

Abstract
RNA dynamics play a fundamental role in many cellular functions. However, a general framework is lacking to describe these complex processes, which typically consist of many structural maneuvers taking place over timescales ranging from picoseconds to seconds. Here we classify RNA dynamics into distinct modes representing transitions between basins on a hierarchical free energy landscape. These include large-scale secondary structural transitions occurring at >0.1 s timescales, base-pair/tertiary dynamics occurring at μs-ms timescales, stacking dynamics at ns-μs and other ‘jittering’ motions occurring at ps-ns timescales. We review various modes within these three different tiers, the different mechanisms by which they are used to regulate function, and how they can be coupled together to achieve greater functional complexity.
Keywords: RNA flexibility, riboswitches, regulatory RNA, molecular adaptation, RNA catalysis
INTRODUCTION
Composed of only four chemically similar nucleotides, RNA was long thought to lack the chemical and structural complexity needed to drive biochemical processes that power living cells, limited instead to a role as a rudimentary messenger. However, discoveries in molecular biology over the past three decades have shown that nothing could be further from the truth. RNA is capable of catalytic activity and can fold into complex 3D structures rivaling those of proteins (1–5). Seventy-five percent of the human genome is now believed to code for RNAs, the functions of which we are only beginning to uncover, whereas less than 2% code for proteins (6, 7). Even classic RNAs such as ribosomal, transfer, and messenger RNAs play surprisingly complex roles in protein synthesis (5, 8).
The functional complexity of RNA and its involvement in a wide range of sophisticated functions can be attributed not only to its ability to fold into complex 3D structures, but perhaps even more importantly, on its ability to undergo precise conformational changes in a biologically specific manner in response to a wide range of cellular cues consisting of proteins, ligands, metals, changes in temperature, and pH (9, 10). These dynamics can be highly complex, often involving many structural maneuvers that take place over timescales spanning from picoseconds to hundreds of seconds. What is lacking is a framework for simplifying this dazzling complexity so that one can begin to see and comprehend the ‘signal buried within the noise’.
In this review, we introduce a framework for deconstructing RNA dynamics into a set of distinct motional modes that have characteristic timescales representing transitions between basins within a hierarchical free energy landscape. This simplifies the description of complex RNA dynamics in terms a set of recurring motional modes, providing a common language that makes it possible to identify similar themes across different RNA functional contexts. This framework is very similar to that first introduced by Frauenfelder, Wolynes, and coworkers to describe protein dynamics in terms of transitions between basins on different tiers of a free energy landscape (11). We review three broad classes of RNA dynamics, their biological significance, and how interdependencies among these classes can be harnessed to achieve yet further functional complexity.
DECOMPOSING RNA DYNAMICS INTO HIERARCHICAL MOTIONS
In solution, a given RNA does not fold into a single structure, but rather forms a statistical distribution, or ensemble, of many inter-converting conformations. As shown for proteins, this statistical ensemble can be described in terms of a continuous free energy landscape, which specifies the free energy of every atomic configuration (11). The population of each configuration depends on its free energy whereas the rates of inter-conversion between individual configurations depend on the height of the barriers separating them. Although the free energy landscape can in principle be arbitrarily complex, in many biomolecules it is hierarchically organized into local energetic minima containing conformational sub-states (CS) that are separated by large barriers, each of which is in turn sub-divided into a larger number of local energetic minima that are separated by lower barriers, and so forth (Figure 1). These hierarchically organized energetic layers form different “Tiers” (Tier 0, Tier 1, etc…), and RNA dynamics can in turn be hierarchically organized in terms of transitions between CSs within different tiers.
Figure 1.

The different tiers of RNA dynamics. At the lowest level of the hierarchy are secondary structure dynamics, which define broad free energy basins with high separating barriers. Within each secondary structure are smaller alternative base pairing arrangements that define Tier 1 dynamics. These include base pair melting (blue, left), reshuffling (middle right, red), and tertiary pairing (green). Each local pairing basin in turn defines a limited set of 3D conformations, transitions between which comprise Tier 2 dynamics. These dynamics include loop dynamics (left, red) and inter-helical dynamics (right, green). Although inter-helical and loop-dynamics have similar barrier heights, due to the larger number of involved coordinates inter-helical dynamics typically proceed more slowly (long rough separating barrier).
The above hierarchical description of free energy landscapes and dynamics was developed originally to explain protein dynamics, and specifically myoglobin dynamics. However, it is also well suited to describe RNA dynamics in general. There are two reasons for this. First, the RNA free energy landscape is strongly hierarchical, and naturally organized into ‘secondary’ and ‘tertiary’ structure levels (12, 13). Unlike proteins, interactions that stabilize secondary structure are much stronger than those that stabilize other aspects of 3D structure, and dynamics at the secondary structure level (Tier 0) occur quite independently of those on lower levels (Tier 1, 2, etc.). Second, the RNA free energy landscape is rugged, with significant barriers separating competing conformations at both the secondary and tertiary structural level. Thus, RNA lends itself to a description in terms of individual CSs within each tier. For our discussion, Tier 0 will represent RNA conformations with distinct secondary structure, Tier 1 conformations that have small differences in base-pairing, and Tier 2 conformations that have similar secondary structures and base-pairing but that differ in other aspects of structure (Figure 1).
Other than being hierarchal, there are three other aspects of the RNA free energy landscape that are worth mentioning. First, there is mounting evidence that cellular cues act to change the energetic balance of pre-existing CSs to trigger specific biological outcomes (10). In other words, the favorable CSs that exist in quiescent RNAs represent the same conformations that nature uses to regulate biological outcomes. Second, as we will illustrate below, nature takes advantage of differences in rates of exchange between CSs on different Tiers to ensure that conformational changes only take place once a given cellular cue is presented, or take place sufficiently rapidly so as to not slow down biochemical processes. Finally, although limited in number, the CSs that populate the free energy landscape can have wildly different conformations, making it possible to effect very large yet highly specific changes in structure.
In what follows, we describe the different Tiers of RNA dynamics and highlight their biological significance.
TIER 0: SECONDARY STRUCTURE DYNAMICS
Overview
Due to the inherent degeneracies of the energetics of base-pairing and stacking, RNA molecules rarely fold into a single secondary structure. Rather, there are additional competing secondary structural forms that can become appreciably populated under the right physiological conditions (14–16). In sequences that have evolved to favor a single functional conformation, these alternative secondary structures present a challenge to RNA folding (17, 18). However, in other cases, this promiscuous pairing ability is deliberately harnessed to create functional transitions between alternative secondary structures (Figure 2).
Figure 2.

(A) Three state secondary structure equilibrium of the add adenine riboswitch. In the adenine-bound conformation both the start codon (green) and ribosome binding site (red) are exposed, upregulating translation. The temperature dependence of the apo-state secondary structure equilibrium offsets the increased ligand affinity of the binding-competent conformation at low temperature (30). (B) Example of a transcriptional acting adenine riboswitch. Ligand binding stabilizes a transient secondary structure, sequestering residues that would otherwise pair with downstream transcribed sequences to form the thermodynamically favored terminator stem. (C) The HIV-1 5’ leader couples exposure of the start codon of the downstream-encoded gag protein to sequestration of the dimerization initiation site (DIS; red), promoting translation while inhibiting dimerization (left). In a process promoted by the nucleocapsid chaperone protein (NC; purple), the leader undergoes a secondary structure switch that exposes the DIS and sequesters the start codon, attenuating translation and promoting dimerization, which initiates genome packaging (right) (40).
Because of the overwhelming stability of RNA duplexes, transitions to conformations possessing just a few less pairs are strongly disfavored. Thus, in theory, secondary structure dynamics can be highly specific, directed to one of a small number of favorable conformations. However, as a transitioning duplex typically must break half of its base-pairs, this stability comes at the cost of slow dynamics timescales (19). For example, transitions of a ‘bistable’ RNA between two alternative 5-bp helices occur at rates of ~0.1 s−1 at 298 K (19). For larger helices, the timescale of interconversion can approach the expected lifetime of the RNA and can be slowed down even further by formation of long-lived intermediates (18, 20–22).
Biological significance
Nature often exploits dynamics between different secondary structures to sequester or expose structural elements that interact with cellular factors in a manner dependent on cellular cues. This gives rise to molecular switches, termed riboswitches, that can be integrated into a wide variety of biological circuits (23). These structural elements can be either a contiguous stretch of nucleotides that are either single-stranded and exposed, or sequestered into hairpins via base-pairing, or be an entire hairpin that is either present or absent from the secondary structure. For example, single stranded mRNA ribosome binding sites (24), degradative endonuclease cleavage sites (25), and splicing sites (26), as well transcription terminator hairpins (27), among others, have all been shown to be exposed or sequestered by secondary structure changes as part of regulatory processes (Figure 2).
Secondary structure dynamics present both a challenge — fast response times are often needed to efficiently respond to biological stimuli — as well as an opportunity — the transitions are unlikely to occur spontaneously in an undirected manner. Nature has evolved several strategies to overcome the first problem, allowing it to take advantage of the second benefit to construct robust regulatory switches. Some secondary structure transitions can be used ‘as is’ without the need for intervening with the rates of inter-conversion. Here, a pre-existing secondary structure equilibrium is precisely tuned by primary sequence to rapidly and ‘reversibly’ respond to changes in small molecule concentration (23, 28), or to temperature in ‘thermosensors’ (29). Many riboswitches that regulate gene expression at the translational level are controlled by such thermodynamic mechanisms. In an interesting example, dynamics between three alternative secondary structures that respond to temperature and small molecule concentration were shown to collaborate in the same adenine riboswitch to maintain robust switching activity across a broad range of temperatures (30). A temperature sensitive ‘pre’ equilibrium that exchanges with rates of ~0.5 s−1 between two translational ‘off’ states sequesters the ligand binding pocket to inhibit switching, serving to compensate for the temperature sensitivity of the ligand modulated equilibrium between translational ‘on’ and ‘off’ states (Figure 2A).
In other cases, the barrier heights between two secondary structures are large enough such that exchange cannot happen within reasonable timescales without some form of intervention. For example, rapid secondary structure transitions are required in riboswitches that regulate gene expression at the transcriptional level, where the structural change has to take place during a short time window dictated by the rates of co-transcriptional folding. An ingenious form of intervention involves altering the co-transcriptional folding pathways, thus acting before a stable secondary structure element has had a chance to fully form. In these cases, a wide range of effectors such as temperature (31), small molecules (23, 28), metals (32), pH (33), proteins (34), or trans-acting RNAs (35), stabilize a metastable secondary structure during co-transcriptional folding that sequesters sequence elements that would otherwise pair with downstream nucleotides emerging from the polymerase (Figure 2B). Not only do such systems allow rapid exchange between conformations that would otherwise be separated by insurmountable energetic barriers, they also ensure that the conformational switch rarely takes place in the absence of effectors.
Nature has also evolved a variety of protein chaperones and helicases that are able to accelerate transitions between more stable secondary structures, as well as clock the transitions so that they take place at specific time points. These proteins act by either destabilizing duplexes or stabilizing unpaired states to lower the effective transition barrier (see melting dynamics below) (21). Such proteins make it possible to efficiently anneal small RNAs (sRNAs) to potentially structured regions of their mRNA targets (36). During assembly of the eukaryotic spliceosome, helicases are used to catalyze successive global secondary structure transitions that serve as a multistep proofreading mechanism that ensure that only optimal substrates are spliced (37). These proteins can also serve as regulatory triggers, with an increase in protein concentration promoting transitions of RNAs to alternative conformations, either by destabilizing a pre-existing state or stabilizing a new conformation. This mechanism is prominently used by retroviruses to regulate genome translation, dimerization and packaging (38–40) (Figure 2C).
TIER 1: BASE-PAIR AND TERTIARY DYNAMICS
Once formed, a given secondary structure may experience smaller more localized changes in base-pairing, or form long-range tertiary interactions between remotely positioned residues involving base-pairing and other interactions. These dynamics do not lead to large-scale changes in secondary structure and can therefore be considered as basins within a given secondary structure CS. We distinguish four different types of dynamics (i) base-pair melting (ii) base-pair reshuffling (iii) base-pair isomerization, and (iv) long-range tertiary interactions (Figure 3).
Figure 3.

Modes of Tier 1 dynamics.
Base-pair Melting
Overview
All base-pairs, including Watson-Crick (WC) base-pairs, transiently break apart (melt) and adopt an ‘open’ conformation that briefly exposes residues to solvent or nearby binding partners. For WC base-pairs, melting occurs on 0.1–50 ms timescales, depending on the identity of the pair as well as on the strength of the stacking interactions with neighboring base-pairs (41, 42). Unlike other forms of base-pair dynamics reviewed below, the open state is strongly energetically disfavored compared to the closed state, by roughly 7–9 kcal/mol for WC base-pairs in duplex RNA. As a result, at room temperature, the open state of WC base-pairs has a minute population of 10−5–10−6 and lifetime of only 1–100 ns (41, 42). However, the population and lifetime of the open state can increase considerably in helix-terminating pairs that only have one set of nearest-neighbor stacking interactions, such as for base-pairs near bulges, apical loops, or internal loops, and in non-canonical base pairs (42, 43). To a lesser degree, instability in a single pair can also increase the melting dynamics of non-nearest-neighbor pairs, though the mechanisms underlying this phenomenon are not fully understood (43).
Biological significance
Sites of increased base opening or transient melting are common trigger points for effecting larger-scale secondary structure transformations. RNA chaperones and helicases operate by lowering the barriers to melting dynamics and then binding with high affinity to the exposed residues (Figure 4) (44, 45). This binding in turn enhances the melting dynamics and thus refolding ability of pairs that neighbor the chaperone/RNA interface.
Figure 4.
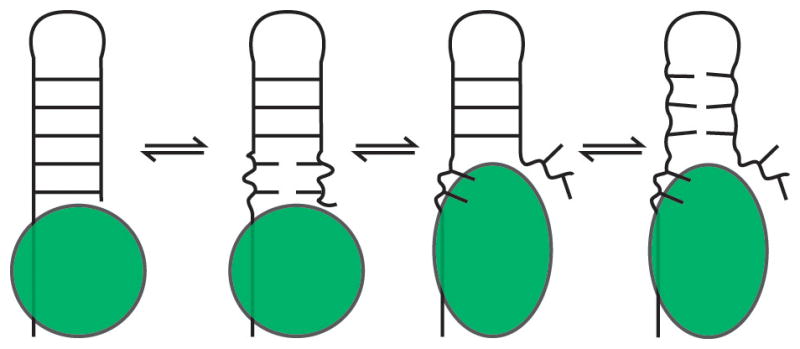
Example of an RNA chaperone. The bound chaperone destabilizes the neighboring RNA helix, promoting melting dynamics, and then binds the exposed nucleotides. The other strand is released and can interact with other RNAs, and the remainder of the helix is also destabilized.
Melting of weak base-pairs can also expose residues that participate in RNA-RNA tertiary interactions and RNA-protein binding motifs (46–49). In an interesting example, in the ribosomal peptidyl transferase center tertiary interactions with the A-site tRNA induces melting of a helix-terminating GU pair in the 23S rRNA that otherwise helps to protect the aminoacyl-linkage of the P-site tRNA from spontaneous hydrolysis (50). Melting dynamics also serve as the basis for several regulatory strategies. For example, the interplay between the helicase activity of the ribosome (51) and the melting dynamics of mRNA secondary structures has been proposed to serve as a second genetic code that regulates the rate of translocation and therefore co-translational protein folding (52), and has also been implicated in the mechanism underlying ribosomal frameshifting (53).
Base-pair Reshuffling
Overview
These dynamics typically involve local rearrangements in base-pairing partners in and around non-canonical structures such as apical and internal loops (54). The transitions typically require the disruption of one or two non-canonical or unstable base-pairs, and therefore typically occur at μs-ms timescales, similar to or slightly faster than base-pair opening (54). In general, the alternative base-pairing is energetically destabilized relative to the more favorable state by <3 kcal/mol and therefore have populations of ≥0.5% and lifetimes on the order of >50 μs (54). Compared to global secondary structure transitions, these more localized changes in base-pairing occur at nearly three orders of magnitude faster rates, without the need for assistance from cellular factors or co-transcriptional folding.
An example of such dynamics in an apical loop is provided by HIV-1 TAR, where two such CSs have been identified using relaxation dispersion NMR spectroscopy (Figure 5A) (54). In the energetically favorable CS, the hexanucleotide apical loop adopts a conformation in which G34 forms a cross-loop Watson-Crick pair with C30, leaving other nucleotides unpaired. By contrast, the energetically less favorable CS, which has a population of 13%, adopts a tetraloop conformation closed by trans-wobble U31-G34 and non-canonical A35+-C30 wobble base pairs (Figure 5A). Prior observations of higher energy CS states involving C-A+ base-pairs in RNA (55–57) and G-C+ Hogsteen base-pairs in DNA (58, 59) suggest that formation of charged base-pairs may be a general feature of Tier 1 dynamics. The ribosomal A-site provides an example of base-pair reshuffling in an internal loop (Figure 5B) (54). Here, adenine and uridine residues alternate between being exposed as a loop or bulge or being sequestered through formation of non-canonical base-pairs.
Figure 5.

Functions of base-pair reshuffling dynamics. (A) In the HIV-1 TAR apical loop, the minor CS sequesters residues involved in HIV Tat and Cyclin T1 binding during transcriptional activation (54). (B) In the major CS of the ribosomal A-site, A92 and A93 are free to interact with and stabilize cognate mRNA/tRNA minihelices during decoding, indicated by the gray dashed arrow and alternative A92/A93 conformation. The minor CS sequesters these residues, inhibiting decoding and also disrupting the B2a inter-subunit bridge (54).
Biological significance
As in global secondary structure transitions, these ‘reshuffled’ CS can differ in whether certain residues are exposed and available for interaction with cellular cues or sequestered through base-pairing interactions. As a result, they can be employed as expose/sequester switches that are much faster than secondary structure transitions. While the function of transient pairing dynamics are still under investigation, several possible biological roles have been proposed.
For example, the higher energy CS in the TAR apical loop discussed above appears to form an auto-inhibited state, as it sequesters residues that are recognized by transcription factors such as Tat (54). Indeed, mutations that stabilize this CS lead to weaker protein binding affinities and inhibit transcriptional activation (Figure 5A). As formation of the A+•C pair in this CS requires protonation, dynamics between the two different CSs are pH-dependent and thus may serve as a regulatory switch. Similar pH-dependent reshuffling of base-pairs have also been observed near the catalytic active sites of the lead-dependent ribozyme (55, 60, 61) and spliceosome (56, 57), and may play important catalytic roles.
For the ribosomal A-site, the higher energy CS sequesters adenine residues that otherwise need to be free to carry out decoding functions (5, 62), and may play a role in processes that bypass decoding such as frameshifting or stop-codon read-through (Figure 5B) (54). In another potential tuning role, a conserved non-canonical motif in one of the helices of the purine riboswitch aptamer was shown to tune ligand affinity and binding kinetics by altering the local pairing dynamics of the ligand-free state (63). More broadly, many internal and apical loops undergo rearrangements in their non-canonical pairs when participating in RNA-RNA tertiary interactions, suggesting that transient pairing dynamics may facilitate these molecular recognition events (64–71).
Base-pair Isomerization
Overview
Two bases can often pair in more than one configuration, representing different sub-states within a secondary structure. For example, there can be a wide variety of G-U, G-G, A-A, and AC base-pairs involving different glycosidic bond angles (syn versus anti), as well as differences in the protonation state such as for A-C base-pairs (Figure 3) (72, 73). Similar to other base-pair dynamics, these different forms can dynamically exchange on μs-ms timescales or can readily adopt different forms dependent on environmental conditions (55, 74–78). These pairs can also involve rare tautomers (79) and in the case of DNA, even Watson-Crick base-pairs have been shown to transiently adopt Hoogsteen base-pairs (58, 59). However, such WC Hoogsteen base-pairs have yet to be reported in A-form RNA.
Biological significance
Isomerizations can significantly alter the chemical presentation of a base-pair by exposing alternative functional groups to the major and minor grooves, and can also affect the overall 3D structure by altering the backbone conformation. These structural changes can play important roles in mediating molecular recognition, such as is observed in binding of the Rev peptide to the HIV Rev Responsive Element (80), RNA tertiary interactions such as in K-turn motifs (81), and specific ion binding in a Group I Intron (82). By changing the local steric profile of the base-pair bordering a junction, these changes may also modulate the inter-helical dynamics across junctions (49). Interestingly, tautomer-driven base-pair isomerizations have been shown to be important in ribosomal decoding (79, 83–85). A recent study reported that uridine tautomerization can allow a non-cognate G-U pair in the mRNA-tRNA minihelix to adopt a WC-like rather than a wobble conformation, changing the steric profile of the pair and circumventing the mechanism used by the ribosome to reject non-cognate codons (79). It should be noted, however, that the high free energy cost of such tautomerizations ensures that decoding accuracy is not significantly comprised (62). Alternatively, post-transcriptional chemical modifications of some tRNA anticodons appear to play an important role in decreasing the energetic cost of tautomerization, allowing the tRNA to form WC-like pairs with multiple different mRNA codons and thus expanding its decoding capacity (83–85).
Tertiary Structure Dynamics
Overview
In many RNAs, distal loops form long-range tertiary contacts that are stabilized by canonical and non-canonical base-pairing, stacking, tightly bound cations, and weaker interactions involving base triples and A-minor motifs (86). Such tertiary interactions play critical roles in stabilizing the overall 3D structure of an RNA and in properly positioning key structural elements that form ligand binding and catalytic sites. The structural elements participating in tertiary interactions can undergo any one of the base-pair dynamic modes, including melting, reshuffling, and isomerization, which result in the dynamic jittering of adjoined stems. In certain cases, these interactions can cooperatively melt, often precipitating large amplitude inter-helical dynamics that lead to global remodeling of 3D structure. Depending on the strength of these interactions, and the extent to which they are disrupted, such motions can occur at timescales ranging between μs-s.
In a growing number of cases it has been shown that tertiary structure dynamics are coupled to other motional modes in Tier 1. As mentioned above, many internal loops undergo reshuffling and melting dynamics upon formation of tertiary contacts. More dramatic changes have also been observed, with the prototypical example being the P5abc domain of the Tetrahymena group I ribozyme. Here, Mg2+ induced folding of tertiary structure is coupled to reshuffling entailing a one base-pair register-shift of the P5c helix. This shift results in a loss of several G-U pairs, but is more than offset by new local non-canonical and long-range tertiary pairs, as well as Mg2+ interactions (Figure 6A) (87). Recent MD and experimental studies have shown that tertiary structure formation and secondary structure reorganization occur concomitantly, with a rate-limiting step that is independent of secondary structure switching (88).
Figure 6.

(A) Coupling of base-reshuffling and tertiary dynamics in the P5abc domain of the Tetrahymena thermophila group I ribozyme. Upon binding of two Mg2+ ions (184), the P5c stem (colored) undergoes a 1-nt register shift, releasing U168 to participate in a long-range pair (right, boxed). Additional tertiary interactions, which are not shown, are also formed upon folding. NMR studies observed the two conformations to be in slow exchange (87), with agreement from recent stopped flow experiments (88). Dashed lines indicate non-canonical pairs. (B) Enzymatic cycle of the hairpin ribozyme (91).
Biological significance
By both modulating access and remodeling the structure of binding and catalytic sites, tertiary structure dynamics can serve a multitude of functions. For example, they play a prominent role in catalytic cycles of ribozymes where they are used to achieve processivity and rapid turnover. In a common strategy, an ‘undocked’ inactive conformation enables rapid substrate binding, which then ‘docks’ into the catalytic active site where it is stabilized and aligned for catalysis by tertiary interactions (Figure 6B) (89–91). Following catalysis, melting of these tertiary interactions precipitates transitions back to the undocked state, where the product is efficiently released. In other catalytic RNAs more local rearrangements involving melting and reshuffling of active site tertiary interactions have been implicated as potential drivers of substrate exchange and catalysis (92–95). In riboswitches, local tertiary melting dynamics such as those observed in the ligand-bound preQ1 riboswitch pseudoknot may help facilitate fast ligand binding and/or unbinding, perhaps tuning switching activity (96).
In addition to facilitating switching between distinct functional states, tertiary dynamics can also serve to toggle a molecule between active and inactive conformations, thus tuning activity. In a unique example, a pH-dependent tertiary folding equilibrium involving formation of base triples between the pseudoknot loop and the pseudoknot helices of the Murine Leukemia Virus read-through element has been shown to dictate the ratio of stop-codon read-through during translation of the MLV mRNA (97). Thus, this equilibrium controls the cellular ratio of the proteins encoded upstream and downstream of the pseudoknot (97). In the Tetrahymena ribozyme, extremely long lived local tertiary structure heterogeneties in the substrate binding site cause docking kinetics to vary as much three orders of magnitude between individual molecules (98). These slow tertiary structure dynamics, which may arise from differentially bound Mg2+ ions (99) and/or alternative sugar pucker conformations (100), do not alter the rate of single-turnover catalysis. However, they may play roles in other aspects of ribozyme function by serving to destabilize the catalytically competent conformation.
Perhaps the most precise use of tertiary structure dynamics are those exhibited in decoding by the ribosome (5, 8). During tRNA initial selection, tertiary structure dynamics involving formation of A-minor interactions between the ribosomal A-site and anticodon-codon minihelix serve to stabilize cognate mRNA-tRNA pairs, preventing tRNA dissociation and driving ‘domain-closure’ conformational changes in the ribosome that activate GTP hydrolysis in EF-Tu (Figure 5B) (62, 101–106). Remarkably, a single incorrect base-pair between the mRNA and tRNA is sufficient to disfavor these conformational changes, forming the basis for the 102–103 selectively of initial selection (107). During the second kinetic proofreading step, a competition between the rates of tertiary structure melting of the tRNA-mRNA minihelix and the rate of accommodation of the tRNA into the ribosome provide a further 10 to 100-fold specificity for cognate tRNAs, as the weaker tertiary interactions of non-cognate tRNAs lead to faster disassociation (105–108).
TIER 2: JITTERING DYNAMICS
Once an RNA structure has formed with well defined secondary structure, local non-canonical pairing, and tertiary interactions, the residues still undergo a wide range of motions including flipping in and out of bulges and internal loops, sugar repuckering, phosphate backbone reorientations, and collective motions of helical domains. These motions cover a relatively broad range of timescales from ps to μs. Base-stacking dynamics take place at slower ns-μs timescales and involve transition states that require disruption of inter-helical stacking across an inter-helical junction, stacking between an unpaired loop residue and a neighboring base-pair, or stacking between two unpaired bases. The extent of these dynamics is highly context dependent, with purine-purine stacks much stronger and thus less dynamic than pyrimidine-pyrimidine stacks (109). Superimposed on top of these dynamics are faster ps-ns jittering dynamics, which can range from small amplitude variations in helical, base pair, and torsion angles in Watson-Crick pairs, to much lager amplitude motions in unstacked and flipped out nucleobases. They can also involve variable amplitude inter-helical motions. Together, these Tier 2 motions span a wider range of timescales as compared to Tiers 0 and 1, but are difficult to decompose into distinct Tiers because the same type of motional mode (e.g. inter-helical dynamics) can take place over the entire range of timescales, and because these distinct motional modes often co-exist and couple to one another.
Inter-helical Dynamics
Overview
Together, local non-canonical pairs and global secondary structure define A-form helical domains that are linked together by various flexible single-stranded junctions. The relative orientation and translation of these helical domains with respect to one another plays an important role in defining the overall RNA architecture and the relative positioning of groups that are involved in long-range tertiary interactions, catalytic activity, and protein binding (9, 110). In many RNAs, however, helices are not pinned down, but rather undergo large collective motions that take place primarily at ns-μs timescales (Figure 7A) (111–122). It is worth noting that slower ms-s timescales have been observed in FRET experiments of isolated four-helix junctions, which likely arise from strong cooperative stacking interactions unique to these junctions (117).
Figure 7.

Modes and functions of Tier 2 jittering dynamics. (A) View of 50 most probable inter-helical conformations for a 3-nt two-helix bulge junction with the lower stem superimposed (green). Most probable conformations were obtained from coarse-grained model simulations that include only steric and connectivity forces (178). Bulge residues were included in coarse-grained modeling but not shown in the figure, instead drawn as orange lines highlighting the possible paths of the bulge. (B) Superposition of classical (green; PDB ID 2WDG) and ratcheted EF-G bound 16S rRNA conformations (grey; PDB IDB 4JUW), highlighting the large inter-helical dynamics associated with ribosomal translocation. The rRNAs were superimposed using residues 1410–1430 and 1470–1490 of H44, with H44 facing the page. (C) Dynamics of the GNRA tetraloop observed by fluorescence spectroscopy (159). Exchange timescales correspond to rates measured by base relaxation (55) and sugar carbon NMR relaxation dispersion experiments (158). (D) Superposition of ligand bound HIV-1 TAR structures (grey) with five conformers from a high-resolution NMR-MD ensemble that have the lowest heavy-atom RMSD to the ligand-bound structures (orange) (123). Left, PDB ID 1LVJ. Right, PDB ID 1UTS.
Inter-helical dynamics have been studied at most depth in the 3-nt bulge of HIV-1 TAR, where a variety of NMR and combined NMR-MD studies have revealed that these dynamics represent a superposition of slower stacking and unstacking transitions on μs timescales, and faster ns motions within a given ‘stacked’ basin (111, 112, 123). Specifically, TAR interconverts between a predominately bent conformation that is stabilized by a stacking interaction between one of the bulge residues and the top of the lower helix, and a lower populated coaxially stacked conformation. On ns timescales, the bent conformation fluctuates between multiple inter-helical bends ranging from 20°–90°, whereas the stacked conformation samples only 0–20° bend angles (123). Increasing the salt concentration, or mutations that increase the strength of inter-helical stacking interactions, increase the population of the stacked conformation (124, 125). However, as stacking is usually expected to provide no more than −3 kcal/mol in stabilizing energy (126, 127), even strongly stacked junctions are likely to exist in unstacked conformations with ≥~1% populations.
An important and universal feature of inter-helical dynamics is that the accessible helical orientations are strongly limited by steric and connectivity constraints, which together are referred to as topological constraints (Figure 7A) (49, 110, 128, 129). These constraints are encoded at the secondary structure and base pair level (Tiers 0 and 1) by the number of unpaired residues within the internal loops that connect a junction’s helices. This makes it possible to construct RNA systems in which helical domains bend in a very directional manner, which can serve a diversity of functions.
Biological significance
Inter-helical motions often allow for optimization of inter-molecular interactions with protein and ligand binding partners. For example, high-resolution structures of tRNA, tRNA-protein, and tRNA-ribosome complexes reveal that binding is often accompanied by significant changes in the relative orientation of the four helical domains (130). Similarly, the two helices of HIV-1 TAR adopt highly varied inter-helical orientations when bound to different small molecules, corresponding to the inter-helical conformations that are sampled in the absence of ligand (112, 123, 128, 131). In more complex RNAs, inter-helical motions have been implicated in the ligand recognition processes of many riboswitch aptamer domains (132–138). Interestingly, cofactor-induced inter-helical changes can also serve as transducers, triggering additional functional events. Specifically, successive changes in inter-helical orientations induced by protein binding are thought to help order the assembly of complex RNP machines, including the 30S ribosome (139, 140), the signal recognition particle (141), and telomerase (142).
The low energy barriers and directionality of inter-helical motions also make them an ideal medium for executing the mechanical motions that underlie the processivity and turnover of ribozymes and RNPs such as the ribosome and telomerase. Examples of some of these motions, such as docking and undocking of ribozyme substrates, were mentioned above. However, the most impressive are those exhibited by the ribosome during tRNA translocation (Figure 7B) (143, 144). Collectively referred to as ‘ratcheting’, these motions involve large allosterically coupled changes in inter-helical conformation of the 30S head and body domains and the 50S L1 stalk, as well as substantial distortions of the tRNA (145–150). These motions remove steric roadblocks to translocation and help transition the ribosome and tRNAs between different intermediates that are stabilized by alternative sets of tertiary interactions. For example, L1 stalk dynamics allow it to form tertiary interactions with P-site tRNAs and then shuttle them to the E-site (151–153). Notably, early theoretical studies demonstrated that ratcheting is inherent to the gross architecture of the ribosome, consistent with a model where these rearrangements are driven by the inherent flexibility of RNA junctions (150). The finding that the inhibitory effects of many antibiotics are in part derived from their ability to alter or arrest ribosomal ratcheting further highlights centrality of these collective motions to ribosome function (154–156).
Loop Dynamics
Overview
RNA secondary structure consists of A-form helical domains that are linked and capped by loops. These single-stranded regions of RNA structure often form important flexible sites for recognition of proteins, RNAs, ligands, and small molecules and for formation of tertiary interactions. Adaptive changes in loop conformation helps optimize these intermolecular interactions, and in the absence of tertiary or ternary stabilizing interactions these regions are among the most dynamic in RNA. Similar to inter-helical dynamics, loop dynamics occur at ps-μs timescales, corresponding to large amplitude jittering dynamics of unstacked residues, smaller jittering of stacked residues, and slower transitions involving exchange between alternatively stacked conformations. Such dynamics can lead to isolated local changes in 3D structure, or, for loops located in inter-helical junctions, can drive global inter-helical dynamics (111, 123).
Loop dynamics are well illustrated by the extensively studied GNRA tetraloop (Figure 7C) (55, 157–162). While the bookending G and A residues form a non-canonical Hoogsteen pair, which transiently melts on μs timescales, the middle N (any base) and R (purine) residues adopt a heterogeneous set of conformations that feature different stacking arrangements on top of the GA pair and that interconvert on μs timescales. In turn, the most solvent exposed residue of each sub-conformation exhibits faster ps-ns unstacking and jittering dynamics, which interestingly appear to be in part dependent on the protonation states of the loop residues (163). A similar separation of timescales between the dynamics of paired and loop residues is observed for the dominant pairing state of the internal loop of the ribosomal A-site (Figure 5B) (54, 62). In the absence of tRNA, the unpaired and weakly stacked A93 undergoes fast ns motions as it rapidly moves in and out of the helical junction. This contrasts with A92, which forms a non-canonical pair with A08 and exhibits slower base-pair melting dynamics.
Biological significance
As mentioned above, the ability of unpaired residues to adopt alternative conformations with low energetic penalties is heavily utilized in RNA recognition processes, allowing the RNA loop to adapt to its molecular partner (164–166). In a recent interesting example, structural changes in an mRNA apical loop induced by binding of either one of two proteins were shown to mediate the cooperative binding of the second protein to the same motif (167). In all of these cases, it is worth emphasizing that the observed adaption corresponds to stabilization of preexisting low free energy conformations. Notably, strongly stacked residues are unlikely to adopt unstacked conformations, which is illustrated by the overwhelming propensity of GNRA tetraloops to adopt fully stacked conformations when participating in tertiary interactions (160). Likewise, studies of the apical loop and 3-nt bulge motif of HIV-1 TAR indicate that the various ligand-bound conformations of these regions strongly correlate with those that are sampled by TAR in the absence of ligand (Figure 7D) (123, 131, 168). Thus, whether weakly stacked and highly dynamic, or more strongly stacked and exhibiting only small local jittering, even at this highest level the RNA free energy landscape is tightly linked to function.
INTERDEPENDENCE OF SUBSTATES ACROSS TIERS
One of the more interesting features of the RNA free energy landscape and dynamics that is just beginning to be explored is the interdependence of CSs across different tiers. For example, a given secondary structure at Tier 0 may only be able to form a single set of tertiary interactions in Tier 1, thus in a sense encoding the properties of Tier 1. Similarly, the number of different loop conformations along Tier 2 can influence the entropic cost associated with forming tertiary interactions along Tier 1. An exciting aspect of these interdependencies is that interactions that stabilize specific CSs in higher order tiers can propagate into stabilization of specific CSs in lower tiers. This can potentially increase the points of entry for effecting RNA conformational change. Below, we discuss some of the better-understood dependencies and their potential connections to biological function. Although not the topic of this review, it is worth noting that the coupling between tiers can be much more complex in the folding of complex RNAs from unstructured states (169).
Secondary Structure and Tertiary Dynamics
One of most important inter-dependencies among tiers is that between tertiary and secondary structure, where free energy supplied by tertiary interactions helps stabilize a secondary structure that would otherwise be unstable. This is exemplified by riboswitches, where ligand binding and subsequent formation of other tertiary interactions provides the necessary interaction energy to stabilize the secondary structure switch either at equilibrium, or transiently during co-transcriptional folding (Figure 8A) (23, 28, 170). In other cases, proteins that stabilize RNA tertiary interactions result in stabilization of specific RNA secondary structures. For example, coupled binding of the maturase and Mrs1 protein cofactors to the RNA of the bI3 group I intron stabilizes native tertiary contacts, promoting a reorganization of a non-native intermediate secondary structure (171). Similar protein induced secondary structure rearrangements play important roles in ribosome assembly (172, 173).
Figure 8.

Interdependencies of CS across Tiers. (A) Aptamer domain of the add adenine riboswitch in complex with adenine (yellow) (PDB ID 1Y26). P1 stem base pairs shown to be unstable in the absence of ligand are shown in red (30), with J2/3 residues that provide stabilizing A-minor interactions shown in green. (B) Stacking interactions limit loop dynamics and pre-organize the 3’ tail for ligand binding and pseudoknot folding in the wild-type Bsu preQ1 riboswitch aptamer (top). An A-to-C mutation distal from the ligand binding pocket disrupts stacking, increasing dynamics and reducing ligand/riboswitch affinity (bottom) (176). The preQ1 ligand, 3’ tail, and mutation are shown in yellow, blue, and red, respectively. (C) Topological constraints preclude a three-way junction from forming two of three possible tertiary interactions. Right, the interaction is precluded due to connectivity. Bottom, the interaction is precluded due to sterics. (D) View of 50 most probable inter-helical conformations for two 1-nt bulge junctions with lower stem superimposed (green). The bulge of the blue junction is located two base-pairs below that of the grey junction. Most probable conformations were obtained from coarse-grained model simulations that include only steric and connectivity forces (178).
Tertiary and Loop Dynamics
Tertiary structure dynamics involving the formation and melting of loop contacts are tightly linked to loop dynamics of the melted state. The extent of these loop dynamics, and their relative order or disorder, encode an entropic penalty for folding. For example, it has been suggested that the extensive loop dynamics of the single-stranded tRNA 3′ CCA play a critical role in resisting tRNA accommodation on the ribosome, a transition which involves formation of several tertiary pairs between the 3′ CCA and the ribosome peptidyl transferase center (174). The entropic barrier presented by these dynamics helps order the accommodation process, preventing premature 3′ CCA entry and peptide transfer, and may also help tune accommodation kinetics, which is important for kinetic proofreading. Another recent example can be found in the preQ1 riboswitch aptamer. In this system, strong stacking interactions in the ligand-free state order the loop that folds to encapsulate the ligand upon binding (Figure 8B) (175, 176). Mutations that decrease stacking, and thus increase loop dynamics, significantly reduce ligand affinity.
Tertiary and Inter-helical Dynamics
As was discussed previously in the inter-helical dynamics section, the basin of inter-helical conformations defined by secondary structure can be quite limited. Emerging research has indicated that these limitations can directly affect tertiary structure dynamics by both modulating the accessibility of the inter-helical conformations needed to form a given tertiary structure, as well as by modulating the entropy of the unfolded state (110). For example, theoretical work of a model two-helix junction has demonstrated that inter-helical dynamics strongly discriminate against the formation of tertiary contacts between some helical faces but allow others (Figure 8C) (129). Subsequent studies have since suggested that this property of inter-helical dynamics is broadly used by RNAs to encode their native folds (49, 128, 177–179). Importantly, such a strategy may be how RNAs are able to overcome the limited information content of tertiary interactions, some of which like A-minor motifs appear to have little sequence specificity (180, 181). Limited inter-helical dynamics may also explain the ability of distal tertiary interactions to cooperatively stabilize each other, a property shown to be crucial to tertiary structure stability (182, 183).
Base Reshuffling and Inter-helical Dynamics
As mentioned above, alternative stacking conformations of junction residues can result in distinct inter-helical orientations. Base-reshuffling dynamics can have even greater effects by redefining junction topology and thereby driving even larger changes in inter-helical orientation. Consider for example the ribosomal A-site RNA system. We previously noted that the A-site internal loop exhibits base-reshuffling dynamics between two alternative local base-pairing CS (Figure 5B) (54). Both states feature effectively a single bulge residue; however, in the dominant state, A93 is the bulge, whereas in the second less energetically favorable state, the bulge is migrated two base-pairs down to U95. This migration of the bulge redirects the topologically allowed inter-helical orientations, allowing certain inter-helical orientations to be sampled that would otherwise be inaccessible in the more energetically favorable junction topology (Figure 8D). Similar topology altering base-pair dynamics induced by tertiary interactions or protein binding may also modify inter-helical dynamics and affect downstream behavior (49, 99). Alternatively, the number of inter-helical conformations available to different CS may influence base-pair reshuffling equilibrium through entropic effects. Although such couplings are only beginning to be uncovered, we predict that they are likely used in RNA to transmit local changes in structure into larger scale changes.
CONCLUSIONS
The past decade alone has seen an astounding explosion in the number of biological roles associated with RNA. While the mechanisms of action and indeed functions of most of these RNAs remain to be elucidated, given our current understanding of RNA biology it is virtually assured that dynamics will serve as a defining feature. As the complexity of the RNA functional universe grows, it will only become more important to establish a common framework for understanding recurrent dynamics strategies.
As laid out above, we suggest that RNA dynamics can be naturally classified in terms of transitions between basins on different tiers of a hierarchical free energy landscape. This description in turn reveals that the same type of motion is often used to effect a particular kind of mechanism which can in turn be wired appropriately into biological circuits to achieve diverse functional outcomes. Thus, secondary structure transitions and base-pair dynamics can serve to expose or sequester key functional elements, while jittering motions play a universal role in conformational adaptation and driving the motions that power RNA and RNP machines. Additional dynamic complexity can be achieved by coupling distinct motions, thus presenting several points of entry for triggering a given type of overall RNA dynamics. Despite the limitations of the above classification – it is not always possible to deconvolute dynamics within a single tier into individual motional modes, and the large range of timescales covered by tertiary and secondary structure dynamics can blur the distinction between the two – it is our hope that such an approach will serve as a first step in facilitating a more universal understanding of the link between RNA function and dynamics.
SUMMARY POINTS.
RNA dynamics can be classified in different motional modes that take place on different tiers of a hierarchical free energy landscape.
RNAs often harness multiple modes to achieve complex functionality.
Functional transitions primarily occur between pre-existing favorable conformational substates of quiescent RNAs.
RNA dynamics can involve very large changes in structure, but these changes are directed to only a limited number of favorable substates.
FUTURE ISSUES.
What is physical basis and functional importance of the long-lived Mg2+-dependent tertiary structure heterogeneities that have now been observed in several different nucleic acids?
How do environmental factors such metal ions and molecular crowders influence the RNA free energy landscape and the dynamic modes?
How important are the inter-dependencies between different dynamics tiers to RNA function?
Are there other motional modes that are yet to be discovered?
Acknowledgments
We thank Isaac Kimsey for his input and assistance in the preparation of figures, and Shan Yang and Dr. Yi Xue for their comments. H.M.A. gratefully acknowledges the Michigan Economic Development Cooperation and the Michigan Technology Tri-Corridor for their support in the purchase of a 600 MHz spectrometer. This work was supported by the National Institutes of Health (R01 AI066975, R01 GM089846, and NIAID R21 AI096985 to H.M.A.; R21 GM096156 to C.L.B. and H.M.A; RR012255 to C.L.B.) and the National Science Foundation (Graduate Research Fellowship to A.M.M.; Career Award CHE0918817 to H.M.A.; PHY0216576 to C.L.B.).
Footnotes
DISCLOSURE STATEMENT
H.M.A. is an advisor to and holds an ownership interest in Nymirum Inc., which is an RNA-based drug discovery company. The research reported in this article was performed by the University of Michigan faculty and students and was funded by an NIH contract to H.M.A.
Contributor Information
Anthony M. Mustoe, Email: amustoe@umich.edu.
Charles L. Brooks, III, Email: brookscl@umich.edu.
Hashim M. Al-Hashimi, Email: hashimi@umich.edu.
LITERATURE CITED
- 1.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–57. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 3.Serganov A, Patel DJ. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu Rev Biophys. 2012;41:343–70. doi: 10.1146/annurev-biophys-101211-113224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiter NJ, Chan CW, Mondragon A. Emerging structural themes in large RNA molecules. Curr Opin Struct Biol. 2011;21:319–26. doi: 10.1016/j.sbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–36. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- 6.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–62. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruz JA, Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136:604–9. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Dethoff EA, Chugh J, Mustoe AM, Al-Hashimi HM. Functional complexity and regulation through RNA dynamics. Nature. 2012;482:322–30. doi: 10.1038/nature10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 12.Tinoco I, Jr, Bustamante C. How RNA folds. J Mol Biol. 1999;293:271–81. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 13.Brion P, Westhof E. Hierarchy and dynamics of RNA folding. Annu Rev Biophys Biomol Struct. 1997;26:113–37. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 14.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–19. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 16.Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–65. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 17.Uhlenbeck OC. Keeping RNA happy. RNA. 1995;1:4–6. [PMC free article] [PubMed] [Google Scholar]
- 18.Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol. 2001;11:309–14. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 19.Furtig B, Wenter P, Reymond L, Richter C, Pitsch S, Schwalbe H. Conformational dynamics of bistable RNAs studied by time-resolved NMR spectroscopy. J Am Chem Soc. 2007;129:16222–9. doi: 10.1021/ja076739r. [DOI] [PubMed] [Google Scholar]
- 20.Grohman JK, Gorelick RJ, Lickwar CR, Lieb JD, Bower BD, et al. A Guanosine-Centric Mechanism for RNA Chaperone Function. Science. 2013;340:190–95. doi: 10.1126/science.1230715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270:20871–4. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 22.Thirumalai D, Woodson SA. Kinetics of folding of proteins and RNA. Acc Chem Res. 1996;29:433–39. [Google Scholar]
- 23.Breaker RR. Riboswitches and the RNA World. Cold Spring Harbor Perspectives in Biology. 2012:4. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–6. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 25.Caron MP, Bastet L, Lussier A, Simoneau-Roy M, Masse E, Lafontaine DA. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc Natl Acad Sci U S A. 2012;109:E3444–53. doi: 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 27.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, et al. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002;111:747–56. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 28.Haller A, Souliere MF, Micura R. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc Chem Res. 2011;44:1339–48. doi: 10.1021/ar200035g. [DOI] [PubMed] [Google Scholar]
- 29.Kortmann J, Sczodrok S, Rinnenthal J, Schwalbe H, Narberhaus F. Translation on demand by a simple RNA-based thermosensor. Nucleic Acids Res. 2011;39:2855–68. doi: 10.1093/nar/gkq1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reining A, Nozinovic S, Schlepckow K, Buhr F, Furtig B, Schwalbe H. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature. 2013;499:355–9. doi: 10.1038/nature12378. [DOI] [PubMed] [Google Scholar]
- 31.Giuliodori AM, Di Pietro F, Marzi S, Masquida B, Wagner R, et al. The cspA mRNA is a thermosensor that modulates translation of the cold-shock protein CspA. Mol Cell. 2010;37:21–33. doi: 10.1016/j.molcel.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 32.Cromie MJ, Shi Y, Latifi T, Groisman EA. An RNA sensor for intracellular Mg(2+) Cell. 2006;125:71–84. doi: 10.1016/j.cell.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 33.Nechooshtan G, Elgrably-Weiss M, Sheaffer A, Westhof E, Altuvia S. A pH-responsive riboregulator. Genes Dev. 2009;23:2650–62. doi: 10.1101/gad.552209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babitzke P, Yanofsky C. Reconstitution of Bacillus subtilis trp attenuation in vitro with TRAP, the trp RNA-binding attenuation protein. Proc Natl Acad Sci U S A. 1993;90:133–7. doi: 10.1073/pnas.90.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundy FJ, Winkler WC, Henkin TM. tRNA-mediated transcription antitermination in vitro: Codon-anticodon pairing independent of the ribosome. Proc Natl Acad Sci U S A. 2002;99:11121–26. doi: 10.1073/pnas.162366799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–89. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci. 2012;37:263–73. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huthoff H, Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7:143–57. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu K, Heng X, Summers MF. Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol. 2011;410:609–33. doi: 10.1016/j.jmb.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu K, Heng X, Garyu L, Monti S, Garcia EL, et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science. 2011;334:242–5. doi: 10.1126/science.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snoussi K, Leroy JL. Imino proton exchange and base-pair kinetics in RNA duplexes. Biochemistry. 2001;40:8898–904. doi: 10.1021/bi010385d. [DOI] [PubMed] [Google Scholar]
- 42.Chen C, Jiang L, Michalczyk R, Russu IM. Structural energetics and base-pair opening dynamics in sarcin-ricin domain RNA. Biochemistry. 2006;45:13606–13. doi: 10.1021/bi060908n. [DOI] [PubMed] [Google Scholar]
- 43.Rinnenthal J, Klinkert B, Narberhaus F, Schwalbe H. Direct observation of the temperature-induced melting process of the Salmonella fourU RNA thermometer at base-pair resolution. Nucleic Acids Res. 2010;38:3834–47. doi: 10.1093/nar/gkq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodson SA. Taming free energy landscapes with RNA chaperones. RNA Biology. 2010;7:677–86. doi: 10.4161/rna.7.6.13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shankar N, Xia T, Kennedy SD, Krugh TR, Mathews DH, Turner DH. NMR reveals the absence of hydrogen bonding in adjacent UU and AG mismatches in an isolated internal loop from ribosomal RNA. Biochemistry. 2007;46:12665–78. doi: 10.1021/bi700802s. [DOI] [PubMed] [Google Scholar]
- 47.Conn GL, Draper DE, Lattman EE, Gittis AG. Crystal structure of a conserved ribosomal protein-RNA complex. Science. 1999;284:1171–74. doi: 10.1126/science.284.5417.1171. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y-X, Huang S, Draper DE. Structure of a U-U pair within a conserved ribosomal RNA hairpin. Nucleic Acids Res. 1996;24:2666–72. doi: 10.1093/nar/24.14.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mustoe AM, Bailor MH, Teixeira RM, Brooks CL, III, Al-Hashimi HM. New insights into the fundamental role of topological constraints as a determinant of two-way junction conformation. Nucleic Acids Res. 2012;40:892–904. doi: 10.1093/nar/gkr751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmeing TM, Huang KS, Strobel SA, Steitz TA. An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature. 2005;438:520–4. doi: 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- 51.Qu X, Wen JD, Lancaster L, Noller HF, Bustamante C, Tinoco I., Jr The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature. 2011;475:118–21. doi: 10.1038/nature10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–6. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mouzakis KD, Lang AL, Vander Meulen KA, Easterday PD, Butcher SE. HIV-1 frameshift efficiency is primarily determined by the stability of base pairs positioned at the mRNA entrance channel of the ribosome. Nucleic Acids Res. 2013;41:1901–13. doi: 10.1093/nar/gks1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A, Al-Hashimi HM. Visualizing transient low-populated structures of RNA. Nature. 2012;491:724–8. doi: 10.1038/nature11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoogstraten CG, Wank JR, Pardi A. Active site dynamics in the lead-dependent ribozyme. Biochemistry. 2000;39:9951–8. doi: 10.1021/bi0007627. [DOI] [PubMed] [Google Scholar]
- 56.Venditti V, Clos L, 2nd, Niccolai N, Butcher SE. Minimum-energy path for a u6 RNA conformational change involving protonation, base-pair rearrangement and base flipping. J Mol Biol. 2009;391:894–905. doi: 10.1016/j.jmb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reiter NJ, Blad H, Abildgaard F, Butcher SE. Dynamics in the U6 RNA intramolecular stem-loop: a base flipping conformational change. Biochemistry. 2004;43:13739–47. doi: 10.1021/bi048815y. [DOI] [PubMed] [Google Scholar]
- 58.Nikolova EN, Kim E, Wise AA, O’Brien PJ, Andricioaei I, Al-Hashimi HM. Transient Hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470:498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikolova EN, Goh GB, Brooks CL, III, Al-Hashimi HM. Characterizing the protonation state of cytosine in transient G.C Hoogsteen base pairs in duplex DNA. J Am Chem Soc. 2013;135:6766–9. doi: 10.1021/ja400994e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yajima R, Proctor DJ, Kierzek R, Kierzek E, Bevilacqua PC. A conformationally restricted guanosine analog reveals the catalytic relevance of three structures of an RNA enzyme. Chem Biol. 2007;14:23–30. doi: 10.1016/j.chembiol.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 61.Kadakkuzha BM, Zhao L, Xia T. Conformational distribution and ultrafast base dynamics of leadzyme. Biochemistry. 2009;48:3807–9. doi: 10.1021/bi900256q. [DOI] [PubMed] [Google Scholar]
- 62.Zeng X, Chugh J, Casiano-Negroni A, Al-Hashimi HM, Brooks CL., III Flipping of the ribosomal A-site adenines provides a basis for tRNA selection. 2013 doi: 10.1016/j.jmb.2014.04.029. In submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoddard CD, Widmann J, Trausch JJ, Marcano-Velazquez JG, Knight R, Batey RT. Nucleotides adjacent to the ligand-binding pocket are linked to activity tuning in the purine riboswitch. J Mol Biol. 2013;425:1596–611. doi: 10.1016/j.jmb.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butcher SE, Dieckmann T, Feigon J. Solution structure of a GAAA tetraloop receptor RNA. EMBO J. 1997;16:7490–9. doi: 10.1093/emboj/16.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shankar N, Kennedy SD, Chen G, Krugh TR, Turner DH. The NMR structure of an internal loop from 23S ribosomal RNA differs from its structure in crystals of 50s ribosomal subunits. Biochemistry. 2006;45:11776–89. doi: 10.1021/bi0605787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schroeder SJ, Fountain MA, Kennedy SD, Lukavsky PJ, Puglisi JD, et al. Thermodynamic stability and structural features of the J4/5 loop in a Pneumocystis carinii group I intron. Biochemistry. 2003;42:14184–96. doi: 10.1021/bi0301587. [DOI] [PubMed] [Google Scholar]
- 67.Rupert PB, Ferre-D’Amare AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–6. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 68.Butcher SE, Allain FH, Feigon J. Solution structure of the loop B domain from the hairpin ribozyme. Nat Struct Biol. 1999;6:212–6. doi: 10.1038/6651. [DOI] [PubMed] [Google Scholar]
- 69.Cai Z, Tinoco I., Jr Solution structure of loop A from the hairpin ribozyme from tobacco ringspot virus satellite. Biochemistry. 1996;35:6026–36. doi: 10.1021/bi952985g. [DOI] [PubMed] [Google Scholar]
- 70.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–85. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 71.Lee JC, Gutell RR, Russell R. The UAA/GAN internal loop motif: a new RNA structural element that forms a cross-strand AAA stack and long-range tertiary interactions. J Mol Biol. 2006;360:978–88. doi: 10.1016/j.jmb.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 72.Davis AR, Kirkpatrick CC, Znosko BM. Structural characterization of naturally occurring RNA single mismatches. Nucleic Acids Res. 2011;39:1081–94. doi: 10.1093/nar/gkq793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao XL, Patel DJ. G(Syn).A(Anti) Mismatch Formation in DNA Dodecamers at Acidic Ph - Ph-Dependent Conformational Transition of G.A Mispairs Detected by Proton Nmr. J Am Chem Soc. 1988;110:5178–82. [Google Scholar]
- 75.Burkard ME, Turner DH. NMR structures of r(GCAGGCGUGC)2 and determinants of stability for single guanosine-guanosine base pairs. Biochemistry. 2000;39:11748–62. doi: 10.1021/bi000720i. [DOI] [PubMed] [Google Scholar]
- 76.Yildirim I, Park H, Disney MD, Schatz GC. A dynamic structural model of expanded RNA CAG repeats: a refined X-ray structure and computational investigations using molecular dynamics and umbrella sampling simulations. J Am Chem Soc. 2013;135:3528–38. doi: 10.1021/ja3108627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen G, Kennedy SD, Qiao J, Krugh TR, Turner DH. An alternating sheared AA pair and elements of stability for a single sheared purine-purine pair flanked by sheared GA pairs in RNA. Biochemistry. 2006;45:6889–903. doi: 10.1021/bi0524464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mathews DH, Case DA. Nudged elastic band calculation of minimal energy paths for the conformational change of a GG non-canonical pair. J Mol Biol. 2006;357:1683–93. doi: 10.1016/j.jmb.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 79.Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–9. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 80.Peterson RD, Feigon J. Structural change in Rev responsive element RNA of HIV-1 on binding Rev peptide. J Mol Biol. 1996;264:863–77. doi: 10.1006/jmbi.1996.0683. [DOI] [PubMed] [Google Scholar]
- 81.Schroeder KT, Lilley DM. Ion-induced folding of a kink turn that departs from the conventional sequence. Nucleic Acids Res. 2009;37:7281–9. doi: 10.1093/nar/gkp791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Znosko BM, Kennedy SD, Wille PC, Krugh TR, Turner DH. Structural features and thermodynamics of the J4/5 loop from the Candida albicans and Candida dubliniensis group I introns. Biochemistry. 2004;43:15822–37. doi: 10.1021/bi049256y. [DOI] [PubMed] [Google Scholar]
- 83.Weixlbaumer A, Murphy FVt, Dziergowska A, Malkiewicz A, Vendeix FA, et al. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat Struct Mol Biol. 2007;14:498–502. doi: 10.1038/nsmb1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vendeix FA, Murphy FVt, Cantara WA, Leszczynska G, Gustilo EM, et al. Human tRNA(Lys3)(UUU) is pre-structured by natural modifications for cognate and wobble codon binding through keto-enol tautomerism. J Mol Biol. 2012;416:467–85. doi: 10.1016/j.jmb.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cantara WA, Murphy FVt, Demirci H, Agris PF. Expanded use of sense codons is regulated by modified cytidines in tRNA. Proc Natl Acad Sci U S A. 2013;110:10964–9. doi: 10.1073/pnas.1222641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc Chem Res. 2011;44:1302–11. doi: 10.1021/ar200098t. [DOI] [PubMed] [Google Scholar]
- 87.Wu M, Tinoco I., Jr RNA folding causes secondary structure rearrangement. Proc Natl Acad Sci U S A. 1998;95:11555–60. doi: 10.1073/pnas.95.20.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koculi E, Cho SS, Desai R, Thirumalai D, Woodson SA. Folding path of P5abc RNA involves direct coupling of secondary and tertiary structures. Nucleic Acids Res. 2012;40:8011–20. doi: 10.1093/nar/gks468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herschlag D. Evidence for processivity and two-step binding of the RNA substrate from studies of J1/2 mutants of the Tetrahymena ribozyme. Biochemistry. 1992;31:1386–99. doi: 10.1021/bi00120a015. [DOI] [PubMed] [Google Scholar]
- 90.Bevilacqua PC, Kierzek R, Johnson KA, Turner DH. Dynamics of ribozyme binding of substrate revealed by fluorescence-detected stopped-flow methods. Science. 1992;258:1355–8. doi: 10.1126/science.1455230. [DOI] [PubMed] [Google Scholar]
- 91.Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–6. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- 92.Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell. 2012;151:497–507. doi: 10.1016/j.cell.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harris DA, Rueda D, Walter NG. Local conformational changes in the catalytic core of the trans-acting hepatitis delta virus ribozyme accompany catalysis. Biochemistry. 2002;41:12051–61. doi: 10.1021/bi026101m. [DOI] [PubMed] [Google Scholar]
- 94.Lee TS, Giambasu G, Harris ME, York DM. Characterization of the Structure and Dynamics of the HDV Ribozyme at Different Stages Along the Reaction Path. J Phys Chem Lett. 2011;2:2538–43. doi: 10.1021/jz201106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ke A, Zhou K, Ding F, Cate JH, Doudna JA. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature. 2004;429:201–5. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Q, Kang M, Peterson RD, Feigon J. Comparison of solution and crystal structures of preQ1 riboswitch reveals calcium-induced changes in conformation and dynamics. J Am Chem Soc. 2011;133:5190–3. doi: 10.1021/ja111769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Houck-Loomis B, Durney MA, Salguero C, Shankar N, Nagle JM, et al. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature. 2011;480:561–4. doi: 10.1038/nature10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Solomatin SV, Greenfeld M, Chu S, Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature. 2010;463:681–4. doi: 10.1038/nature08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hyeon C, Lee J, Yoon J, Hohng S, Thirumalai D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat Chem. 2012;4:907–14. doi: 10.1038/nchem.1463. [DOI] [PubMed] [Google Scholar]
- 100.Mortimer SA, Weeks KM. C2’-endo nucleotides as molecular timers suggested by the folding of an RNA domain. Proc Natl Acad Sci U S A. 2009;106:15622–7. doi: 10.1073/pnas.0901319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–32. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 102.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 103.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–71. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 104.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FVt, et al. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–94. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, et al. Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J Mol Biol. 2010;399:576–95. doi: 10.1016/j.jmb.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol. 2004;11:1008–14. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 107.Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell. 2006;21:369–77. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 108.Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999;18:3800–7. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Turner DH, Sugimoto N, Freier SM. RNA structure prediction. Annu Rev Biophys Biophys Chem. 1988;17:167–92. doi: 10.1146/annurev.bb.17.060188.001123. [DOI] [PubMed] [Google Scholar]
- 110.Bailor MH, Mustoe AM, Brooks CL, III, Al-Hashimi HM. Topological constraints: using RNA secondary structure to model 3D conformation, folding pathways, and dynamic adaptation. Curr Opin Struct Biol. 2011;21:296–305. doi: 10.1016/j.sbi.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–6. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 112.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–7. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 113.Sun X, Zhang Q, Al-Hashimi HM. Resolving fast and slow motions in the internal loop containing stem-loop 1 of HIV-1 that are modulated by Mg2+ binding: role in the kissing-duplex structural transition. Nucleic Acids Res. 2007;35:1698–713. doi: 10.1093/nar/gkm020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bailor MH, Musselman C, Hansen AL, Gulati K, Patel DJ, Al-Hashimi HM. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat Protoc. 2007;2:1536–46. doi: 10.1038/nprot.2007.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Getz MM, Andrews AJ, Fierke CA, Al-Hashimi HM. Structural plasticity and Mg2+ binding properties of RNase P P4 from combined analysis of NMR residual dipolar couplings and motionally decoupled spin relaxation. RNA. 2007;13:251–66. doi: 10.1261/rna.264207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Olsen GL, Bardaro MF, Jr, Echodu DC, Drobny GP, Varani G. Intermediate rate atomic trajectories of RNA by solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:303–8. doi: 10.1021/ja907515s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hohng S, Wilson TJ, Tan E, Clegg RM, Lilley DM, Ha T. Conformational flexibility of four-way junctions in RNA. J Mol Biol. 2004;336:69–79. doi: 10.1016/j.jmb.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 118.Melcher SE, Wilson TJ, Lilley DM. The dynamic nature of the four-way junction of the hepatitis C virus IRES. RNA. 2003;9:809–20. doi: 10.1261/rna.5130703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reblova K, Sponer J, Lankas F. Structure and mechanical properties of the ribosomal L1 stalk three-way junction. Nucleic Acids Res. 2012;40:6290–303. doi: 10.1093/nar/gks258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Besseova I, Reblova K, Leontis NB, Sponer J. Molecular dynamics simulations suggest that RNA three-way junctions can act as flexible RNA structural elements in the ribosome. Nucleic Acids Res. 2010;38:6247–64. doi: 10.1093/nar/gkq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grant GP, Boyd N, Herschlag D, Qin PZ. Motions of the substrate recognition duplex in a group I intron assessed by site-directed spin labeling. J Am Chem Soc. 2009;131:3136–7. doi: 10.1021/ja808217s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Q, Kim NK, Peterson RD, Wang Z, Feigon J. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc Natl Acad Sci U S A. 2010;107:18761–8. doi: 10.1073/pnas.1013269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salmon L, Bascom G, Andricioaei I, Al-Hashimi HM. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: the basis for interhelical motions revealed. J Am Chem Soc. 2013;135:5457–66. doi: 10.1021/ja400920w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stelzer AC, Kratz JD, Zhang Q, Al-Hashimi HM. RNA dynamics by design: biasing ensembles towards the ligand-bound state. Angew Chem Int Ed Engl. 2010;49:5731–3. doi: 10.1002/anie.201000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Casiano-Negroni A, Sun X, Al-Hashimi HM. Probing Na(+)-induced changes in the HIV-1 TAR conformational dynamics using NMR residual dipolar couplings: new insights into the role of counterions and electrostatic interactions in adaptive recognition. Biochemistry. 2007;46:6525–35. doi: 10.1021/bi700335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Walter AE, Turner DH, Kim J, Lyttle MH, Muller P, et al. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc Natl Acad Sci U S A. 1994;91:9218–22. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tyagi R, Mathews DH. Predicting helical coaxial stacking in RNA multibranch loops. RNA. 2007;13:939–51. doi: 10.1261/rna.305307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science. 2010;327:202–6. doi: 10.1126/science.1181085. [DOI] [PubMed] [Google Scholar]
- 129.Chu VB, Lipfert J, Bai Y, Pande VS, Doniach S, Herschlag D. Do conformational biases of simple helical junctions influence RNA folding stability and specificity? RNA. 2009;15:2195–205. doi: 10.1261/rna.1747509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alexander RW, Eargle J, Luthey-Schulten Z. Experimental and computational determination of tRNA dynamics. FEBS Lett. 2010;584:376–86. doi: 10.1016/j.febslet.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 131.Frank AT, Stelzer AC, Al-Hashimi HM, Andricioaei I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: new insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009;37:3670–9. doi: 10.1093/nar/gkp156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heppell B, Blouin S, Dussault AM, Mulhbacher J, Ennifar E, et al. Molecular insights into the ligand-controlled organization of the SAM-I riboswitch. Nat Chem Biol. 2011;7:384–92. doi: 10.1038/nchembio.563. [DOI] [PubMed] [Google Scholar]
- 133.Haller A, Altman RB, Souliere MF, Blanchard SC, Micura R. Folding and ligand recognition of the TPP riboswitch aptamer at single-molecule resolution. Proc Natl Acad Sci U S A. 2013;110:4188–93. doi: 10.1073/pnas.1218062110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fiegland LR, Garst AD, Batey RT, Nesbitt DJ. Single-molecule studies of the lysine riboswitch reveal effector-dependent conformational dynamics of the aptamer domain. Biochemistry. 2012;51:9223–33. doi: 10.1021/bi3007753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY, Batey RT. Free state conformational sampling of the SAM-I riboswitch aptamer domain. Structure. 2010;18:787–97. doi: 10.1016/j.str.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baird NJ, Ferre-D’Amare AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA. 2010;16:598–609. doi: 10.1261/rna.1852310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brenner MD, Scanlan MS, Nahas MK, Ha T, Silverman SK. Multivector fluorescence analysis of the xpt guanine riboswitch aptamer domain and the conformational role of guanine. Biochemistry. 2010;49:1596–605. doi: 10.1021/bi9019912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, et al. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J Mol Biol. 2007;365:1393–406. doi: 10.1016/j.jmb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, et al. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science. 2010;330:673–7. doi: 10.1126/science.1193220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Adilakshmi T, Bellur DL, Woodson SA. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature. 2008;455:1268–72. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Menichelli E, Isel C, Oubridge C, Nagai K. Protein-induced conformational changes of RNA during the assembly of human signal recognition particle. J Mol Biol. 2007;367:187–203. doi: 10.1016/j.jmb.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 142.Stone MD, Mihalusova M, O’Connor CM, Prathapam R, Collins K, Zhuang X. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature. 2007;446:458–61. doi: 10.1038/nature05600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen J, Tsai A, O’Leary SE, Petrov A, Puglisi JD. Unraveling the dynamics of ribosome translocation. Curr Opin Struct Biol. 2012;22:804–14. doi: 10.1016/j.sbi.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Frank J, Gonzalez RL., Jr Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu Rev Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ratje AH, Loerke J, Mikolajka A, Brunner M, Hildebrand PW, et al. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature. 2010;468:713–6. doi: 10.1038/nature09547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–33. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 147.Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. Locking and unlocking of ribosomal motions. Cell. 2003;114:123–34. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 148.Zhang W, Dunkle JA, Cate JH. Structures of the ribosome in intermediate states of ratcheting. Science. 2009;325:1014–7. doi: 10.1126/science.1175275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, et al. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–4. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tama F, Valle M, Frank J, Brooks CL., III Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc Natl Acad Sci U S A. 2003;100:9319–23. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fei J, Kosuri P, MacDougall DD, Gonzalez RL., Jr Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol Cell. 2008;30:348–59. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 152.Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL., Jr Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc Natl Acad Sci U S A. 2009;106:15702–7. doi: 10.1073/pnas.0908077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cornish PV, Ermolenko DN, Staple DW, Hoang L, Hickerson RP, et al. Following movement of the L1 stalk between three functional states in single ribosomes. Proc Natl Acad Sci U S A. 2009;106:2571–6. doi: 10.1073/pnas.0813180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tsai A, Uemura S, Johansson M, Puglisi EV, Marshall RA, et al. The impact of aminoglycosides on the dynamics of translation elongation. Cell Rep. 2013;3:497–508. doi: 10.1016/j.celrep.2013.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, et al. Allosteric control of the ribosome by small-molecule antibiotics. Nat Struct Mol Biol. 2012;19:957–63. doi: 10.1038/nsmb.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ermolenko DN, Spiegel PC, Majumdar ZK, Hickerson RP, Clegg RM, Noller HF. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat Struct Mol Biol. 2007;14:493–7. doi: 10.1038/nsmb1243. [DOI] [PubMed] [Google Scholar]
- 157.Menger M, Eckstein F, Porschke D. Dynamics of the RNA hairpin GNRA tetraloop. Biochemistry. 2000;39:4500–7. doi: 10.1021/bi992297n. [DOI] [PubMed] [Google Scholar]
- 158.Johnson JE, Jr, Hoogstraten CG. Extensive backbone dynamics in the GCAA RNA tetraloop analyzed using 13C NMR spin relaxation and specific isotope labeling. J Am Chem Soc. 2008;130:16757–69. doi: 10.1021/ja805759z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhao L, Xia T. Direct revelation of multiple conformations in RNA by femtosecond dynamics. J Am Chem Soc. 2007;129:4118–9. doi: 10.1021/ja068391q. [DOI] [PubMed] [Google Scholar]
- 160.DePaul AJ, Thompson EJ, Patel SS, Haldeman K, Sorin EJ. Equilibrium conformational dynamics in an RNA tetraloop from massively parallel molecular dynamics. Nucleic Acids Res. 2010;38:4856–67. doi: 10.1093/nar/gkq134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jucker FM, Heus HA, Yip PF, Moors EH, Pardi A. A network of heterogeneous hydrogen bonds in GNRA tetraloops. J Mol Biol. 1996;264:968–80. doi: 10.1006/jmbi.1996.0690. [DOI] [PubMed] [Google Scholar]
- 162.Zhang YF, Zhao X, Mu YG. Conformational Transition Map of an RNA GCAA Tetraloop Explored by Replica-Exchange Molecular Dynamics Simulation. J Chem Theory Comput. 2009;5:1146–54. doi: 10.1021/ct8004276. [DOI] [PubMed] [Google Scholar]
- 163.Goh GB, Knight JL, Brooks CL., III pH-dependent dynamics of complex RNA macromolecules. J Chem Theory Comput. 2013;9:935–43. doi: 10.1021/ct300942z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Leulliot N, Varani G. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40:7947–56. doi: 10.1021/bi010680y. [DOI] [PubMed] [Google Scholar]
- 165.Xia T. Taking femtosecond snapshots of RNA conformational dynamics and complexity. Curr Opin Chem Biol. 2008;12:604–11. doi: 10.1016/j.cbpa.2008.08.033. [DOI] [PubMed] [Google Scholar]
- 166.Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science. 2000;287:820–5. doi: 10.1126/science.287.5454.820. [DOI] [PubMed] [Google Scholar]
- 167.Tan D, Marzluff WF, Dominski Z, Tong L. Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3’hExo ternary complex. Science. 2013;339:318–21. doi: 10.1126/science.1228705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7:553–9. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Woodson SA. Compact intermediates in RNA folding. Annu Rev Biophys. 2010;39:61–77. doi: 10.1146/annurev.biophys.093008.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Frieda KL, Block SM. Direct observation of cotranscriptional folding in an adenine riboswitch. Science. 2012;338:397–400. doi: 10.1126/science.1225722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Duncan CD, Weeks KM. Nonhierarchical ribonucleoprotein assembly suggests a strain-propagation model for protein-facilitated RNA folding. Biochemistry. 2010;49:5418–25. doi: 10.1021/bi100267g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Stern S, Changchien LM, Craven GR, Noller HF. Interaction of proteins S16, S17 and S20 with 16 S ribosomal RNA. J Mol Biol. 1988;200:291–9. doi: 10.1016/0022-2836(88)90241-0. [DOI] [PubMed] [Google Scholar]
- 173.Ramaswamy P, Woodson SA. S16 throws a conformational switch during assembly of 30S 5’ domain. Nat Struct Mol Biol. 2009;16:438–45. doi: 10.1038/nsmb.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Whitford PC, Geggier P, Altman RB, Blanchard SC, Onuchic JN, Sanbonmatsu KY. Accommodation of aminoacyl-tRNA into the ribosome involves reversible excursions along multiple pathways. RNA. 2010;16:1196–204. doi: 10.1261/rna.2035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Eichhorn CD, Feng J, Suddala KC, Walter NG, Brooks CL, III, Al-Hashimi HM. Unraveling the structural complexity in a single-stranded RNA tail: implications for efficient ligand binding in the prequeuosine riboswitch. Nucleic Acids Res. 2012;40:1345–55. doi: 10.1093/nar/gkr833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Suddala KC, Rinaldi AJ, Feng J, Mustoe AM, Eichhorn CD, et al. Single transcriptional and translational preQ1 riboswitches adopt similar pre-folded ensembles that follow distinct folding pathways into the same ligand-bound structure. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt798. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sim AY, Levitt M. Clustering to identify RNA conformations constrained by secondary structure. Proc Natl Acad Sci U S A. 2011;108:3590–5. doi: 10.1073/pnas.1018653108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mustoe AM, Al-Hashimi HM, Brooks CL., III . Coarse Grained Models Reveal Essential Contributions of Topological Constraints to the Conformational Free Energy of RNA Bulges. 2013. In submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mustoe AM, Al-Hashimi HM, Brooks CL., III Topological Constraints Specify tRNA 3D Structure and Provide Basis for Tertiary Folding Cooperativity. 2013 doi: 10.1093/nar/gku807. In submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc Natl Acad Sci U S A. 2001;98:4899–903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Doherty EA, Batey RT, Masquida B, Doudna JA. A universal mode of helix packing in RNA. Nat Struct Biol. 2001;8:339–43. doi: 10.1038/86221. [DOI] [PubMed] [Google Scholar]
- 182.Sattin BD, Zhao W, Travers K, Chu S, Herschlag D. Direct measurement of tertiary contact cooperativity in RNA folding. J Am Chem Soc. 2008;130:6085–7. doi: 10.1021/ja800919q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Behrouzi R, Roh JH, Kilburn D, Briber RM, Woodson SA. Cooperative tertiary interaction network guides RNA folding. Cell. 2012;149:348–57. doi: 10.1016/j.cell.2012.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Das R, Travers KJ, Bai Y, Herschlag D. Determining the Mg2+ stoichiometry for folding an RNA metal ion core. J Am Chem Soc. 2005;127:8272–3. doi: 10.1021/ja051422h. [DOI] [PMC free article] [PubMed] [Google Scholar]