

Abstract
Chronic cocaine administration induces a number of biochemical alterations within the mesolimbic dopamine system that may mediate various aspects of the addictive process such as sensitization, craving, withdrawal, and relapse. In the present study, rats were allowed to self-administer cocaine (0.5 mg/infusion) for 1 or 20 days. Tyrosine hydroxylase immunopositive cells were microdissected from the ventral tegmental area (VTA) using laser capture microdissection, and changes in the abundances of 95 mRNAs were assessed using cDNA macroarrays. Five GABA-A receptor subunit mRNAs (α4, α6, β2, γ2, and δ) were down-regulated at both 1 and 20 days of cocaine self-administration. In contrast, the catalytic subunit of protein phosphatase 2A (PP2α), GABA-A α1, and Gαi2 were significantly increased at both time points. Additionally, calcium/calmodulin-dependent protein kinase IIα mRNA levels were increased initially followed by a slight decrease after 20 days, whereas neuronal nitric-oxide synthase mRNA levels were initially decreased but returned to near control levels by day 20. These results indicate that alterations of specific GABA-A receptor subtypes and other signal transduction transcripts seem to be specific neuroadaptations associated with cocaine self-administration. Moreover, as subunit composition determines the functional properties of GABA-A receptors, the observed changes may indicate alterations in the excitability of dopamine transmission underlying long-term biochemical and behavioral effects of cocaine.
Repeated cocaine use induces biochemical adaptations in reinforcement-relevant brain regions (White and Kalivas, 1998; Koob and Le Moal, 2001; Nestler, 2001). These neuro-adaptations are important in that they may underlie processes of sensitization, craving, withdrawal, and relapse (Nestler and Aghajanian, 1997). Numerous studies have examined biochemical changes that occur within the mesolimbic dopamine system (Nestler and Aghajanian, 1997), a pathway considered to be a primary substrate for various types of reinforcing stimuli, including cocaine (Koob, 1996; Wise, 1998). The mesolimbic dopamine pathway originates in the ventral tegmental area (VTA) and projects to several fore-brain regions, including the nucleus accumbens (NAc) (Ungerstedt, 1971), considered a primary substrate for the reinforcing effects of cocaine. Acute and chronic cocaine administration produces significant elevations in NAc extra-cellular dopamine concentrations in animal models (Pettit and Justice, 1989; Hemby et al., 1997a,b), effects that contribute to the abuse liability of cocaine (Ritz et al., 1987).
In addition to the effects of acute administration, chronic cocaine administration induces neuroadaptations that may represent persistent or even permanent alterations in neuronal function (Nestler and Aghajanian, 1997; White and Kalivas, 1998). For example, cocaine induces a generalized up-regulation of the cAMP pathway (Nestler and Aghajanian, 1997) as well as activator protein 1 family members (Hope et al., 1992; Nye et al., 1995; Haile et al., 2001). Specifically, chronic cocaine administration increases activity of cAMP-dependent protein kinase (PKA) and adenylate cyclase in the NAc of rats (Terwilliger et al., 1991), as well as mRNA and protein levels of the α-catalytic subunit of PKA in the NAc of cynomolgus monkeys (Freeman et al., 2001). Furthermore, manipulation of specific components of these biochemical pathways has been shown to alter subsequent responsivity to the drug (Carlezon et al., 1998; Self et al., 1998). Numerous studies have also indicated that chronic cocaine induces hyperglutamatergic function in the VTA that seems to mediate the behavioral and neurochemical effects of cocaine (Fitzgerald et al., 1996; Ghasemzadeh et al., 1999; Loftis and Janowsky, 2000; Ungless et al., 2001; Tang et al., 2003). Given that chronic cocaine administration also alters the abundance of GABA-A receptor subunit mRNAs (Suzuki et al., 2000; Yamaguchi et al., 2000, 2002) and proteins (Lilly and Tietz, 2000; Jung and Peris, 2001) in the NAc and elsewhere, decreased GABA neurotransmission may complement the hyperglutamatergia in the VTA to increase cell excitability.
At present, there is a paucity of literature demonstrating the cellular specificity of cocaine-induced changes in gene expression in the mesolimbic dopamine system or other areas. Studies using in situ hybridization allow the analysis of mRNAs in defined cell populations, yet the sensitivity may not allow analysis of low-abundance mRNAs, the means to evaluate numerous transcripts, or the ability to reliably quantify such changes. Studies using regional assessment of gene expression emphasize transcripts contained in the majority of neuronal and glial populations and/or transcripts in highest abundance in the region but may not adequately reflect alterations in gene expression in target neuronal populations. Such limitations can be overcome by the combination of laser capture microdissection and array technology that enables precise localization of coordinate changes in gene expression within defined cell types (Eberwine et al., 1992; Hemby et al., 2002, 2003; Kamme et al., 2003).
Whereas the majority of studies investigating the molecular adaptations associated with chronic drug use have relied on noncontingent administration, several studies indicate pronounced biochemical differences between the contingent and noncontingent administration of drugs (Hemby et al., 1995, 1997a; Mark et al., 1999; McFarland et al., 2003). The concept of contingency is critical to interpreting changes associated with cocaine administration as they relate to the reinforcing effects of the drug. The present study was undertaken to evaluate differences in gene expression in tyrosine hydroxylase immunopositive neurons in the VTA after acute and chronic cocaine self-administration in rats. To this end, custom-designed macroarrays were used to simultaneously assess 95 genes and determine whether functional classes of genes were differentially expressed as a function of cocaine self-administration. The present study sought to expand upon previous findings in three ways: 1) examine gene expression changes in the VTA after self-administered rather than experimenter-administered cocaine, 2) monitor gene expression changes occurring specifically in VTA dopaminergic neurons, and 3) evaluate glutamate and GABA-A receptor subunits to delineate cocaine’s effects on excitatory as well as inhibitory neurotransmission in the VTA.
Materials and Methods
Subjects and Surgical Procedures
Male Sprague-Dawley rats (60–90 days; 225–275 g; Charles River Laboratories, Inc., Wilmington, MA) were randomly assigned to one of three groups: acute cocaine (n = 9), chronic cocaine (n = 8), and control (n = 9). Animals were housed in pairs under a reverse 12-h light/dark cycle (lights on 8:00 PM) and fed ad libitum before surgery. Rats in the cocaine groups were implanted with chronic indwelling jugular catheters under isofluorane anesthesia, as described previously (Hemby et al., 1995, 1997b). Infusions of methohexital (100 μl; 10 mg/kg i.v.) were administered as needed to assess catheter patency. Health of the rats was monitored twice daily by the experimenter and biweekly by institutional veterinarians according to the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Self-Administration
After surgery, subjects were housed in standard operant conditioning chambers (24.5 × 23.5 × 21 cm; MED Associates; St. Albans, VT) containing a retractable lever (0.25 N to operate) and a stimulus light mounted directly above the lever. The chambers were enclosed in sound-attenuating boxes containing an exhaust fan, a house light, a tone source, and a water bottle. A motor-driven syringe pump was located on the side of the external chamber. Extraneous noise was masked by the exhaust fan. Immediately after surgery, rats were placed in their respective chambers and received infusions of heparinized 0.9% bacteriostatic saline (1.7 U/ml; 200 μl/30 min) for 48 h. The next day, the self-administration procedure began. Rats assigned to the acute cocaine self-administration group were allowed to self-administer cocaine (0.5 mg/infusion) during one 8-h self-administration session under a fixed ratio (FR) 1, time-out 20-s schedule of reinforcement. Upon completion of the response requirement, a cocaine infusion was delivered and a 20-s time-out was in effect. During the time-out, the lever light was extinguished, the house light illuminated, and a tone was generated. The end of the time-out was signaled by illumination of the lever light and extinguishing of the house light and tone. During the time-out period, responses on levers were recorded but had no scheduled consequence. For rats in the chronic cocaine self-administration group, the ratio was gradually increased to FR5 (the terminal ratio) over 7 days. Rats self-administered cocaine under 8 h limited access conditions for 20 days after attainment of the terminal ratio. Rats assigned to the control group were drug naïve and did not receive jugular catheter implants. IBM-compatible computers were used for session programming and data collection.
After completion of the experimental procedures, rats were sacrificed using CO2 and transcardially perfused with 0.1 M phosphate-buffered saline followed by 4% paraformaldehyde in phosphate-buffered saline. After removal, brains were placed in a brain matrix, and 3-mm coronal blocks were taken from the rostral to the caudal portion of the brain. The blocks were placed in cassettes, post-fixed in 4% paraformaldehyde for 1 h and then transferred to 70% ethanol/150 mM NaCl for 2 h. All blocks were then placed in a Tissue Tek VIP5 vacuum infiltration processor (Torrance, CA) and processed in 70% ethanol (30 min at 40°C), 80% ethanol (30 min at 40°C), twice in 95% ethanol (45 min at 40°C), twice in 100% ethanol (45 min at 40°C), twice in xylenes (45 min at 40°C), and four times in paraffin (Paraplast; 30 min at 58°C). Afterward, tissue was transferred from the tissue processor to a Tissue Tek TEC tissue embedding console where each cassette containing processed tissue was transferred to an orientation platform (58°C). The tissue was removed and placed in a base mold before paraffin was dispensed into the mold. The top of the cassette was placed on top of the base mold, and the cassette and tissue were moved onto the chiller (3°C) to solidify the tissue block.
Immunocytochemistry and Laser Capture Microdissection (LCM)
Tissue blocks that included the mesencephalon were cut into 8-μm sections and then deparaffinized in xylenes (2 × 5 min) followed by descending concentrations of ethanol (1 min, 70, 80, and 2 × 95%; 5 min, 2 × 100%) and RNase-free water (22°C for 30 min). Next, sections were immersed in Tris buffer (0.1 M, pH 7.4) for 5 min at 22°C followed by immersion in 0.1 M Tris buffer (pH 7.4)/3% denatured horse serum for 5 min at 22°C. Sections were then incubated in primary monoclonal antibody against tyrosine hydroxylase (TH; 1:1000; T-1209; Sigma-Aldrich, St. Louis, MO) in 0.1 M Tris buffer (pH 7.4)/3% denatured horse serum for 15 min at 22°C. Sections were rinsed with 0.1 M Tris buffer (pH 7.4) and incubated with a horse anti-mouse secondary antibody in 0.1M Tris buffer (pH 7.4) for 10 min at 22°C, followed by a rinse with Tris buffer. Next, the conjugate was labeled using the avidin-biotin method (ABC Vectastain; Vector Laboratories; Burlingame, CA) in 0.1 M Tris buffer (pH 7.4) for 10 min at 22°C, followed by a rinse with Tris buffer. Immunolabeling was visualized with 3,5′-diaminobenzidine. After the peroxidase reaction, sections were rinsed in RNase-free water for 15 min and dehydrated in ascending ethanol concentrations (1 min each in 70, 80, 95, and 100%) and xylenes for 5 min.
Slides were removed from xylene and air-dried before microdissection. Laser dissection parameters for the Arcturus Pix Cell IIe system (Arcturus, Mountain View, CA) were as follows: diameter, 7 μm; power, 30 to 100 mW; and duration, 400 to 1200 μs. Approximately 200 TH-immunopositive cells were dissected from the VTA (−5.8 to −6.3 mm caudal to bregma, ventral to the red nucleus, and dorsomedial to the medial lemniscus (Paxinos and Watson, 1998). Cells close to the boundary between the VTA and the substantia nigra were not dissected to ensure distinction between the regions (Fig. 1). After dissection, the cap was removed and placed on a CapSure Pad (Arcturus) to remove excess tissue.
Fig. 1.
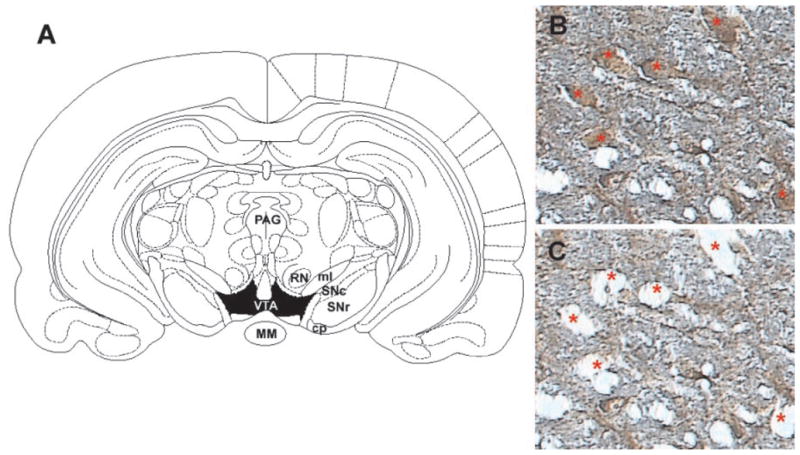
A, schematic of region for laser capture microdissection. Rat coronal midbrain section at −5.30 mm (relative to bregma) showing boundaries for VTA (shaded in black) from which tyrosine hydroxylase immunopositive cells were dissected. Midbrain section immunolabeled with anti-TH antibody (B) and the same section after microdissection of the indicated neurons (C) under 20× magnification. Note the specificity of the dissections and the minimal disruption of surrounding neuropil. MM, mammillary nucleus; PAG, periaqueductal gray area; SNc, substantia nigra-pars compacta; SNr, substantia nigra-pars reticulata; RN, red nucleus; ml, medial lemniscus; cp, cerebral peduncle.
RNA Extraction and Amplification
Each cap was then secured to the top of a 0.5-ml Eppendorf microcentrifuge tube containing 200 μl of TRIzol reagent (Invitrogen, Carlsbad, CA). The tube was inverted, vortexed, and incubated at 22°C for 15 min. RNA was then extracted with phenol/chloroform and precipitated with 100% ethanol (−80°C) in the presence of 10 μg of linear acrylamide. After extraction, a 66-base pair oligo(dT)-T7-primer/promoter (Tecott et al., 1988) was hybridized to poly(A+) mRNA for 7 min at 70°C and then quick cooled on ice for 5 min. cDNA was synthesized using reverse transcriptase (AMVRT, Seikagaku; Falmouth, MA) in 1× reverse transcriptase buffer containing 7 mM dithiothreitol; 250 μM each of dATP, dCTP, dGTP, and dTTP; and 0.12 U/μl of RNAsin and incubated at 42°C for 90 min.
The amplification and reamplification procedures have been described in detail elsewhere (Van Gelder et al., 1990; Hemby et al., 2002, 2003). During the second round of amplification, [33P]UTP was incorporated into the aRNA probes for hybridization with reverse Northern blots. Under optimal conditions, the first round of aRNA amplification results in approximately 1000-fold yield, and two rounds of amplification result in approximately 106-fold yield versus the original amount of poly(A+) mRNA. The aRNA procedure is a linear amplification process with minimal change in the relative abundance of the mRNA population in the native state of the neuron.
cDNA Macroarray Construction
Macroarrays were prepared on nylon membranes and consisted of 95 cDNAs selected for likely involvement in the effects of cocaine, including dopamine receptors, glutamate receptors, GABA receptors, synaptic proteins, and intra-cellular signaling molecules. Inserts were amplified from plasmid stocks in 96-well plates using PCR with GF200 primers under the following conditions: 95°C for 5 min (1 cycle); 95°C for 20 s, 55°C for 20 s, and 72°C for 2 min (40 cycles); and 72°C for 7 min (1 cycle). Aliquots of PCR samples were electrophoresed on a 1% agarose gel (1× Tris-acetate-EDTA, 0.05% ethidium bromide) at 5 V/cm for PCR band size verification. Gel images were captured by digital camera and archived. PCR product concentration was determined by spectrofluorometry (Gemini; Molecular Devices Corp., Sunnyvale, CA) using a 1:5000 dilution of SYBR 1 Green/Tris-EDTA and an aliquot of the PCR product. Values were compared with known concentrations of DNA standards for quantitation. Approximately 700 ng of each amplified insert was spotted on Nytran SuPerCharge nylon transfer membrane (Schleicher & Schuell, Keene, NH) using a 96-well dot blot apparatus (Minifold I; Schleicher & Schuell). DNA was crosslinked to the membrane by ultraviolet radiation at 120,000 μJ/cm2.
Macroarrays were prehybridized with UltraHyb solution (Ambion, Austin, TX) in hybridization bottles for 1 h at 42°C. Next, 33P-labeled antisense RNA probes from the VTA for each subject were heat denatured for 5 min at 70°C, placed on ice for 5 min, and hybridized to their respective arrays overnight at 42°C in a rotisserie hybridization oven. Samples from the VTA of each rat were hybridized to separate macroarrays and were not pooled within groups. After hybridization, membranes were washed once briefly with 2× SSC/0.1% SDS, twice with 2× SSC/0.1% SDS for 15 min each at 42°C, and once with 0.1× SSC/0.1% SDS for 10 min at 42°C. Labeled hybridized products were detected using PhosphorImager cassettes, and hybridization signal intensities were analyzed using ImageQuant software (Amersham Biosciences Inc., Sunnyvale, CA).
Data Analysis
Densitometry values (hybridization intensities) were obtained for each clone and for background (nonspecific) hybridization on the array. Background hybridization values were obtained from spots on the macroarrays in which no clone was loaded as well as regions between spotted clones. Only clones with spot symmetry and lack of significant artifactual signal intensity as assessed by visual identification were accepted for analysis. Values below background were not included in the analysis. The background value for each macroarray was subtracted from the densitometry value for each clone on that array. This “signal – background” value was then divided by the summed values for all of the clones on the array (global normalization) to yield a normalized value for each clone, thereby minimizing variations due to differences in the specific activity of the probe and the absolute quantity of probe present (Ginsberg et al., 2000; Hemby et al., 2002, 2003; Tang et al., 2003). Genes were grouped into nine classes (dopamine-related, glutamate-related, GABA-related, G proteins, kinases, peptides and lipids, phosphatases, structural and signaling, and transcription factors), and a two-factor analysis of variance (Transcript and Group) with repeated measures on one factor (Transcript) was then used for each group of genes. Post hoc comparisons were then conducted as needed using Tukey’s test. The null hypothesis was rejected when p < 0.05.
Results
Behavior
Rats in the acute cocaine group self-administered an average of 40 (± 10 S.E.M.) infusions (20 ± 5 mg of cocaine) over the 8-h self-administration session (Fig. 2). In the chronic cocaine group, rats self-administered an average of 31 (± 3) infusions (15 ± 3 mg of cocaine) per day (Fig. 2). As expected, the mean total cocaine intake for the chronic cocaine self-administration group (317.94 ± 32.19 mg) was significantly greater than the acute cocaine group (20.06 ± 5.01 mg; t = −9.707, df = 15, p < 0.001) Four rats in the chronic cocaine group received sham injections in the NAc on day 15 as part of another study. Animals were then returned to their operant chambers and allowed to self-administer cocaine for a normal 8-h self-administration session. However, there was no significant difference in the number of infusions self-administered or in the gene expression pattern compared with the rest of the group.
Fig. 2.
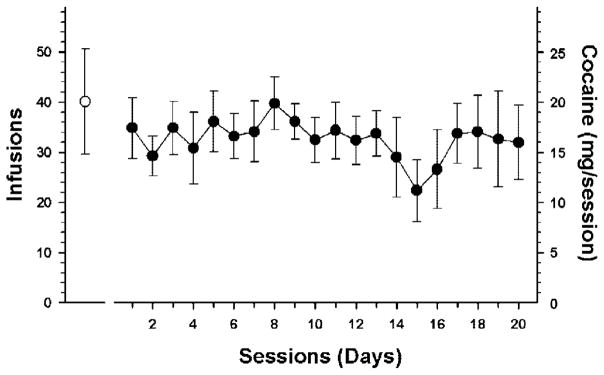
The mean (± S.E.M.) number of infusions for cocaine self-administration for 1 (open circle) or 20 days (filled circles). There was no significant difference in the number of infusions on day one between the two groups.
Gene Expression
Analysis of the macroarrays revealed distinct patterns of expression for the majority of transcripts investigated (Fig. 3). Of the 95 mRNAs examined, 11 were found to be differentially expressed among the groups (Fig. 4; Table 1), and at least one transcript was differentially expressed in five of the nine categories examined: GABA-related, G proteins, kinases, phosphatases, and structural/signaling molecules.
Fig. 3.

Representative cDNA macroarray (95 clones and one blank spot) demonstrating the expression profile of VTA dopamine neurons. The signal intensity of each spot is proportional to the abundance of the particular mRNA in the sample. CCK, cholecystokinin; D1, dopamine receptor 1; D2, dopamine receptor 2; D3, dopamine receptor 3; D4, dopamine receptor 4; D1b, dopamine receptor 1b; DAT, dopamine transporter; BDNF, brain-derived neurotrophic factor; CART, cocaine and amphetamine regulated transcript; GAD65, glutamate decarboxylase 65 kDa; GAD67, glutamate decarboxylase 65 kDa; Gαi1, G protein á inhibiting activity polypeptide 1; Gαi2, G protein á inhibiting activity polypeptide 2; Gαi3, G protein á inhibiting activity polypeptide 3; Gαs, G protein á stimulating activity polypeptide; Gαo, G protein á activating activity polypeptide O; Gαz, G protein á z polypeptide; Gα15, G protein á 15 (Gq class); Gβ1, G protein β polypeptide 1; Gβ2, G protein β polypeptide 2; Gβ3, G protein β polypeptide 3; Gαq, G protein Q polypeptide; Gγ5, = G protein γ polypeptide 5; PP1α, protein phosphatase 1, catalytic subunit, á isoform; PP1β, protein phosphatase 1, catalytic subunit, β isoform; PP2α, protein phosphatase 2, catalytic subunit, á isoform; PP1γ, protein phosphatase 1, catalytic subunit, γ isoform; PP2β, protein phosphatase 2, catalytic subunit, β isoform; ABP, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor binding protein; GRM1a, metabotropic glutamate receptor 1a; GRM3, metabotropic glutamate receptor 3; GRM4, metabotropic glutamate receptor 4; GRM5, metabotropic glutamate receptor 5; GluRδ1, glutamate receptor subunit delta1; GluRδ2, glutamate receptor subunit; GRIN1, glutamate receptor subunit, ionotropic, N-methyl-D-aspartate 1; GRIN2A, glutamate subunit receptor, ionotropic, N-methyl-D-aspartate 2A; GRIN2B, glutamate subunit receptor, ionotropic, N-methyl-D-aspartate 2B; GRIN2C, glutamate subunit receptor, ionotropic, N-methyl-D-aspartate 2C; GRIN2D, glutamate subunit receptor, ionotropic, N-methyl-D-aspartate 2D; GRIA1, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepro-pionic acid 1; GRIA2, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 2; GRIA3, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 3; GRIA4, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 4; GRIK1, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 5; GRIK2, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 6; GRIK3, glutamate receptor subunit, ionotropic, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid 7; GRIK4, glutamate receptor subunit, ionotropic, kainate 4; GRIK5, glutamate receptor subunit, ionotropic, kainate 5; SPCT, spectrin; citron; cript, cysteine-rich interactor of PDZ3; hom1c, homer 1c; PSD95, postsynaptic density protein SAPAP =/PSD-95-associated protein; GRIP1, glutamate receptor interacting protein 1; FRA1, fos-related antigen 1; FRA2, fos-related antigen 2; pDYN, prodynorphin; CaMKIIα, calcium/calmodulin-dependent protein kinase 2á; CaMKIIβ3, calcium/calmodulin-dependent protein kinase 2β 3 subunit; CaMKIIδ, calcium/calmodulin-dependent protein kinase 2δ subunit; CaMKIIγ, calcium/calmodulin-dependent protein kinase 2γ subunit; CaMKIV, calcium/calmodulin-dependent protein kinase; CaMKKα, calcium/calmodulin-dependent protein kinase kinase α subunit; PKR1α, cAMP-dependent protein kinase type I-α regulatory chain; PKR1β, cAMP-dependent protein kinase type I-β regulatory chain; PKR2β, cAMP-dependent protein kinase type II-β regulatory chain; GABAγ1, GABA-A receptor subunit γ1; GABAγ2, GABA-A receptor subunit γ2; GABAγ3, GABA-A receptor subunit γ3; GABAα1, GABA-A receptor subunit α1; GABAα3, GABA-A receptor subunit α3; GABAα4, GABA-A receptor subunit α4; GABAα6, GABA-A receptor subunit α6; GABAβ1, GABA-A receptor subunit β1; GABAβ2, GABA-A receptor subunit β2; GABAβ3, GABA-A receptor subunit β3; GABAδ, GABA-A receptor subunit δ; GABAε, GABA-A receptor subunit ε; PLD, phospholipase D; AKAP, protein kinase A anchor protein; synj2, synaptojanin 2; syntx5, syntaxin 5; synbv2, synaptobrevin 2; stat5b, signal transducer and activator of transcription 5B; RLZF-Y, rat lung zinc finger-Y; CB1, cannabinoid receptor 1; GralA, GTP-binding protein ral A; GralB, GTP-binding protein ral B; GRK4, G protein-dependent receptor kinase 4; grk5, G protein-dependent receptor kinase 5; CREB, blank.
Fig. 4.

Comparisons of gene expression changes in VTA tyrosine-hydroxylase immunopositive neurons after 1 or 20 days of cocaine self-administration. mRNA expression values correspond to hybridization intensity for individual transcripts and represent the relative abundance of mR-NAs normalized to the summated signal intensities for all spots on the blot (minus background; see Materials and Methods).*, p < 0.05; **, p < 0.01, compared with controls; #, p < 0.05; ##, p < 0.01 compared with 1-day cocaine.
TABLE 1.
Summary of results for differences in relative gene expression following cocaine self-administration
Normalized expression values for designated transcript classes were analyzed using two way ANOVA (Group × Transcript) with repeated measures (Transcript). Post hoc analyses were conducted as needed using Tukey’s test, and the null hypothesis was rejected when P < 0.05. P values are indicated for the main effect, interaction, and post hoc analyses.
| Transcript | Main Effect | Interaction | Post Hoc Analysis
|
||
|---|---|---|---|---|---|
| Control vs. 1 Day | Control vs. 20 Days | 1 Day vs. 20 Days | |||
| Dopamine-related | 0.990 | 0.738 | |||
| Tyrosine hydroxylase (TH) | |||||
| Dopamine receptor 1 (D1) | |||||
| Dopamine receptor 2 (D2) | |||||
| Dopamine receptor 3 (D3) | |||||
| Dopamine receptor 4 (D4) | |||||
| Dopamine receptor 5 (D5) | |||||
| Dopamine transporter (DAT) | |||||
| Transcription factors | 0.266 | 0.763 | |||
| Fos-related antigen 1 (FRA-1) | |||||
| Fos-related antigen 2 (FRA-2) | |||||
| Transcription factor stat 5b (TFstat5b) | |||||
| Rat lung zinc finger protein Y1 (RLZF-Y) | |||||
| cAMP response element binding protein (CREB) | |||||
| GABA-related | <0.001 | <0.001 | |||
| Glutamic acid decarboxylase 65 (GAD 65) | 0.014 | ||||
| Glutamic acid decarboxylase 67 (GAD 67) | |||||
| GABA receptor type A, γ1 subunit (GABA-A γ1) | |||||
| GABA receptor type A, γ2 subunit (GABA-A γ2) | 0.017 | ||||
| GABA receptor type A, γ3 subunit (GABA-A γ3) | |||||
| GABA receptor type A, α1 subunit (GABA-A α1) | <0.001 | <0.001 | |||
| GABA receptor type A, α3 subunit (GABA-A α3) | |||||
| GABA receptor type A, α4 subunit (GABA-A α4) | 0.033 | 0.030 | |||
| GABA receptor type A, α6 subunit (GABA-A α6) | 0.025 | 0.017 | |||
| GABA receptor type A, β1 subunit (GABA-A β1) | |||||
| GABA receptor type A, β2 subunit (GABA-A β2) | 0.030 | 0.023 | |||
| GABA receptor type A, β3 subunit (GABA-A β3) | |||||
| GABA receptor type A, δ subunit (GABA-A δ) | 0.042 | 0.020 | |||
| GABA receptor type A, ε subunit (GABA-A ε) | |||||
| Glutamate-related | 0.844 | 0.245 | |||
| AMPA receptor binding protein (AMPAbp) | |||||
| Metabotropic glutamate receptor 1a (mGluR1a) | |||||
| Metabotropic glutamate receptor 3 (mGluR3) | |||||
| Metabotropic glutamate receptor 4 (mGluR4) | |||||
| Metabotropic glutamate receptor 5 (mGluR5) | |||||
| Glutamate receptor δ1 (GluRδ1) | |||||
| Glutamate receptor δ2 (GluRδ2) | |||||
| NMDA receptor 1 (NR1) | |||||
| NMDA receptor 2A (NR2A) | |||||
| NMDA receptor 2B (NR2B) | |||||
| NMDA receptor 2C (NR2C) | |||||
| NMDA receptor 2D (NR2D) | |||||
| Glutamate receptor 1 (GluR1) | |||||
| Glutamate receptor 2 (GluR2) | |||||
| Glutamate receptor 3 (GluR3) | |||||
| Glutamate receptor 4 (GluR4) | |||||
| Glutamate receptor 5 (GluR5) | |||||
| Glutamate receptor 6 (GluR6) | |||||
| Glutamate receptor 7 (GluR7) | |||||
| Kainate receptor 1 (KA1) | |||||
| Kainate receptor 2 (KA2) | |||||
| G proteins | 0.136 | <0.001 | |||
| GTP-binding protein, α inhibiting polypeptide 1 (Gαi1) | |||||
| GTP-binding protein, α inhibiting polypeptide 2 (Gαi2) | <0.001 | <0.001 | |||
| GTP-binding protein, α inhibiting polypeptide 3 (Gαi3) | |||||
| GTP-binding protein, α stimulating polypeptide 1 (Gαs1) | |||||
| GTP-binding protein, αo polypeptide (Gαo) | |||||
| GTP-binding protein, αz polypeptide (Gαz) | |||||
| GTP-binding protein, α15 plypeptide (Gα15) | |||||
| GTP-binding protein, β1 polypeptide (Gβ1) | |||||
| GTP-binding protein, β2 polypeptide (Gβ2) | |||||
| GTP-binding protein, β3 polypeptide (Gβ3) | |||||
| GTP-binding protein, q polypeptide (Gαq) | |||||
| GTP-binding protein, γ polypeptide (Gγ5) | |||||
| GTP-binding protein ral A (GralA) | |||||
| GTP-binding protein ral B (GralB) | |||||
| Kinases | 0.043 | <0.001 | |||
| Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) | <0.001 | <0.001 | 0.042 | ||
| Ca2+/calmodulin-dependent protein kinase IIβ3 (CaMKIIβ3) | |||||
| Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ) | |||||
| Ca2+/calmodulin-dependent protein kinase IIγ (CaMKIIγ) | |||||
| Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) | |||||
| CaMKII kinase α (CaMKKα) | |||||
| Protein kinase A, regulatory subunit 1A (PRKAR1A) | |||||
| Protein kinase A, regulatory subunit 1B (PRKAR1B) | |||||
| Protein kinase A, regulatory subunit 2B (PRKAR2B) | |||||
| G protein-coupled receptor kinase 4 (GRK4) | |||||
| G protein-coupled receptor kinase 5 (GRK5) | |||||
| Phosphatases | 0.026 | 0.001 | |||
| Protein phosphatase 1A, catalytic subunit (PP1CA) | |||||
| Protein phosphatase 1B, catalytic subunit (PPP1CB) | |||||
| Protein phosphatase 2A, catalytic subunit (PP2α) | <0.001 | <0.001 | |||
| Protein phosphatase 1, γ subunit (PP1γ) | |||||
| Protein phosphatase 2A, β subunit (PP2Aβ) | |||||
| Structural/signaling | 0.003 | <0.001 | |||
| Spectrin α subunit (spectrin) | |||||
| nNOS | 0.001 | 0.019 | 0.001 | ||
| Rho-interacting, serine/threonine kinase 21 (citron) | |||||
| Cysteine-rich interactor of PDZ3 (CRIPT) | |||||
| Homer, neural immediate early, gene 1C (homer1c) | |||||
| Post-synaptic density protein 95 kDa (PSD-95) | |||||
| PSD-95/SAP90-associated protein-1 (SAPAP) | |||||
| Glutamate receptor interacting protein 1 (GRIP1) | |||||
| Phospholipase D (PLD) | |||||
| A-kinase anchoring protein (AKAP) | |||||
| Synaptojanin II (SYNJN II) | |||||
| Syntaxin 5 (SYNTX 5) | |||||
| Synaptobrevin 2 (SYNBREV 2) | |||||
| Peptide-related | 0.413 | 0.208 | |||
| Precursor to cholecystokinin (pCCK) | |||||
| Brain-derived neurotrophic factor (BDNF) | |||||
| Cocaine/amphetamine-related transcript (CART) | |||||
| Prodynorphin (PDYN) | |||||
| Cannabinoid receptor 1 (CB1) | |||||
Dopamine-Related Transcripts and Transcription Factors
There was no significant difference for seven dopamine-related transcripts (Table 1) in either Group [F(2181) = 0.00958; P = 0.990] or Group and Transcript interaction [F(12,181) = 0.712; P = 0.738]. Likewise, there was no significant effect of cocaine on the levels of transcription factor mRNAs [F(2129) = 1.403; P = 0.266] and no significant interaction [F(8129) = 0.615; P = 0.763] (Table 1).
GABA-Related Transcripts
The most extensively affected by cocaine self-administration, the GABA-related transcripts exhibited both a significant effect of Group [F(2363) = 10.864; p < 0.001] and an interaction between Group and Transcript [F(26,363) = 2.869; p < 0.001]. Of the 14 GABA-related transcripts examined, post hoc analysis revealed that expression of six GABA-A receptor subunits was altered after cocaine self-administration (Table 1; Fig. 4). In all cases where receptor subunit mRNAs were altered after acute cocaine self-administration, expression remained altered after 20 days of self-administration. mRNA levels of four transcripts (GABA-A α4, GABA-A α6, GABA-A β2, and GABA-A δ) were significantly decreased after both acute and chronic cocaine self-administration. The GABA-A γ2 subunit was significantly decreased after 20 days, but not 1 day, of self-administration. In contrast, α1 subunit mRNA was up-regulated with cocaine self-administration at both time points. Additionally, expression of the enzyme GAD 65 was significantly decreased after chronic cocaine self-administration compared with acute self-administration.
Glutamate-Related Transcripts
Previous studies have indicated significant involvement of VTA glutamate receptors in the behavioral effects of cocaine (Pierce et al., 1996; White and Kalivas, 1998). However, in the present study, there was no significant difference in glutamate receptor subunit mRNA expression between acute and chronic cocaine self-administration and controls [F(2,545) = 0.171; P = 0.844]. Furthermore, there was no significant Group and Transcript interaction [F(40,545) = 1.154; P = 0.245] for the glutamate-related transcripts (Table 1). However, GRIN2C, GRIA1, and GRIA3 were decreased slightly after acute cocaine but returned to control levels after chronic cocaine self-administration. In an opposite manner, GRIN2B, GRIA2, and GRIK4 were increased after 1 day and decreased toward control levels by day 21.
Other Transcripts
A significant main effect of Group was observed for kinases [F(2285) = 3.611; P = 0.043], phosphatases [F(2129) = 4.299; P = 0.026], and structural/signaling [F(2337) = 7.840; P = 0.003] transcripts. Additionally, G proteins, kinases, phosphatases, and structural/signaling transcripts all showed a significant interaction between Group and Transcript [F(26,363) = 3.651; P < 0.001; F(20,285) = 4.252; P < 0.001; F(8129) = 3.6; P = 0.001; and F(24,337) = 2.671; P < 0.001, respectively]. Post hoc analyses revealed a significant increase in Gαi2 mRNA after acute and chronic cocaine self-administration. Although not significant, Gαi2 continued increase after chronic self-administration relative to acute self-administration. Both CaMKIIα and neuronal nitric-oxide synthase (nNOS) were significantly different for all pairwise comparisons between groups. After an initial increase in mRNA expression with acute cocaine self-administration, CaMKIIα expression declined after 20 days of self-administration, although levels did not return to control levels. nNOS followed the opposite pattern of expression with an initial decrease after acute cocaine self-administration followed by an increase that did not return to control levels. PP2α mRNA levels were up-regulated over control levels after both 1 day and 20 days of self-administration. Although the peptide-related category showed no main effect of Group [F(2129) = 0.918; P = 0.413] and no interaction between Group and Transcript [F(8129) = 1.399; P = 0.208], there was a trend toward increased levels of precursor to cholecystokinin after both acute and chronic self-administration.
Discussion
The present study was undertaken to identify coordinate changes in the expression of multiple genes in VTA dopamine neurons as a function of cocaine self-administration. A membrane-based macroarray platform with radioactive hybridization was used to query 95 cDNAs of interest. Significant alterations were observed for a variety of protein families, including GABA-A receptor subunits, G protein subunits, kinases, phosphatases, other miscellaneous transcripts, and a trend toward significance in iGlur receptor subunits. A variety of effects on gene expression were observed between acute and chronic cocaine administration and are likely due to the chronicity of drug administration. However, similar effects between acute and chronic cocaine self-administration where observed for several mRNAs and likely reflect the direct pharmacological actions of cocaine on the dopamine neurons assessed in the present study. Previously, our laboratory and others have used similar approaches to identify molecular profiles of cocaine in rats (Ang et al., 2001; Freeman et al., 2002; Toda et al., 2002), monkeys (Freeman et al., 2001), and humans (Tang et al., 2003). Regional assessments of gene expression create an informative mosaic of expression level changes; however, determining the cellular origins of the gene expression has been complicated by cellular heterogeneity of subcortical brain regions and the ability to assess multiple genes in discrete neuronal populations. Single cell gene expression methodology combined with array technology can overcome some of these limitations by assessing multiple transcripts in specific target neuronal populations, providing a level of assessment heretofore unattainable (Ginsberg et al., 2000; Hasenkamp and Hemby 2002; Hemby et al., 2002, 2003; Fasulo and Hemby, 2003; Kamme et al., 2003). Future studies directed toward protein confirmation and functional relevance of the observed mRNA change are warranted. The present study provides the first molecular profile of VTA dopamine neurons after acute and chronic cocaine self-administration in rats.
The major finding of the present study was a significant alteration in the expression of GABA-A receptor subunit mRNAs. GABA-A receptors are composed of α, β, γ, δ, ε, θ, π, and ρ subunits that form a pentameric structure with an integrated chloride channel. The expression and assembly of different subunits confer different physiological and pharmacological properties to the receptor complex. In the present study, α1 and γ2 subunits were of similarly high abundance relative to the other subunits, yet the α1 subunit was up-regulated and the γ2 subunit was down-regulated with cocaine self-administration. These data are supported by previous studies demonstrating significant decreases in α1, α6, β2, β3, and γ2 mRNAs in the striatum and NAc (Suzuki et al., 2000; Yamaguchi et al., 2000), along with decreased receptor number and function in this region (Peris, 1996; Yamaguchi et al., 2002). Cocaine-induced alterations in these subunits alter subsequent responsivity to the drug (Peris et al., 1998). Decreased expression of the γ2 subunit could also have a potentially profound effect on normal receptor function, because γ2 subunits are essential for receptor clustering (Essrich et al., 1998). Although of lower relative abundance, α4, α6, β2, and δ-subunit mRNAs were also down-regulated in response to cocaine self-administration. The regulation of α and γ subunits in the present study and their involvement in benzodiazepine modulation of the GABA-A receptors provides a possible explanation of the utility of benzodiazepine therapy for cocaine-related anxiety.
Although several scenarios have been proposed, growing evidence suggests that GABA-A receptors contribute to the tonic inhibition of dopamine neurons in the VTA, especially projections to the NAc. For example, intra-VTA infusions of GABA decrease VTA dopaminergic cell activity as well as dopamine release in the NAc (Suaud-Chagny et al., 1992). Other studies have demonstrated that induction of hyperpolarization in VTA neurons by focal stimulation was reversed by the GABA-A antagonists picrotoxin and bicuculline (Johnson and North, 1992b). Additionally, bicuculline administration stimulated VTA dopaminergic neurons (Johnson and North, 1992a) and supported intra-VTA self-administration (Ikemoto et al., 1997a,b). Together, these data suggest that decreased function or abundance of GABA-A receptors may result in increased firing of mesoaccumbal dopamine neurons, an effect that would likely contribute to cocaine reinforcement.
Previous studies have indicated that chronic cocaine administration significantly alters the abundance of iGluR subunit protein, but not mRNA, levels within the mesolimbic dopamine system in rats, which may lead to increased excitability of dopamine neurons (White et al., 1995; Fitzgerald et al., 1996; Ghasemzadeh et al., 1999; Loftis and Janowsky 2000). Chronic cocaine administration in rats failed to alter mRNA levels of GRIA1-4, GRIN1 or mGluR5 in the VTA (Ghasemzadeh et al., 1999; Lu et al., 2002), although specific subunits were significantly up-regulated in the VTA of human cocaine overdose victims (Tang et al., 2003). The present study supported and extended the findings in rats by demonstrating no significant alteration in iGluR subunits in VTA dopamine neurons in rats with acute and chronic self-administration histories. However, significant elevations have been demonstrated in NMDAR 1 and GluR1 protein levels in the VTA after chronic cocaine administration (Fitzgerald et al., 1996; Churchill et al., 1999; Loftis and Janowsky, 2000) as well as NMDAR 1, GluR2, GluR5, and kainate receptor 2 protein levels in cocaine overdose victims (Tang et al., 2003). The possibility of cocaine-induced alterations in iGluR protein but not mRNA levels in rats should be evaluated further with particular attention to quantification and localization of the subunits with respect to VTA dopamine neurons. The inability to recapitulate the changes observed in cocaine overdose victims in rats self-administering cocaine is likely due to several factors, including differences in anatomical complexity between the rat and human and the chronicity and regimen of cocaine administration. Further studies are warranted to investigate the changes in the levels of these and other transcripts in rats with histories of binge self-administration and abstinence from cocaine as well as similar studies in rhesus monkeys.
Often activated in response to glutamatergic transmission, nNOS was also differentially regulated in response to cocaine self-administration. nNOS produces nitric oxide, which can be either neurotoxic or neuroprotective, depending upon experimental conditions (Khaldi et al., 2002). nNOS inhibitors have been shown to protect against methamphetamine-induced dopamine neurotoxicity, but prolonged exposure to nNOS inhibitors may also lead to apoptosis. In the current experiment, nNOS mRNA decreased with acute cocaine and returned to control levels after chronic cocaine self-administration. For example, previous studies have found that nNOS knockout mice were resistant to locomotor sensitization to cocaine (Itzhak et al., 1998); however, the nNOS knockout was not restricted to the VTA and the results could reflect a nitric oxide deficiency in other brain regions or in nondopaminergic cells of the VTA. Recent studies suggest nNOS is increased by NMDA activation (Khaldi et al., 2002), and nNOS may increase CREB activity (Ciani et al., 2002), proving a mechanism by which nNOS may contribute to cocaine: induced plasticity in the VTA. However, as the activity of nNOS varies among different brain regions, a specific role(s) for nNOS in the VTA as a function of cocaine self-administration remains to be elucidated.
Both acute and chronic cocaine self-administration increased mRNA levels of Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα), a predominant protein in the post synaptic density which is necessary for LTP and is hypothesized to be a molecular correlate of memory (Lisman et al., 2002). The present data support previous studies demonstrating that both cocaine-induced behavioral sensitization and increased dopamine levels in the NAc are dependent upon activation of CaMKII-dependent mechanisms (Pierce and Kalivas, 1997; Pierce et al., 1998). Although the present study found no alterations in the expression levels of iGluR subunits, CaMKII is an important modulator of glutamate transmission, acting as a sensor of Ca2+ current when bound to NMDA receptors (Lisman et al., 2002). In addition, CaMKII phosphorylates AMPA receptor subunits, thereby increasing calcium conductance through the AMPA channels, stimulating CREB and possibly contributing to long-term potentiation in the VTA observed in response to cocaine (Liu and Anand, 2001; Ungless et al., 2001).
Investigators have demonstrated cocaine-induced neuroadaptations in the cAMP pathway in mesolimbic brain regions (Nestler and Aghajanian, 1997). Assessment of various α, β, and γ G protein subunits, CREB, and various protein kinase A subunits revealed only a significant increase in Gαi2 subunit mRNA after acute and chronic cocaine self-administration. The present data contrast previous studies showing decreased ADP ribosylation and immunoreactivity Gαi and Gαo in the VTA of cocaine-treated rats (Nestler et al., 1990; Striplin and Kalivas, 1993). Increased Gαi2 mRNA levels in the present study may serve to decrease adenylate cyclase activity and thereby decrease PKA phosphorylation of CREB. In addition, increased expression of PP2α which dephosphorylates a number of different kinases and their substrates, including CaMKII (Millward et al., 1999), may further serve to regulate the activity of CREB and other constituents of the cAMP pathway in response to cocaine.
In summary, the present study provides direct evidence of cocaine-induced adaptations in VTA dopamine neurons after cocaine self-administration. Understanding alterations in the functional integrity of neurotransmission within the mesocorticolimbic dopamine system is a critical step in the development and/or refinement of pharmacotherapies for cocaine addiction. Future characterization of altered gene and protein expression will provide a panoramic view of the potential molecular underpinnings of cocaine addiction. Future studies should include a comparison of genomic and proteomic alterations, examination of gene expression in sub-populations of VTA dopaminergic neurons based on axonal targets, assessment of binge cocaine access on gene and protein expression in these populations. Efforts should also include attempts to recapitulate biochemical alterations identified in human cocaine overdose victims in animal models to refine/generate more appropriate biological models of the cocaine addictive process.
Acknowledgments
This research was supported by the National Institute on Drug Abuse Grants DA13234 and DA13772 (to S.E.H.).
ABBREVIATIONS
- VTA
ventral tegmental area
- NAc
nucleus accumbens
- PKA
cAMP-dependent protein kinase
- FR
fixed ratio
- LCM
laser capture microdissection
- TH
tyrosine hydroxylase
- PCR
polymerase chain reaction
- SSC
sodium chloride-sodium citrate
- nNOS
neuronal nitric-oxide synthase
- PP2α
protein phosphatase 2A, catalytic subunit α
- iGluR
ionotropic glutamate receptor
- mGluR
metabotropic glutamate receptor
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- CREB
cAMP response element-binding protein
- CaMKII
calcium/calmodulin-dependent protein kinase II
References
- Ang E, Chen J, Zagouras P, Magna H, Holland J, Schaeffer E, Nestler EJ. Induction of nuclear factor-kappaB in nucleus accumbens by chronic cocaine administration. J Neurochem. 2001;79:221–224. doi: 10.1046/j.1471-4159.2001.00563.x. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science (Wash DC) 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem. 1999;72:2397–2403. doi: 10.1046/j.1471-4159.1999.0722397.x. [DOI] [PubMed] [Google Scholar]
- Ciani E, Guidi S, Bartesaghi R, Contestabile A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem. 2002;82:1282–1289. doi: 10.1046/j.1471-4159.2002.01080.x. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci USA. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Fasulo WH, Hemby SE. Time-dependent changes in gene expression profiles of midbrain dopamine neurons following haloperidol administration. J Neurochem. 2003 doi: 10.1046/j.1471-4159.2003.01986.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WM, Brebner K, Patel KM, Lynch WJ, Roberts DC, Vrana KE. Repeated cocaine self-administration causes multiple changes in rat frontal cortex gene expression. Neurochemical Research. 2002;27:1181–1192. doi: 10.1023/a:1020929526688. [DOI] [PubMed] [Google Scholar]
- Freeman WM, Nader MA, Nader SH, Robertson DJ, Gioia L, Mitchell SM, Daunais JB, Porrino LJ, Friedman DP, Vrana KE. Chronic cocaine-mediated changes in non-human primate nucleus accumbens gene expression. J Neurochem. 2001;77:542–549. doi: 10.1046/j.1471-4159.2001.00252.x. [DOI] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Nelson LC, Lu XY, Kalivas PW. Neuroadaptations in ionotropic and metabotropic glutamate receptor mRNA produced by cocaine treatment. J Neurochem. 1999;72:157–165. doi: 10.1046/j.1471-4159.1999.0720157.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- Haile CN, Hiroi N, Nestler EJ, Kosten TA. Differential behavioral responses to cocaine are associated with dynamics of mesolimbic dopamine proteins in Lewis and Fischer 344 rats. Synapse. 2001;41:179–190. doi: 10.1002/syn.1073. [DOI] [PubMed] [Google Scholar]
- Hasenkamp W, Hemby SE. Functional genomics and psychiatric illness. In: Hofman MA, Boer GJ, Holtmaat AJGD, Van Someren EJW, Verhaagen J, Swaab DF, editors. Progress in Brain Research: Plasticity in the Adult Brain. Vol. 138. Elsevier; Amsterdam: 2002. pp. 277–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI. Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology. 1997a;133:7–16. doi: 10.1007/s002130050365. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Ginsberg SD, Brunk B, Trojanowski JQ, Eberwine JH. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry. 2002;59:631–640. doi: 10.1001/archpsyc.59.7.631. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Johnson BA, Dworkin SI. Neurobiological basis of drug reinforcement. In: Johnson BA, Roache JD, editors. Drug Addiction and Its Treatment: Nexus of Neuroscience and Behavior. Lippincott Williams & Wilkins; Philadelphia: 1997b. pp. 137–169. [Google Scholar]
- Hemby SE, Martin TJ, Co C, Dworkin SI, Smith JE. The effects of intravenous heroin administration on extracellular nucleus accumbens dopamine concentrations as determined by in vivo microdialysis. J Pharmacol Exp Ther. 1995;273:591–598. [PubMed] [Google Scholar]
- Hemby SE, Trojanowski JQ, Ginsberg SD. Neuron specific age related decreases in dopamine receptor subtype mRNAs. J Comp Neurol. 2003;456:176–183. doi: 10.1002/cne.10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope B, Kosofsky B, Hyman SE, Nestler EJ. Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci USA. 1992;89:5764–5768. doi: 10.1073/pnas.89.13.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S, Kohl RR, McBride WJ. GABA(A) receptor blockade in the anterior ventral tegmental area increases extracellular levels of dopamine in the nucleus accumbens of rats. J Neurochem. 1997a;69:137–143. doi: 10.1046/j.1471-4159.1997.69010137.x. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, Murphy JM, McBride WJ. Self-infusion of GABA(A) antagonists directly into the ventral tegmental area and adjacent regions. Behav Neurosci. 1997b;111:369–380. doi: 10.1037//0735-7044.111.2.369. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Ali SF, Martin JL, Black MD, Huang PL. Resistance of neuronal nitric oxide synthase-deficient mice to cocaine-induced locomotor sensitization. Psychopharmacology. 1998;140:378–386. doi: 10.1007/s002130050779. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992a;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992b;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung BJ, Peris J. Lack of allosteric modulation of striatal GABA(A) receptor binding and function after cocaine sensitization. Pharmacol Biochem Behav. 2001;70:55–63. doi: 10.1016/s0091-3057(01)00580-9. [DOI] [PubMed] [Google Scholar]
- Kamme F, Salunga R, Yu J, Tran DT, Zhu J, Luo L, Bittner A, Guo HQ, Miller N, Wan J, Erlander M. Single-cell microarray analysis in hippocampus CA1: demonstration and validation of cellular heterogeneity. J Neurosci. 2003;23:3607–3615. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaldi A, Chiueh CC, Bullock MR, Woodward JJ. The significance of nitric oxide production in the brain after injury. Ann NY Acad Sci. 2002;962:53–59. doi: 10.1111/j.1749-6632.2002.tb04055.x. [DOI] [PubMed] [Google Scholar]
- Koob GF. Drug addiction: the yin and yang of hedonic homeostasis. Neuron. 1996;16:893–896. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Lilly SM, Tietz EI. Chronic cocaine differentially affects diazepam’s anxiolytic and anticonvulsant actions. Relationship to GABA(A) receptor subunit expression. Brain Res. 2000;882:139–148. doi: 10.1016/s0006-8993(00)02858-4. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu JG, Anand KJ. Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev. 2001;38:1–19. doi: 10.1016/s0165-0173(01)00057-1. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Regulation of NMDA receptor subunits and nitric oxide synthase expression during cocaine withdrawal. J Neurochem. 2000;75:2040–2050. doi: 10.1046/j.1471-4159.2000.0752040.x. [DOI] [PubMed] [Google Scholar]
- Lu W, Monteggia LM, Wolf ME. Repeated administration of amphetamine or cocaine does not alter AMPA receptor subunit expression in the rat midbrain. Neuropsychopharmacology. 2002;26:1–13. doi: 10.1016/S0893-133X(01)00272-X. [DOI] [PubMed] [Google Scholar]
- Mark GP, Hajnal A, Kinney AE, Keys AS. Self-administration of cocaine increases the release of acetylcholine to a greater extent than response-independent cocaine in the nucleus accumbens of rats. Psychopharmacology. 1999;143:47–53. doi: 10.1007/s002130050918. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science (Wash DC) 1997;278:58– 63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Terwilliger RZ, Walker JR, Sevarino KA, Duman RS. Chronic cocaine treatment decreases levels of the G protein subunits Gi alpha and Go alpha in discrete regions of rat brain. J Neurochem. 1990;55:1079–1082. doi: 10.1111/j.1471-4159.1990.tb04602.x. [DOI] [PubMed] [Google Scholar]
- Nye HE, Hope BT, Kelz MB, Iadarola M, Nestler EJ. Pharmacological studies of the regulation of chronic FOS-related antigen induction by cocaine in the striatum and nucleus accumbens. J Pharmacol Exp Ther. 1995;275:1671–1680. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4. Academic Press; San Diego: 1998. [Google Scholar]
- Peris J. Repeated cocaine injections decrease the function of striatal gamma-aminobutyric acid(A) receptors. J Pharmacol Exp Ther. 1996;276:1002–1008. [PubMed] [Google Scholar]
- Peris J, Jung BJ, Resnick A, Walker P, Malakhova O, Bokrand Y, Wielbo D. Antisense inhibition of striatal GABAA receptor proteins decreases GABA-stimulated chloride uptake and increases cocaine sensitivity in rats. Brain Res Mol Brain Res. 1998;57:310–320. doi: 10.1016/s0169-328x(98)00102-8. [DOI] [PubMed] [Google Scholar]
- Pettit HO, Justice JB., Jr Dopamine in the nucleus accumbens during cocaine self-administration as studied by in vivo microdialysis. Pharmacol Biochem Behav. 1989;34:899–904. doi: 10.1016/0091-3057(89)90291-8. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. J Neurosci. 1997;17:3254–3261. doi: 10.1523/JNEUROSCI.17-09-03254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Quick EA, Reeder DC, Morgan ZR, Kalivas PW. Calcium-mediated second messengers modulate the expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1998;286:1171–1176. [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science (Wash DC) 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striplin CD, Kalivas PW. Robustness of G protein changes in cocaine sensitization shown with immunoblotting. Synapse. 1993;14:10–15. doi: 10.1002/syn.890140103. [DOI] [PubMed] [Google Scholar]
- Suaud-Chagny MF, Chergui K, Chouvet G, Gonon F. Relationship between dopamine release in the rat nucleus accumbens and the discharge activity of dopaminergic neurons during local in vivo application of amino acids in the ventral tegmental area. Neuroscience. 1992;49:63–72. doi: 10.1016/0306-4522(92)90076-e. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Abe S, Yamaguchi M, Baba A, Hori T, Shiraishi H, Ito T. Effects of cocaine administration on receptor binding and subunits mRNA of GABA(A)-benzodiazepine receptor complexes. Synapse. 2000;38:198–215. doi: 10.1002/1098-2396(200011)38:2<198::AID-SYN11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Tang WX, Fasulo WH, Mash DC, Hemby SE. Molecular profiling of midbrain dopamine regions in cocaine overdose victims. J Neurochem. 2003;85:911–924. doi: 10.1046/j.1471-4159.2003.01740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecott LH, Barchas JD, Eberwine JH. In situ transcription: specific synthesis of complementary DNA in fixed tissue sections. Science (Wash DC) 1988;240:1661–1664. doi: 10.1126/science.2454508. [DOI] [PubMed] [Google Scholar]
- Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- Toda S, McGinty JF, Kalivas PW. Repeated cocaine administration alters the expression of genes in corticolimbic circuitry after a 3-week withdrawal: a DNA macroarray study. J Neurochem. 2002;82:1290–1299. doi: 10.1046/j.1471-4159.2002.01083.x. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl. 1971;367:1–48. doi: 10.1111/j.1365-201x.1971.tb10998.x. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature (Lond) 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ, Hu XT, Zhang XF, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther. 1995;273:445–454. [PubMed] [Google Scholar]
- White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- Wise RA. Drug-activation of brain reward pathways. Drug Alcohol Depend. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Suzuki T, Abe S, Baba A, Hori T, Okado N. Repeated cocaine administration increases GABA(B(1)) subunit mRNA in rat brain. Synapse. 2002;43:175–180. doi: 10.1002/syn.10037. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Suzuki T, Abe S, Baba A, Ito T, Okado N. Time-course effects of a single administration of cocaine on receptor binding and subunit mRNAs of GABA(A) receptors. Brain Res Mol Brain Res. 2000;81:155–163. doi: 10.1016/s0169-328x(00)00166-2. [DOI] [PubMed] [Google Scholar]