] [PubMed] [Google Scholar]
- Patwardhan P.; Resh M. D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 2010, 30, 4094–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M. D. Targeting protein lipidation in disease. Trends Mol. Med. 2012, 18, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Sun J.; Deschamps A. M.; Kim G.; Liu C.; Murphy E.; Levine R. L. Myristoylated methionine sulfoxide reductase A protects the heart from ischemia–reperfusion injury. Am. J. Physiol.: Heart Circ. Physiol. 2011, 301, H1513–H1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin B.; Patzewitz E. M.; Brady D.; Silvie O.; Wright M. H.; Ferguson D. J.; Wall R. J.; Whipple S.; Guttery D. S.; Tate E. W.; Wickstead B.; Holder A. A.; Tewari R. Unique apicomplexan IMC sub-compartment proteins are early markers for apical polarity in the malaria parasite. Biol. Open 2013, 2, 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möskes C.; Burghaus P. A.; Wernli B.; Sauder U.; Dürrenberger M.; Kappes B. Export of Plasmodium falciparum calcium-dependent protein kinase 1 to the parasitophorous vacuole is dependent on three N-terminal membrane anchor motifs. Mol. Microbiol. 2004, 54, 676–691. [DOI] [PubMed] [Google Scholar]
- Rees-Channer R. R.; Martin S. R.; Green J. L.; Bowyer P. W.; Grainger M.; Molloy J. E.; Holder A. A. Dual acylation of the 45kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 2006, 149, 113–116. [DOI] [PubMed] [Google Scholar]
- Pino P.; Sebastian S.; Kim E. A.; Bush E.; Brochet M.; Volkmann K.; Kozlowski E.; Llinás M.; Billker O.; Soldati-Favre D. A tetracycline-repressible transactivator system to study essential genes in malaria parasites. Cell Host Microbe 2012, 12, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Clough B.; Rackham M. D.; Rangachari K.; Brannigan J. A.; Grainger M.; Moss D. K.; Bottrill A. R.; Heal W. P.; Broncel M.; Serwa R. A.; Brady D.; Mann D. J.; Leatherbarrow R. J.; Tewari R.; Wilkinson A. J.; Holder A. A.; Tate E. W. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat. Chem. 2014, 6, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham M. D.; Brannigan J. A.; Moss D. K.; Yu Z.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivaxN-myristoyltransferase. J. Med. Chem. 2013, 56, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A.; Brand S.; McElroy S. P.; Cleghorn L. A. T.; Smid O.; Stojanovski L.; Price H. P.; Guther M. L. S.; Torrie L. S.; Robinson D. A.; Hallyburton I.; Mpamhanga C. P.; Brannigan J. A.; Wilkinson A. J.; Hodgkinson M.; Hui R.; Qiu W.; Raimi O. G.; van Aalten D. M. F.; Brenk R.; Gilbert I. H.; Read K. D.; Fairlamb A. H.; Ferguson M. A. J.; Smith D. F.; Wyatt P. G. N-Myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung-Chi C.; Prusoff W. H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Hann M. M. Molecular obesity, potency and other addictions in drug discovery. MedChemComm 2011, 2, 349–355. [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- Keseru G. M.; Makara G. M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 2009, 8, 203–212. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Brannigan J. A.; Moss D. K.; Brzozowski A. M.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Design and synthesis of inhibitors of Plasmodium falciparumN-myristoyltransferase, a promising target for antimalarial drug discovery. J. Med. Chem. 2012, 55, 8879–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watjen F.; Baker R.; Engelstoff M.; Herbert R.; MacLeod A.; Knight A.; Merchant K.; Moseley J.; Saunders J. Novel benzodiazepine receptor partial agonists: oxadiazolylimidazobenzodiazepines. J. Med. Chem. 1989, 32, 2282–2291. [DOI] [PubMed] [Google Scholar]
- Griffen E.; Leach A. G.; Robb G. R.; Warner D. J. Matched molecular pairs as a medicinal chemistry tool. J. Med. Chem. 2011, 54, 7739–7750. [DOI] [PubMed] [Google Scholar]
- Goldberg K.; Groombridge S.; Hudson J.; Leach A. G.; MacFaul P. A.; Pickup A.; Poultney R.; Scott J.; Svensson P. H.; Sweeney J. B. Oxadiazole isomers: all bioisosteres are not created equal. MedChemComm 2012, 3, 600–604. [Google Scholar]
- Bostrom J.; Hogner A.; Llinas A.; Wellner E.; Plowright A. T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 2012, 55, 1817–1830. [DOI] [PubMed] [Google Scholar]
- Dossetter A. G.; Charles O. D.; Douglas A. A matched molecular pair analysis of in-vitro human microsomal metabolic stability measurements for heterocyclic replacements of di-substituted benzene containing compounds—identification of those isosteres more likely to have beneficial effects. MedChemComm 2012, 3, 1164–1169. [Google Scholar]
- Bird C. W. Heteroaromaticity, 5, a unified aromaticity index. Tetrahedron 1992, 48, 335–340. [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunitz J. D. The entropic cost of bound water in crystals and biomolecules. Science 1994, 264, 670–671. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Macdonald S. J. F.; Peace S.; Pickett S. D.; Luscombe C. N. The developability of heteroaromatic and heteroaliphatic rings—Do some have a better pedigree as potential drug molecules than others?. MedChemComm 2012, 3, 1062–1069. [Google Scholar]
- Böhm H.-J.; Brode S.; Hesse U.; Klebe G. Oxygen and nitrogen in competitive situations: Which is the hydrogen-bond acceptor?. Chem.—Eur. J. 1996, 2, 1509–1513. [Google Scholar]
- Brand S.; Cleghorn L. A. T.; McElroy S. P.; Robinson D. A.; Smith V. C.; Hallyburton I.; Harrison J. R.; Norcross N. R.; Spinks D.; Bayliss T.; Norval S.; Stojanovski L.; Torrie L. S.; Frearson J. A.; Brenk R.; Fairlamb A. H.; Ferguson M. A. J.; Read K. D.; Wyatt P. G.; Gilbert I. H. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2011, 55, 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines for Malaria Venture Compound Progression Criteria. http://www.mmv.org/sites/default/files/uploads/docs/essential_info_for_scientists/Compound_progression_criteria.pdf (accessed Feb 8, 2014).
- Kenny P. W.; Montanari C. A. Inflation of correlation in the pursuit of drug-likeness. J. Comput.-Aided Mol. Des. 2013, 27, 1–13. [DOI] [PubMed] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Thinon E.; Olaleye T. O.; Serwa R.; Lanzarone S.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. A fluorescence-based assay for N-myristoyltransferase activity. Anal. Biochem. 2012, 421, 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Whalley D.; Ansell K. H.; Saxty B.; Holder A. A.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. Discovery of Plasmodium vivax N-myristoyltransferase inhibitors: screening, synthesis, and structural characterization of their binding mode. J. Med. Chem. 2012, 55, 3578–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins R. E.; Canfield C. J.; Haynes J. D.; Chulay J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan J. A.; Smith B. A.; Yu Z.; Brzozowski A. M.; Hodgkinson M. R.; Maroof A.; Price H. P.; Meier F.; Leatherbarrow R. J.; Tate E. W.; Smith D. F.; Wilkinson A. J. N-Myristoyltransferase from Leishmania donovani: structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010, 396, 985–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.