

Abstract
Diabetes mellitus (DM) is a complex metabolic disorder which leads to development of various long-term complications including cardiomyopathy. Oxidative stress due to hyperglycemia plays a key role in the development and progression of diabetic cardiomyopathy (DC). Oxidative stress causes the opening of mitochondrial permeability transition pore (mPTP) eventually leading to myocardium dysfunction. The Ginkgo biloba extract (EGb 761) has antioxidant and mitochondrial membrane potential stabilizing property. Therefore, this study was designed to evaluate the effect of EGb 761 and its possible mechanism of action in DC.
Materials and Methods:
DM was induced by single injection of Streptozotocin (STZ) (50 mg/kg, i.p.) and cardiac dysfunction was developed on 8th weeks after STZ injection. Cardiac dysfunction was assessed by measuring left ventricle weight/body weight (LVW/BW) ratio, left ventricle (LV) collagen content, LV protein content, serum lactate dehydrogenase (LDH) level.
Results:
EGb 761 treatment (started after 7th week of STZ injection and continued for 3 weeks) attenuated cardiac dysfunction in diabetic rats as evidenced by a decrease in LV collagen content, protein content, LVW/BW ratio, serum LDH level. Moreover, EGb 761 attenuated the oxido-nitrosative stress (thiobarbituric acid reactive substances, superoxide anion generation, myocardium nitrite) and concomitantly improved the antioxidant enzyme (reduced glutathione) level as compared to untreated diabetic rats. However, protective effect of EGb 761 was inhibited by atractyloside (mPTP opener) that was given for 3 weeks, 30 min before the EGb 761 treatment. These results indicate that EGb 761 corrects diabetic cardiac dysfunction probably by its direct radical scavenging activity and its ability to inhibit the opening of mPTP channel since the cardioprotective effect of EGb 761 was completely abolished by atractyloside.
Keywords: Cardioprotective, diabetes mellitus, EGb 761, mPTP channel, oxidative stress
INTRODUCTION
Diabetes mellitus (DM) is a metabolic disorder resulting from a defect in insulin secretion and/or insulin action, which results in hyperglycemia with disturbances of carbohydrate, fat, and protein metabolism[1] which gradually lead to chronic complications such as nephropathy,[2] neuropathy,[3] retinopathy, and cardiomyopathy.[4] Among them, diabetic cardiomyopathy (DC) increases the risk of heart failure independently of underlying coronary artery disease and hypertension.[5] The pathogenesis of DC is complex and yet to be explored. Various hypotheses have been proposed, including autonomic dysfunction, metabolic derangements, abnormalities in ion homeostasis, alteration in structural proteins, and interstitial fibrosis.[4]
Hyperglycemia induces oxidative stress via several mechanisms plays the key role in the development and progression of DC.[6] These include activation of xanthine and NADPH oxidases, cyclooxygenase, uncoupled nitric oxide synthase (NOS), glucose autoxidation, polyol pathway and formation of advanced glycation end products (AGE).[5,6] Activation of all these pathways causes increased production of superoxide anion (.O2-) which further activates stress-signaling pathways, leading to increased expression of inflammatory cytokines, Ang II, endothelin-1 and NADPH oxidases and also favors increased expression of NOS through the activation of Nuclear factor-kB (NF-kB), which increases the generation of nitric oxide (NO).[7] Superoxide anion quenches and combine with NO to formed peroxynitrite, a potent oxidant, thereby reducing the efficacy of a potent endothelium-derived vasodilator system. Further, the generated oxido-nitrosative stress causes over-activation of GSK-3β and the opening of mitochondrial permeability transition pore (mPTP), a non-selective pore that opens in the mitochondrial membrane.[8] As a consequence of mPTP opening, uncoupling occurs because of the increased permeability to protons which lead to dissipation of the two components of the proton motive force, the pH gradient and the membrane potential.[9] In the absence of a proton motive force gradient mitochondria cannot synthesize ATP via oxidative phosphorylation. As a result, myocytes in which a significant number of mitochondria have undergone the mPTP opening cannot maintain their ATP levels and the resulting disruption of metabolism and ionic homeostasis leads to necrotic and apoptotic cell death.[10,11]
Recently, herbal therapy has been gaining popularity among clinicians due to their beneficial effects with minimum toxicity. The Ginkgo biloba- EGb 761 is a standardized extracts of dried leaves that contains 24% flavonol glycosides and 6% terpene lactones such as ginkgolides A, B, C, J and bilobalide. It has been used in the treatment of various cardiovascular diseases.[12,13] Moreover, several studies have been reported the antioxidant and anti-apoptotic properties of EGb 761 and its constituents.[13,14,15] Additionally, bilobalide upregulate the anti-apoptotic protein Bcl-2 while attenuates ROS-induced elevation of the pro-apoptotic protein like Bax. It can stabilize the changes in mitochondrial membrane potential occurred due to excessive ROS and peroxynitrite generation.[16,17] However, the pharmacological potential and mechanism of action of EGB-761 in late complication of diabetes i.e. DC is yet to be explored. Therefore, the present study was designed to evaluate the protective effect of EGb 761 and its possible mechanism of action in STZ-DC in rats.
MATERIALS AND METHODS
Age matched Wistar rats (160-260 g) of either sex were employed. Rats were fed on standard chow diet and water was provided ad libitum. They were acclimatized in animal house and were exposed to day and light cycle. The experimental protocol used in the present study was approved by the Institutional Animal Ethics Committee. Every effort was made to minimize the pain and suffering of animals.
Drugs and chemicals
EGb 761 was obtained Ex-gratia from Dr. Willmar Schwabe Pharmaceuticals, Germany and was dissolved in 3% ethanol and 7% saline. STZ and Atractyloside were purchased from Sigma Aldrich Ltd. (St. Louis, USA). All other reagents used in the present study were of analytical grade and freshly prepared.
Induction of DM and DC
Diabetes mellitus was induced by single injection of STZ (50 mg/kg, i.p.) dissolved in freshly prepared ice cold citrate buffer (pH 4.5) and animals having random serum glucose more than 240 mg/dl were considered as diabetic. Persistent hyperglycemia, after 8 weeks of STZ administration, has been reported to induce DC,[18] therefore, EGb 761 treatment was started on 7th week of diabetic animals and was continued by 10 weeks. Dose selection was based on our previous study[19] as well by the pilot study that shown maximum protective effect at a dose of 50 mg/kg. Therefore, 50 mg/kg dose of EGb 761 was administered in all treated group.
Experimental design
The animals were divided into five groups and each group consisted of six rats.
Group I (Normal Control): Rats were maintained on standard food and water regime with no treatment and were sacrificed after 10th weeks.
Group II (Diabetic Control): Rats were administered STZ (50 mg/kg, i.p., once) dissolved in citrate buffer (pH 4.5) and were sacrificed after 10th week of STZ administration for accessing the biochemical parameters.
Group III (EGb 761 treated-Diabetic rats): EGb 761 (50 mg/kg, i.p.) treatment was started on 7th week after STZ injection and was continued by 10th week. Animals were sacrificed after 10th weeks.
Group IV (Atractyloside in EGb 761 treated Diabetic rats): The atractyloside (5 mg/kg, i.p.) dissolved in normal saline was given for 3 week, starting after 7th week of STZ-diabetic rats, 30 min before EGb 761 administration.
Group V (Lisinopril treated group): The diabetic rats, after 7th weeks of STZ injection, were given lisinopril (1 mg/kg, p.o.) for 3 weeks, used as standard group for comparison.
Methods
Estimation of serum glucose level
The blood was collected from animals and serum was separated and stored at 4-6°C for estimation of various biochemical parameters. The glucose concentration was estimated by glucose oxidase-peroxidase (GOD-POD) method[20] using commercially available kit (Coral Clinical System, Goa, India).
Assessment of Cardiomyopathy
The cardiomyopathy was assessed by measuring the left ventricle (LV) collagen content, LVW/BW ratio, LV protein content, serum LDH level and level of oxidative parameters such as TBARS, myocardium nitrite level, superoxide anion generation (SAG) and glutathione (GSH) in the myocardium.
Tissue homogenate preparation
For the estimation of protein and oxidative stress left ventricle (LV) from freshly excised heart was minced and homogenized in 0.1-M Tris HCl buffer (pH 7.4, 10% w/v) using Teflon homogenizer. The clear supernatant of homogenate was used to estimate total protein content,[21] lipid peroxidation[22] and reduced glutathione[23] after centrifugation at 800 g for 10 min.
Estimation of LVW/BW ratio
The LV was properly separated from the whole heart firstly by separating the auricles from the ventricles and then separating the LV including the inter-ventricular septum from the right ventricle. The weight of LV was measured and expressed as ratio, determined by dividing with the body weight of rat just before sacrifice.
Estimation of left ventricle collagen content
LV collagen content was determined by measuring the hydroxyproline concentration.[24]
Estimation of left Ventricle protein content
LV protein content was determined by using bovine serum albumin (BSA) as a standard as previously reported by Lowry[21]
Estimation of TBARS
Lipid peroxidation in the supernatant was determined by measuring the amount of malondialdehyde (MDA) produced primarily or TBARS, according to the method of Ohkawa et al., (1979).[22]
Estimation of reduced glutathione
GSH content in tissue was estimated by the method of Beutler et al., (1963).[23]
Estimation of SAG
The LV superoxide anion generation was estimated in terms of measuring reduced nitroblue tetrazolium (NBT) according to the Wang et al., (1998) method.[25]
Estimation of myocardium nitrite level
To determine total nitrite and nitrate, acidic Griess reaction for color development after the reduction of nitrate with cadmium alloy was used.[26]
Estimation of serum LDH
LDH activity was estimated in serum using commercially available kit, spectrophotometrically at 340-nm wavelength.[27]
STATISTICAL ANALYSIS
All values are expressed as mean ± S.D. Statistical analysis was performed using the Graph Pad Prism 5 Software. The data obtained from various groups were statistically analyzed by using one way analysis of variance (ANOVA) followed by Tukey's multiple comparison test. The P value <0.05 was considered to be statistically significant.
RESULTS
Effect of various pharmacological intervention on serum glucose level
Administration of STZ produced marked increase in serum glucose levels as compared to normal control (saline treated) rats [Table 1]. The administration of EGb 761 (50 mg/kg/day) per se did not affect the serum glucose level in control and diabetic rats. Further, the mPTP opener atractyloside (5 mg/kg/day, for 3 weeks) per se, also did not affect the glucose level in STZ diabetic rats.
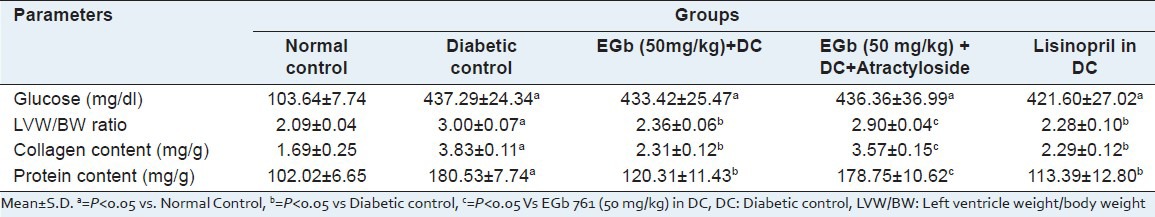
Table 1.
Effect of EGb 761 on Glucose, LVW/BW ratio, collagen content and protein content
Effect of various pharmacological interventions on left ventricle weight/body weight (LVW/BW) ratio
A significant (P < 0.05) increase in LVW/BW ratio was observed in diabetic rats, as compared to normal control. Pre-emptive treatment with EGb 761 (50 mg/kg) for 3 weeks, starting after 7th weeks of STZ-injection, significantly attenuated the LVW/BW ratio as compared to the untreated diabetic rats (Table 1, P < 0.05). However, administration of atractyloside (5 mg/kg/day, for 3 weeks), a mPTP opener, abolished the protective effect of EGb 761. The protective effect shown by the EGb 761 at a dose of 50 mg/kg in diabetic treated rats is similar to standard lisinopril (1 mg/kg, p.o., 3 weeks).
Effect of various pharmacological interventions on left ventricle collagen content
A significant increase in collagen content was observed in diabetic rats, as compared to normal control rats. Administration of EGb 761 (50 mg/kg) for 3 weeks, starting after 7th weeks of STZ injection, significantly attenuated the increased level of collagen content as compared to the untreated diabetic rats (Table 1, P < 0.05). However, administration of atractyloside (5 mg/kg, i.p. 3 week), an opener of mPTP in EGb 761 (50 mg/kg) treated diabetic rat abolished the protective effect of EGb 761 (Table 1, P < 0.05). The protective effect shown by EGb 761 at a high dose in diabetic animals was similar as produced by the standard lisinopril (1 mg/kg, p.o.) in DC.
Effect of various pharmacological interventions on LV protein content
A significant increase in the LV protein content was observed in diabetic rats as compared to normal control rats (Table 1: P <0.05). Administration of EGb 761 at a dose of 50 mg/kg in diabetic animals significantly attenuated LV protein content as compared to untreated diabetic rats. However, administration of atractyloside (5 mg/kg) abolished the protective effect shown by EGb 761 (50 mg/kg) in treated diabetic rats (Table 1, P < 0.05).
Effect of various pharmacological interventions on TBARS, GSH and SAG
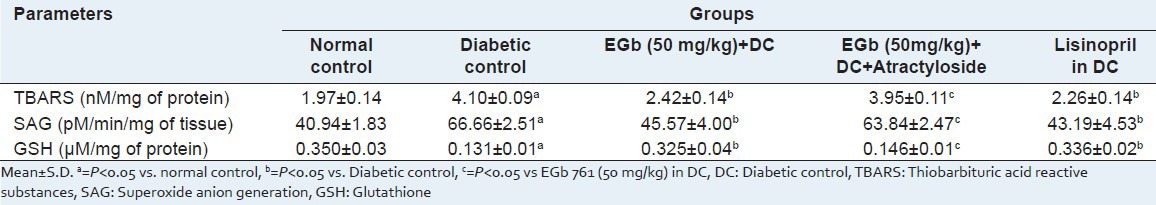
A significant increase in the levels of TBARS and SAG was observed in STZ-treated diabetic rats, as compared to normal control rats (Table 2: P <0.05). However, administration of EGb 761 at a higher dose (50 mg/kg) in DC significantly attenuated TBARS and SAG levels as compared to untreated rats. The level of antioxidant enzyme GSH was significantly decreased in the myocardium of diabetic rats as compared to normal control rats which was restored by EGb 761 treatment [Table 2]. However, administration of atractyloside (5 mg/kg), an opener of mPTP, abolished the protective effect shown with EGb 761 (50 mg/kg) treated diabetic rats.
Table 2.
Effect of EGb 761 on TBARS, SAG and GSH
Effect of various pharmacological interventions on myocardium nitrite level
A significant increase in myocardium nitrite was observed in diabetic rats, as compared to normal control rats (Figure 1, P < 0.05). Administration of EGb 761 at a dose of 50 mg/kg in DC significantly attenuated nitrite level, as compared to untreated diabetic rats. However, administration of atractyloside (5 mg/kg) abolished the protective effect shown by EGb 761 (50 mg/kg) treated diabetic rats [Figure 1]. The protective effect shown by EGb 761 at a dose of 50 mg/kg in DC group was similar to that of standard lisinopril (1 mg/kg, p.o.) in DC group [Figure 1].
Figure 1.
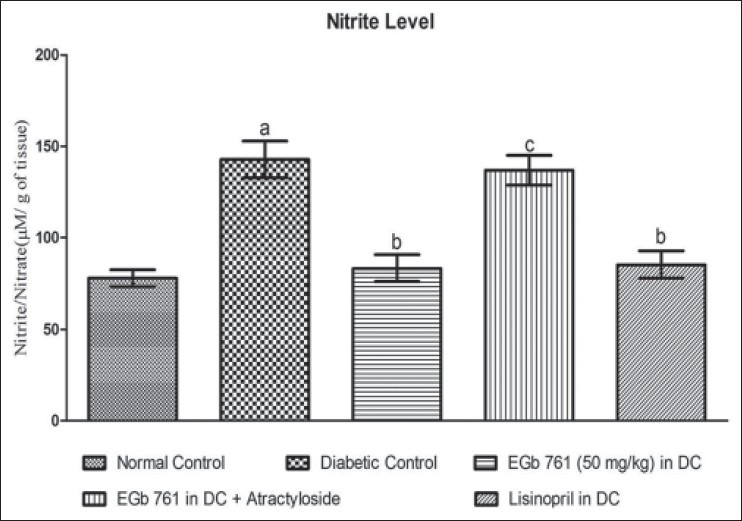
Effect of Various pharmacological interventions on myocardial nitrite level. Results are expressed as Mean ± S.D. a = p<0.05 Vs Normal Control, b = p<0.05 Vs Diabetic Control, c = p<0.05 Vs EGb 761 (50 mg/kg) in DC. DC= diabetic control
Effect of various pharmacological interventions on serum LDH level
A significant increase in serum LDH level was observed in STZ-diabetic rats, as compared to normal control rats (Figure 2, P < 0.05). Administration of EGb 761 (50 mg/kg, i.p.) significantly attenuated LDH level, as compared to untreated diabetic rats (Figure 2, P < 0.05). However, administration of atractyloside (5 mg/kg/day, i.p., for 1 week) abolished the protective effect shown by EGb 761 (50 mg/kg) treated diabetic rats. The protective effect shown by EGb 761 at a dose of 50 mg/kg in DC group was comparable to that of standard lisinopril (1 mg/kg, p.o.) in DC group [Figure 2].
Figure 2.
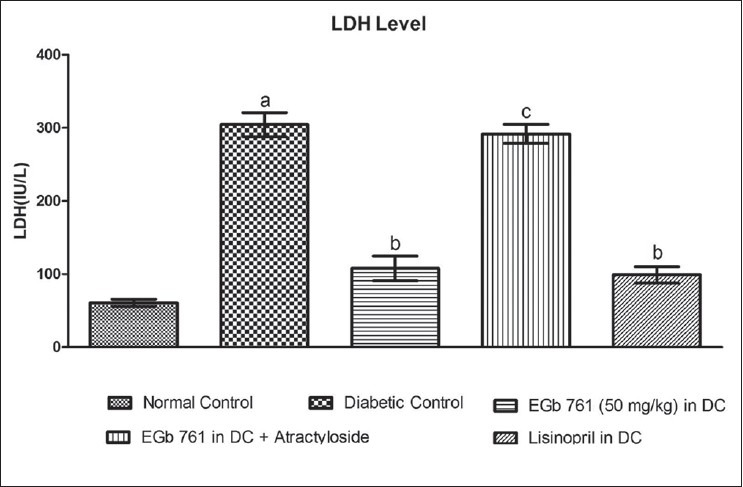
Effect of Various pharmacological interventions on LDH level. Results are expressed as Mean ± S.D. a = P < 0.05 vs Normal Control, b = P < 0.05 vs Diabetic control, c = P < 0.05 vs EGb 761 (50 mg/kg) in DC, DC = Diabetic Control
DISCUSSION
This study demonstrated the protective effect of EGb-761 against cardiac dysfunction in diabetes that may occurs through ROS inhibition and its ability to inhibit mPTP opening in cardiomyocyte. Studies in the experimental animal models such as the STZ-induced diabetic model had helped to define the pathophysiology of cardiac dysfunction and that may mimic the aspects of in humans and for this reason, STZ-diabetic rats have been increasingly used as a model of DC. However, important caveats exist when extrapolating findings obtained in animal models of diabetes to humans.[28] For example, cardiac physiology such as heart rate is different in rodents and humans, and rodent models are less likely to develop atherosclerotic disease in coronary arteries and spontaneous ischemia, which exists in many humans with DM.[29] Nevertheless, STZ animal models provide the opportunity to conduct mechanistic studies, which in some cases have been confirmed in human studies.
In this study, we observed that single injection of STZ caused significant increase in the serum glucose level as compared to normal control. However, EGb 761 showed no effect on the serum glucose level. Moreover, diabetes induced cardiac dysfunction and cardiomyopathy was appeared after 8 week following STZ-injection, which was assessed by measuring the LVW/BW ratio, collagen content, LV protein content, LDH and oxidative stress markers. The nitrite level, and oxidative parameters i.e. TBARS and SAG was significantly increase while the level of antioxidant enzyme reduced glutathione was reduce in STZ-diabetic rats. Therefore, it seems that oxidative stress and mitochondrial dysfunction is a key mechanism involved in the DC.
Hyperglycemia may mediate its damaging effects through excess generation of AGEs,[28] ROS and NO,[30] angiotension-II formation with deactivation of NO,[31] myocardial collagen deposition and fibrosis,[32] activation of DAG/PKC signal transduction pathway leads to reduction in tissue blood flow, increased vascular permeability, alterations in neovascularization and enhanced extracellular matrix deposition.[33] An increase in mitochondrial ROS generation, decreases NO levels, leads to myocardial inflammation and endothelial dysfunction via PARP [poly (ADP-ribose) polymerase], inhibition of which has been shown to reverse diabetic endothelial dysfunction.[34] The excess generation of mitochondrial ROS and mitochondria-dependent caspase-3 pathway is one of the critical signal pathways in apoptotic cell death.[9] Apoptosis-inducing factor and cytochrome-C released from mitochondria is associated with apoptosis protease-activating factor (Apaf-1) and pro-caspase-9, triggering the activation of caspase-3 and resulting in cell death.[35] Oxidative stress accompanied by calcium overload, ATP depletion, and elevated phosphate levels induces mitochondrial permeability transition with formation of nonspecific pores in the inner mitochondrial membrane.[36,37] mPTP opening results in mitochondrial dysfunction with uncoupled oxidative phosphorylation and ATP hydrolysis, ultimately leading to cell death. Pore opening has been demonstrated in heart failure induced by myocardial infarction in rats as well as in Ca2+- induced cardiomyopathy and DC.[37,38] Antioxidative treatment in addition to the usual insulin substitution would seem sensible in preventing or delaying long-term diabetic complications. Recent studies have shown cardioprotective effects of EGb761 in various preclinical and clinical studies.[13] The flavonoid components of EGb761 scavenge superoxide, hydroxyl radicals, NO and protect myocardium from ischemia-reperfusion injury.[39] Moreover, EGb761 can effectively and extensively counteract the cardial toxicity of doxorubicin via preventing the activation of the p53-mediated, mitochondrion-dependent apoptotic signaling pathway.[40,41]
Administration of EGb 761 at dose of 50 mg/kg significantly attenuated the LV protein content and LV collagen content, the serum LDH level, a biomarker of myocardial injury [Figure 2] and significantly restored the LVW/BW ratio [Table 1]. Further, administration of EGb 761 attenuated the various oxidative stress markers. TBARS, an index of lipid peroxidation, and SAG levels were significantly attenuated by EGb 761. The antioxidant enzyme GSH level was significantly restored to normal level by EGb 761 [Table 2] in diabetic rat. Hence, it seems that EGb 761 might have cardioprotective action by reducing the oxidative stress and concomitantly by restoring the antioxidant enzyme level.
Although in this study, we did not measure the direct peroxynitrite or nitrotyrosine level, but NO, an indirect marker of peroxynitrite formation, was estimated in the myocardium of diabetic rat with or without EGb 761 treatment. EGb 761 administration significantly attenuated the oxidative stress and level of nitrite [Figure 1] and it is in accordance with previous report which shows oxido-nitrosative inhibiting potential of EGb 761.[39] Further, it was known that generated oxido-nitrosative stress causes over-activation of GSK-3β and the opening of mPTP, a non-selective pore, that opens in the mitochondrial membrane. To further explore the involvement of mPTP which is seems to be involved in cardioprotection observed with EGb 761 in DC, we administered, atractyloside, a mPTP opener, for 3 weeks, to access mitochondrial dysfunction and confirm the mitochondrial membrane stabilizing potential of EGb 761, as reported earlier.[40,41]
Our study for the first time showed that administration of atractyloside (mPTP opener) abolished the cardio-protective effect of EGb 761 (50 mg/kg) in STZ-diabetic rats. The cardio-protective effect of EGb 761 seems due to its antioxidant properties along with its ability to inhibit the opening of mPTP since the cardioprotective effect of EGb 761 was completely abolished by the mPTP opener atractyloside.
CONCLUSIONS
The results suggest that EGb 761 has a cardioprotective effects against late complication of diabetes. EGb 761 cardioprotective effect may be mediated by its direct free radical scavenging activity and its ability to inhibit the opening of mPTP channel, since the cardioprotective effect of EGb 761 was completely abolished by atractyloside. Thus EGb 761 may be useful as an adjuvant therapy for the prevention of diabetic cardiomyopathy.
ACKNOWLEDGMENT
We express our gratitude to Shri Parveen Garg, Chairman, ISF College of Pharmacy, Moga, Punjab, India, for his constant support to this study. We are also indebted to Dr. Willmar Schwabe Pharmaceuticals, Germany, for providing EGb 761 as a gift sample for studies.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Marwick TH. The diabetic myocardium. Curr Diab Rep. 2006;6:36–41. doi: 10.1007/s11892-006-0049-0. [DOI] [PubMed] [Google Scholar]
- 2.Parchwani ND, Upadhyah AA. Diabetic nephropathy: Progression and pathophysiology. Int J Med Sci Public Health. 2012;1:59–70. [Google Scholar]
- 3.Chauhan N, Taliyan R, Sharma PL. Effect of dipyrone and thalidomide alone and in combination on STZ-induced diabetic neuropathic pain. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:527–38. doi: 10.1007/s00210-011-0724-9. [DOI] [PubMed] [Google Scholar]
- 4.Jay D, Hitomi H, Griendling KK. Oxidative stress and diabetic cardiovascular complications. Free Radic Biol Med. 2006;40:183–92. doi: 10.1016/j.freeradbiomed.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 5.Opie LH. Cardiomyopathies and heart failure. Heart Metab. 2010;46:17–24. [Google Scholar]
- 6.Spitaler MM, Graier WF. Vascular targets of redox signalling in diabetes mellitus. Diabetologia. 2102;45:476–94. doi: 10.1007/s00125-002-0782-0. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci. 2005;26:302–10. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, et al. NF-kB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res. 2010;85:473–83. doi: 10.1093/cvr/cvp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2010;192:1001–4. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halestrap AP. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem Soc Trans. 2006;34:232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;5:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 12.Drieu K. Preparation and definition of Ginkgo biloba extract. In: Funfgeld EW, editor. Rokan (Ginkgo biloba) Recent results in pharmacology and clinic. Berlin: Springer-Verlag; 1988. pp. 32–6. [Google Scholar]
- 13.Pietri S, Maurelli E, Drieu K, Culcasi M. Cardioprotective and anti-oxidant effects of the terpenoid constituents of Ginkgo biloba extract (EGb 761) J Mol Cell Cardiol. 1997;29:733–42. doi: 10.1006/jmcc.1996.0316. [DOI] [PubMed] [Google Scholar]
- 14.Smith JV, Burdick AJ, Golik P, Khan I, Wallace D, Luo Y. Anti-apoptotic properties of Ginkgo biloba extract EGb 761 in differentiated PC12 cells. Cell Mol Biol. 2002;48:699–707. [PubMed] [Google Scholar]
- 15.Eckert A, Keil U, Kressmann S, Schindowski K, Leutner S, Leutz S, et al. Effects of EGb 761 Ginkgo biloba extract on mitochondrial function and oxidative stress. Pharmacopsychiatry. 2003;36:S15–23. doi: 10.1055/s-2003-40449. [DOI] [PubMed] [Google Scholar]
- 16.Zhou LJ, Zhu XZ. Reactive oxygen species-induced apoptosis in PC12 cells and protective effect of bilobalide. J Pharmacol Exp Ther. 2000;293:982–8. [PubMed] [Google Scholar]
- 17.Abdel-Kader R, Hauptmann S, Keil U, Scherping I, Leuner K, Eckert A, et al. Stabilization of mitochondrial function by Ginkgo biloba extract (EGb 761) Pharmacol Res. 2007;56:493–502. doi: 10.1016/j.phrs.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Zhu XX, Zhou XP, Zhong XL, Zhong CS, Yu YF. Streptozotocin induced cardiomyopathy in diabetic rats. Chin Med J (Engl) 1993;106:463–6. [PubMed] [Google Scholar]
- 19.Taliyan R, Sharma PL. Protective effect and potential mechanism of Ginkogo-biloba extract-EGb-761 on STZ-induced neuropathic pain behavior in rats. Pytother Res. 2012;26:1823–9. doi: 10.1002/ptr.4648. [DOI] [PubMed] [Google Scholar]
- 20.Trinder P. Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann Clin Biochem. 1969;6:24. [Google Scholar]
- 21.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with folin-phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 22.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–8. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 23.Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–8. [PubMed] [Google Scholar]
- 24.McAnulty RJ. Methods for measuring hydroxyproline and estimating in vivo rates of collagen synthesis and degradation. Methods Mol Med. 2005;117:189–207. doi: 10.1385/1-59259-940-0:189. [DOI] [PubMed] [Google Scholar]
- 25.Wang HD, Pagano PJ, Du Y, Cayatte AJ, Quinn MT, Brecher P, et al. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ Res. 1998;82:810–8. doi: 10.1161/01.res.82.7.810. [DOI] [PubMed] [Google Scholar]
- 26.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite and [15 N] nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 27.Lum G, Gambino SR. A comparison of serum versus heparinised plasma for routine chemistry tests. Am J Clin Pathol. 1974;61:108. doi: 10.1093/ajcp/61.1.108. [DOI] [PubMed] [Google Scholar]
- 28.Eizirik DL, Pipeleers DG, Ling Z, Welsh N, Hellerstrom C, Andersson A. Major species differences between humans and rodents in the susceptibility to pancreatic beta-cell injury. Proc Natl Acad Sci U S A. 1994;91:9253–6. doi: 10.1073/pnas.91.20.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med. 2009;13:1751–64. doi: 10.1111/j.1582-4934.2008.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Taliyan R, Sharma PL. Possible mechanism of protective effect of thalidomide in STZ-induced-neuropathic pain behavior in rats. Inflammopharmacology. 2012;20:89–97. doi: 10.1007/s10787-011-0106-4. [DOI] [PubMed] [Google Scholar]
- 31.Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells-implications in cardiovascular disease. Braz J Med Biol Res. 2004;37:1263–73. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- 32.Marijianowski MM, Teeling P, Mann J, Becker A. Dilated cardiomyopathy is associated with an increase in the type I/type III collagen ratio: A quantitative assessment. J Am Coll Cardiol. 1995;25:1263–72. doi: 10.1016/0735-1097(94)00557-7. [DOI] [PubMed] [Google Scholar]
- 33.Geraldes P, King GL. Activation of protein Kinase C Isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiu J, Farhangkhoee H, Xu BY, Chen S, George B, Chakrabarti S. PARP mediates structural alterations in diabetic cardiomyopathy. J Mol Cell Cardiol. 2008;45:385–93. doi: 10.1016/j.yjmcc.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 36.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther. 2009;330:670–8. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 37.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–44. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira PJ, Seiça R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, et al. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–4. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 39.Shen J, Wang J, Zhao B, Hou J, Gao T, Xin W. Effects of EGb 761 on nitric oxide and oxygen free radicals, myocardial damage and arrhythmia in ischemia-reperfusion injury in vivo. Biochim Biophys Acta. 1998;1406:228–36. doi: 10.1016/s0925-4439(98)00007-6. [DOI] [PubMed] [Google Scholar]
- 40.Shen J, Lee W, Gu Y, Tong Y, Fung P, Tong L. Ginkgo biloba extract (EGb761) inhibits mitochondria-dependent caspase pathway and prevents apoptosis in hypoxia-reoxygenated cardiomyocytes. Chin Med. 2011;6:1–8. doi: 10.1186/1749-8546-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh YC, Liu TJ, Wang LC, Lee HW, Ting CT, Lee WL, et al. A standardized extract of Ginkgo biloba suppresses doxorubicin- induced oxidative stress and p53-mediated mitochondrial apoptosis in rat testes. Br J Pharmacol. 2009;156:48–61. doi: 10.1111/j.1476-5381.2008.00042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]