

Abstract
Background and Aims
A strong correlation exists between smoking and the use of alcohol and cannabis. This paper uses polygenic risk scores to explore the possibility of overlapping genetic factors. Those scores reflect a combined effect of selected risk alleles for smoking.
Methods
Summary-level p-values were available for smoking initiation, age at onset of smoking, cigarettes per day and smoking cessation from the Tobacco and Genetics Consortium (N between 22,000 and 70,000 subjects). Using different p-value thresholds (.1, .2 and .5) from the meta-analyses, sets of ‘risk alleles’ were defined and used to generate a polygenic risk score (weighted sum of the alleles) for each subject in an independent target sample from the Netherlands Twin Register (N=1583). The association between polygenic smoking scores and alcohol/cannabis use was investigated with regression analyses.
Results
The polygenic scores for ‘cigarettes per day’ were significantly associated with, the number of glasses alcohol per week (p=.005, R2=.4–.5%) and cannabis initiation (p=.004, R2=0.6–.9%). The polygenic scores for ‘age at onset of smoking’ were significantly associated with ‘age at regular drinking’ (p=.001, R2=1.1–1.5%), while the scores for ‘smoking initiation’ and ‘smoking cessation’ did not significantly predict alcohol or cannabis use.
Conclusions
Smoking, alcohol and cannabis use are influenced by aggregated genetic risk factors shared between these substances. The many common genetic variants each have a very small individual effect size.
Introduction
A strong correlation exists between smoking and the use of other substances like alcohol and cannabis. Smoking is positively correlated with alcohol consumption, the severity of alcohol dependence (1) and with the use of cannabis (2). Twin and family studies have shown that smoking behavior (3–5), alcohol consumption (6–8) and cannabis use (9–11) are influenced by genetic factors. Heritability estimates range from low to moderate for initiation of substance use, to rather high for quantity and dependence (3, 5, 11–13). The co-morbidity of tobacco-, alcohol-, and cannabis use is mediated by common genetic influences (14–16).
In the past years, genome-wide association (GWA) studies to smoking behavior revealed several regions and candidate genes (17–20). However, none of these GWA studies reported genome-wide significant results because of the limited sample sizes. It is now recognized that a well-powered GWA needs to include ten thousands and possibly hundred-thousands of subjects. In 2010, three large consortia, the Oxford-GlaxoSmithKline (Ox-GSK), Tobacco and Genetics Consortium (TAG) and ENGAGE consortium, each carried out meta-analyses for smoking phenotypes. They also combined their analyses for smoking initiation and cigarettes per day (21–24). The most significant finding was the association between the number of cigarettes per day and a cluster of nicotinic receptor genes on chromosome 15 (21–24).
For cannabis use several candidate genes are suggested based on linkage and association studies (25), but a GWA meta-analyses based on 2 samples (effective sample size 4312) (26) a GWA analyses of cannabis dependence did not reveal genome-wide significant results (27).
Rietschel and Treutlein (2013) reviewed the current literature on alcohol GWAS and concluded that few genome-wide significant findings have been reported. Among the top-hits are often alcohol dehydrogenase genes (ADH and ALDH2) although a variety of other genes is also reported (28).
Twin-family studies suggested a genetic overlap between use of different substances, but so far none of the top-results in GWA-studies for smoking, alcohol and cannabis overlapped.
Some examples exist of well-know substance specific genes that are also associated with another substance. Mouse studies showed for example that polymorphisms located within the Chrna5-Chrna3-Chrnb4 cluster on mouse chromosome 9 (well-know smoking genes) co-segregate with alcohol preference in mice (29). This suggest there is some overlap in risk genes for substance use or abuse.
The effect sizes of individual risk alleles underlying substance use are small, with most genotype relative risks in the range of 1.1–2.0. The joint effect of all measured DNA variants explained 19–28% of the variance in smoking initiation, 24–44% in current smoking (30) and 6% in cannabis use (26). These findings suggest that individuals may be at risk for substance use through multiple genetic variants each with a small contribution.
Polygenic risk scores have been used to summarize genetic effects among a group of genetic variants that do not individually achieve significance in a large-scale association study. First a meta-analyses on GWA results is conducted on an initial discovery sample, and the markers are ranked by their evidence for association, usually based on their P-values. An independent target sample is then analyzed by constructing a polygenic score consisting of the weighted sum of the associated alleles within each subject. Association between a trait and this score implies a genetic effect of the trait in the discovery sample on the trait in the target sample. The first successful application of a polygenic risk score analyses was in a GWA study to schizophrenia (31). A polygenic risk score based on the GWA for schizophrenia was associated to the risk of bipolar disorder, but not to several non-psychiatric diseases (which suggested disease specificity). The polygenic risk score method is used in several studies, with mixed results. Some studies report positive associations (for example (32–35)) while others did not find evidence that common genetic risk variation is shared between two traits (for example (36, 37)). This might be due to the size of the discovery sample (because the accuracy of the prediction score increases with the size of the discovery sample), or it may indicate a lack of genetic overlap.
In the present study, polygenic risk scores for smoking were identified based on the large meta-analyses of the Tobacco and Genetics (TAG) Consortium including 20,000–70,000 subjects. Four phenotypes were included in the TAG GWA meta-analyses: ever versus never regular smoking (Ever), age at onset of smoking (AOS), cigarettes per day (CPD) and smoking cessation (Former). The risk alleles from TAG were used to calculate a polygenic risk scores in an unrelated sample of the Netherlands Twin Register (N=1583) and the association between this risk score for smoking and alcohol/cannabis use was explored.
Methods
Discovery sample from TAG consortium
The TAG consortium reported summary-level p-values of the GWA meta-analyses of 4 smoking phenotypes based on 20,000–70,000 subjects (38). 16 studies contributed to the meta-analyses and performed their own genotyping, quality control and imputation. Studies ranged in size from n=585 to n=22,037 and were genotyped on six different GWAS platforms.
Four dimensions of smoking behavior were analyzed (38):
Ever versus never regular smokers (Ever): Regular smokers (1) were defined as those who reported having smoked ≥100 cigarettes during their lifetime and never regular smokers (0) were defined as those who reported having smoked between 0 and 99 cigarettes during their lifetime. Total sample size N=69.409.
Age at onset of smoking (AOS): Age of smoking initiation was the reported age the participant started smoking cigarettes. Total sample size N=22.438.
Cigarettes per day (CPD): the average or maximum (depending on study) number of cigarettes smoked per day. Total sample size N=38.181.
Smoking cessation (Former): Smoking cessation contrasted former (=0) versus current (=1) smokers.
Each study conducted uniform cross-sectional analyses for each smoking phenotype using an additive genetic model. Linear regression was used for quantitative traits (CPD and AOS), and logistic regression was used for dichotomous traits (Ever and Former). Total sample size N=35.845.
The analyses were run separately for males and females. Since the TAG consortium did not detect significant interactions by sex, data were analyzed together. Age was not included as covariate. Case-control studies included case/control status as a covariate, cohort studies did not include an additional covariate.
Target sample from NTR
The target sample consisted of subjects from the Netherlands Twin Register (NTR) who were not part of the TAG meta-analyses. The NTR collects longitudinal data in twin-families (39,40). In total, 8 waves of survey data on personality, health and lifestyle are collected in 1991, 1993, 1995, 1997, 2000, 2002, 2004 and 2009.
Age at regular alcohol use: Answer options: <11 years, 12, 13, 14, 15, 16, 17, 18 years or older, never (survey 2,3,4,8). When longitudinal data were available (also for age first cannabis): with a discrepancy of 1 or 2 years, the youngest age is selected, with a discrepancy of more than 2 years, the variable is set to missing 0–5% of cases).
Glasses alcohol per week: Answer options: <1 glass, 1–5 glasses, 6–10 glasses, 11–15 glasses, 21–40 glasses, > 40 glasses. When longitudinal data were available we used the highest number of glasses reported in all available data (survey 2–8). No survey data on alcohol use were available for 203 subjects.
Age at first time cannabis: answer options: <11 years, 12, 13, 14, 15, 16, 17, 18 years or older, never (survey 2,3,8).
Ever cannabis: The question age at first cannabis is collapsed into ever (1) and never (0).
We have chosen alcohol and cannabis phenotypes as similar as possible to smoking phenotypes from the TAG study (CPD -> glasses alcohol per week, AOS -> age at regular alcohol use/age at cannabis initiation, Ever smoked -> ever used cannabis).
DNA samples (41) were genotyped in different projects and genotyping was performed on Affymetrix 6.0 (N=298), Affymetrix Perlegen 5.0 (N=3697), Illumina 370 (N=290), Illumina 660 (N=1439), Illumina Omni Express 1M (N=455) platforms. Calls were made with platform specific software (Genotyper, Beadstudio). The quality control thresholds for SNPs were MAF > 1%, HWE > 0.00001, call rate >95% and 0.30 <Heterozygosity <0.35. Samples were excluded from the data if their expected sex and IBD status did not match, or if the genotype missing rate was >10%. SNPs were aligned to the positive strand of the Hapmap-2-Build 36-release-24 CEU reference set. Alignment was checked using individuals and family members tested on multiple platforms. SNPs were excluded if allele frequencies differed more than 15% with the reference set and/or the other platforms. The data of the platforms were merged into a single dataset (N=5856). This merged set was imputed against the reference set using IMPUTE v2. After imputation, genotype dosage was calculated if the highest genotype probability was above 90%. Badly imputed SNPs were removed based on HWE < 0.00001, proper info < 0.40, MAF < 1%, allele frequency difference >.15 against reference.
NTR subjects who participated in the GAIN-NTR study were excluded because those subjects were included in the original TAG meta-analyses. Family members of subjects in the GAIN-NTR study were also excluded (except non-biological members like spouses of twins). This resulted in a sample of 1583 subjects with genotype data, and 72% of the sample was female. The year of birth ranged between 1915–1994 (median 1958). Subject were from European decent.
Polygenic risk scores and statistical analyses
The polygenic risk scores reflect a combined effect of a number of selected Single Nucleotide Polymorphisms (SNPs) (38). Different p-value thresholds (Pt) of 0.1, 0.2 and 0.5 were used to define large sets of ‘risk alleles’ in the discovery sample (from TAG meta-analyses summary-level data). Those sets of risk alleles are used to generate a polygenic risk score for individuals in an independent target sample from the NTR. The individual risk score is calculated by multiplying the number of risk alleles per SNP (0,1,2) with the regression coefficient from the GWA meta-analyses, summed over all SNPs in the considered set of SNPs (42). The individual polygenic risk scores for the NTR participants were calculated using PLINK, with commands: --bfile NTRfile --maf 0.01 --mind 0.1 --geno 0.1 --hwe 0.000001 --score TAG_AOS_P5.dat --out TAG_AOSp5. Only SNPs that overlapped between the TAG sample and the NTR sample were included (Table 1).
Table 1.
Overview of the number of available SNPs in the TAG sample (All SNPs TAG), the number of overlapping SNPs between the TAG sample and the NTR sample, and the number of SNPs selected with the different p-value selection criteria.
| p-value thresholds (Pt): | N SNPs CPD | N SNPs AOS | N SNPs Ever | N SNPs former |
|---|---|---|---|---|
|
| ||||
| All SNPs TAG | 2, 502,107 | 2,500,547 | 2,498,833 | 2,499,522 |
| SNPs TAG & NTR | 2,123,025 | 2,122,544 | 2,121,558 | 2,121,558 |
| Pt =.5 | 1,088,808 | 1,079,361 | 1,103,228 | 1,085,301 |
| Pt=.2 | 450,210 | 442,816 | 474,407 | 449,091 |
| Pt=.1 | 230,447 | 224,460 | 252,924 | 233,788 |
Regression models were used to test the association with the polygenic risk scores based on smoking (predictor variable) and alcohol- and cannabis variables (independent variables). Linear regression models were used for continuous variables and logistic regression models for the dichotomous outcome variables. Regression analyses were carried out in STATA (version 9.0) and corrected for family clustering by employing the robust cluster option. Sex and birth cohort were added as covariates. To make clear how much variance is explained by the risk score itself and how much by the covariates, the R2 will be presented of the regression models including only the polygenic risk score (model 1), the regression model with risk score and sex (model 2) and the regression model with risk score, sex and age.
An association between a polygenic risk score and an outcome variable was considered significant if p <0.005 (we used a more stringent p-value than 0.05 to correct for multiple testing). We considered the results with 0.05<p<0.005 as marginally significant and discuss the results in this context.
Because correlations between the 4 different risk scores were (relatively) low, we also analyzed the 4 risk scores simultaneously in a regression analyses to explore whether the risk scores have an independent effect when corrected for the other risk scores.
Results
Table 2 shows the distribution of the alcohol and cannabis variables for the NTR target sample. About 3.5% of the sample never initiated alcohol use and those subjects were excluded for age at regular drinking. From the subjects who ever tried alcohol, 22.6% never started to drink regularly and more than half of the subjects (58.7%) started regular drinking after the age of 17. Almost 9% reported to drink more than 20 glasses alcohol per week. In the total sample, 85% never tried cannabis. Most of the subjects who tried cannabis started at 18 years or older.
Table 2.
Distribution of the alcohol and cannabis variables in the NTR target sample (N=1583).
| Variable | Categories | N (%) |
|---|---|---|
|
| ||
| Age regular drinking (among subjects who ever tried alcohol) | 11 or younger | 1 (0,1%) |
| 12 years | 1 (0,1%) | |
| 13 years | 1 (0,1%) | |
| 14 years | 14 (1.5%) | |
| 15 years | 41 (4.4%) | |
| 16 years | 97 (10.4%) | |
| 17 years | 69 (7.4%) | |
| 18 years or older | 497 (51.3%) | |
| Never | 210 (22.6%) | |
| Missing | 396 | |
|
| ||
| Glasses alcohol per week (among subjects who ever tried alcohol) | < weekly | 290 (21.2%) |
| 1–5 glasses | 271 (19.8%) | |
| 6–10 glasses | 276 (20.2%) | |
| 11–15 glasses | 243 (17.8%) | |
| 16–20 glasses | 166 (12.2%) | |
| 21–40 glasses | 104 (7.6%) | |
| >40 glasses | 15 (1.1%) | |
| Missing | 13 | |
|
| ||
| Age at first cannabis* | 11 or younger | 0 (0%) |
| 12 years | 1 (0.1%) | |
| 13 years | 0 (0%) | |
| 14 years | 10 (0.9%) | |
| 15 years | 15 (1.4%) | |
| 16 years | 28 (2.6%) | |
| 17 years | 16 (1.5%) | |
| 18 years or older | 93 (8.5%) | |
| Never | 925 (85.0%) | |
| Missing | 495 | |
This variable is also collapsed into ever/never cannabis use.
All polygenic risk scores for smoking showed a marginally significant association with 1 or more smoking variables (0.005<p<0.05) in our independent target sample, except the polygenic risk score for age at smoking onset (Supplemental Tables S1).
The polygenic risk score based on age at onset of smoking was significantly associated with age at which regular drinking started. This risk score was not associated with any of the other alcohol or cannabis phenotypes (Table 3A). The polygenic risk scores ever and former smoking did not significantly predict alcohol- or cannabis use (Table 3B en 3C). The risk scores based on CPD were significantly associated with the number of glasses alcohol per week and cannabis initiation, but not with age at regular drinking or age at first cannabis (Table 3D).
Table 3.
(A,B,C,D) Overview of results from linear (continuous variables) and logistic (dichotomous variables) regression analyses to predict alcohol and cannabis use with polygenic risk scores for smoking. Polygenic scores are based on meta-analyses summary-data of the Tobacco and Genetics consortium. Regression analyses are carried out on independent sample from the NTR. Sex was used as covariate in the regression analyses. p= p-value from the regression analyses; R2 = the explained variance; β = regression coefficient ; standardized β/OR = standardized beta from linear regression analyses or Odds Ratio from logistic regression. The standardized beta coefficients are the estimates resulting from the analysis carried out on the independent variables that have been standardized so that their variances are 1. Therefore, standardized coefficients refer to how many standard deviations a dependent variable will change, per standard deviation increase in the predictor variable. A positive beta reflects a positive association (for example the higher the polygenic risk score for age at smoking onset, the higher the age at regular drinking)
|
3A. Age at smoking initiation (AOS)
| ||||||
|---|---|---|---|---|---|---|
| Age at regular drinking | N=721, 561 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | 4.923 | .103 | .001 | .015 | .029 | .202 |
| Pt =.2 | 6.957 | .098 | .001 | .011 | .028 | .201 |
| Pt =.5 | 11.610 | .087 | .002 | .011 | .024 | .200 |
|
| ||||||
| Glasses alcohol per week | N=1069, 819 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 | |||
|
| ||||||
| Pt =.1 | 1.791 | .011 | .717 | .015 | .108 | .117 |
| Pt =.2 | .165 | .002 | .950 | .000 | .108 | .117 |
| Pt =.5 | −.982 | −.006 | .843 | .000 | .108 | .117 |
|
| ||||||
| Age at first cannabis | N=163, 154 families (ever cannabis = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 | |||
|
| ||||||
| Pt =.1 | −1.975 | −.032 | .655 | .000 | .002 | .101 |
| Pt =.2 | −1.581 | −.017 | .814 | .001 | .003 | .100 |
| Pt =.5 | .621 | .004 | .961 | .002 | .004 | .100 |
|
| ||||||
| Ever cannabis | N=1088, 820 families | |||||
|
| ||||||
| β | OR | P | R2 | |||
|
| ||||||
| Pt =.1 | −3.275 | .038 | .382 | .001 | .004 | .116 |
| Pt =.2 | −4.090 | .017 | .442 | .000 | .004 | .116 |
| Pt =.5 | −3.625 | .0266 | .716 | .000 | .004 | .116 |
|
3B Ever/never smoked (ever)
| ||||||
|---|---|---|---|---|---|---|
| Age regular drinking | N=721, 561 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .026 | .004 | .911 | .000 | .014 | .192 |
| Pt =.2 | −.098 | −.010 | .785 | .001 | .014 | .192 |
| Pt =.5 | −.288 | −.015 | .661 | .001 | .014 | .192 |
|
| ||||||
| Glasses alcohol per week | N=1069, 819 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .108 | .014 | .638 | .000 | .108 | .118 |
| Pt =.2 | −.038 | −.003 | .910 | .000 | .108 | .117 |
| Pt =.5 | −.216 | −.009 | .737 | .000 | .108 | .118 |
|
| ||||||
| Age at first cannabis | N=163, 154 families (ever cannabis = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .755 | .090 | .196 | .009 | .010 | .108 |
| Pt =.2 | 1.103 | .086 | .208 | .010 | .011 | .107 |
| Pt =.5 | 2.503 | .101 | .125 | .012 | .013 | .110 |
|
| ||||||
| Ever cannabis | N=1088, 820 families | |||||
|
| ||||||
| β | OR | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .427 | 1.533 | .606 | .001 | .005 | .116 |
| Pt =.2 | .488 | 1.628 | .689 | .001 | .004 | .116 |
| Pt =.5 | .461 | 1.586 | .836 | .000 | .004 | .116 |
|
3C. Smoking cessation (former)
| ||||||
|---|---|---|---|---|---|---|
| Age regular drinking | N=931, 712 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| P=.1 | .026 | .004 | .911 | .000 | .014 | .192 |
| P=.2 | −.098 | −.010 | .785 | .000 | .014 | .192 |
| P=.5 | −.288 | −.015 | .66 | .001 | .014 | .192 |
|
| ||||||
| Glasses alcohol per week | N=1313, 1004 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 | |||
|
| ||||||
| P=.1 | .108 | .014 | .638 | .000 | .108 | .118 |
| P=.2 | −.038 | −.003 | .910 | .000 | .108 | .118 |
| P=.5 | −.216 | −.009 | .737 | .000 | .108 | .118 |
|
| ||||||
| Age at first cannabis | N=163, 154 families (ever cannabis = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 | |||
|
| ||||||
| P=.1 | .755 | .090 | .169 | .009 | .010 | .108 |
| P=.2 | 1.103 | .087 | .204 | .010 | .011 | .107 |
| P=.5 | 2.503 | .101 | .125 | .012 | .013 | .110 |
|
| ||||||
| Ever cannabis | N=1087, 829 families | |||||
|
| ||||||
| β | OR | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| P=.1 | −.015 | .985 | .979 | .001 | .004 | .116 |
| P=.2 | −.509 | .601 | .573 | .000 | .003 | .116 |
| P=.5 | −.990 | .371 | .526 | .000 | .003 | .116 |
|
3D. Cigarettes per day (CPD)
| ||||||
|---|---|---|---|---|---|---|
| Age at regular drinking | N=721, 561 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .007 | .005 | .886 | .000 | .014 | .192 |
| Pt =.2 | .004 | .002 | .958 | .000 | .014 | .192 |
| Pt =.5 | −.035 | −.009 | .783 | .000 | .014 | .192 |
|
| ||||||
| Glasses alcohol per week | N=1069, 819 families (ever alcohol = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .111 | .068 | .016 | .004 | .113 | .122 |
| Pt =.2 | .191 | .078 | .005 | .005 | .114 | .124 |
| Pt =.5 | .342 | .075 | .007 | .004 | .114 | .123 |
|
| ||||||
| Age at first cannabis | N=163, 154 families (ever cannabis = yes) | |||||
|
| ||||||
| β | β standardized | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .039 | .026 | .758 | .000 | .002 | .101 |
| Pt =.2 | .029 | .013 | .874 | .000 | .002 | .100 |
| Pt =.5 | −.144 | −.033 | .690 | .002 | .003 | .101 |
|
| ||||||
| Ever cannabis (yes/no) | N=1088, 820 families | |||||
|
| ||||||
| β | OR | P | R2 model 1 |
R2 model 2 |
R2 model 3 |
|
|
| ||||||
| Pt =.1 | .278 | 1.321 | .015 | .006 | .010 | .123 |
| Pt =.2 | .481 | 1.617 | .004 | .009 | .013 | .125 |
| Pt =.5 | .897 | 2.453 | .004 | .007 | .011 | .125 |
Figures 1, 2 and 3 show the regression coefficients or odds ratio’s of the significant associations as well as the proportion of explained variance. The polygenic risk score for age at smoking onset explained 1.1–1.5% of the variance in age at regular drinking. When sex and birth cohort were included in the model, the explained variance was higher (around 20% for model 3, see Table 3A).
Figure 1.
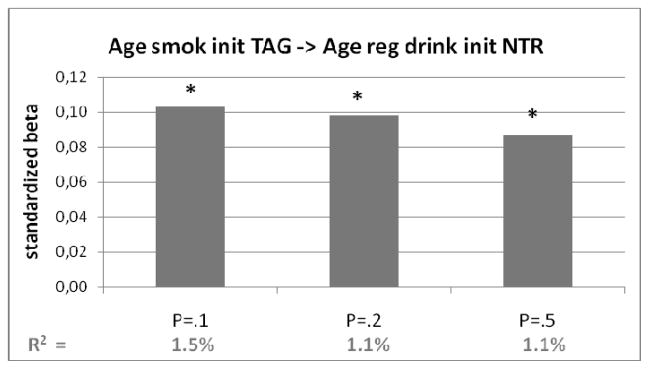
Polygenic risk score of age at smoking initiation (with different p-value thresholds) predicting age at regular drinking in target sample from NTR. On vertical axis the standardized regression coefficients (beta) from the regression analyses. In bottom row the explained variance (R2). Bars marked with a *: p<.05
Figure 2.

Polygenic risk score of cigarettes per day (with different p-value thresholds) predicting daily drinking in target sample from NTR. On vertical axis the odds ratio’s from the regression analyses. In bottom row the explained variance (R2). Bars marked with a *: p<.05
Figure 3.
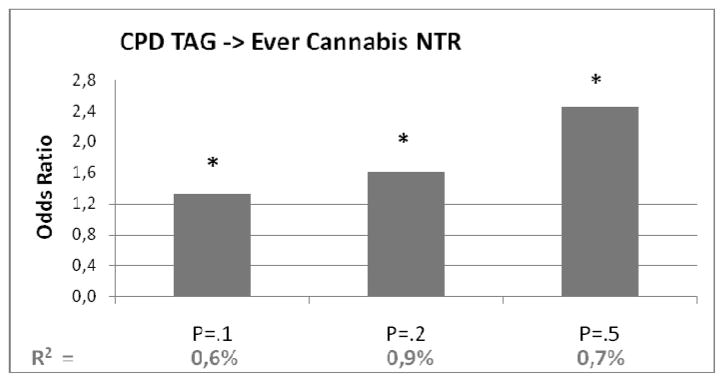
Polygenic risk score of cigarettes per day (with different p-value thresholds) predicting cannabis initiation in target sample from NTR. On vertical axis the odds ratio’s from the regression analyses. In bottom row the explained variance (R2). Bars marked with a *: p<.05
The polygenic risk score for CPD based, predicted .4–.5% of the variance in the number of glasses alcohol per week (see Table 3D) in the target sample. The polygenic risk score for CPD explained .6–.9% of the variance in cannabis use.
The correlation between the 4 different risk scores is moderate to low (Table 4). We compared the risk scores based on Pt=.2. The score for ever/never smoking was not significantly associated with the scores for former smoking or CPD. The highest correlation was found between the polygenic scores for CPD and former smoking (−.20).
Table 4.
Correlation between polygenic risk scores at P-value threshold of Pt=.2.
| Riskprofile at Pt=.2 | AOS | Ever | Former | CPD |
|---|---|---|---|---|
| AOS | 1 | |||
| Ever | −.1183* | 1 | ||
| Former | −.1562* | −.0346 | 1 | |
| CPD | .0831* | .0200 | −.2044* | 1 |
Significant correlations (p<.05) are flagged with a *. AOS= age at onset of smoking; Ever = ever/never regular smoker; Former = smoking cessation yes/no; CPD=Cigarettes per day
Because the correlations between the different risk scores were low, we also analyzed the 4 polygenic risk scores simultaneously in a regression analyses. The risk scores for CPD still predicted glasses alcohol per week and cannabis initiation, even when correcting for the other risk scores while the risk score for age at smoking onset still predicted age at regular alcohol use (Table 5).
Table 5.
Best fitting models when predicting alcohol- and cannabis use (dependent variables) with all 4 polygenic risk scores at Pt=.2 (AOS, Ever, Former, CPD) simultaneously and sex and birth cohort as predictors in a regression analyses. A backward method is used, with p-value threshold of .05. Only the variables with at least 1 significant risk score are shown: A. Glasses alcohol per week, B. Ever cannabis and C. Age at regular drinking. The variable age at first cannabis was not significantly associated with 1 or more polygenic risk scores.
| A. Glasses alcohol per week | β | β standardized | P | R2 |
|
| ||||
| CPD risk score | .192 | .313 | .005 | |
| Sex | −1.137 | −.099 | .000 | |
| Year of birth | −.009 | .078 | .001 | .124 |
|
| ||||
| B. Ever cannabis | β | OR | P | R2 |
|
| ||||
| CPD risk score | .480 | 1.617 | .004 | |
| Sex | .744 | 2.104 | .000 | |
| Year of birth | .063 | 1.065 | .000 | .125 |
|
| ||||
| C. Age at regular drinking | ||||
|
| ||||
| AOS risk score | 6.958 | .098 | .001 | |
| Sex | −.443 | −.200 | .000 | |
| Year of birth | −.032 | −.426 | .000 | .202 |
Discussion
The aim of this study was to investigate the overlap in polygenic risk factors between smoking behavior and alcohol- and cannabis use. Using polygenic risk scores derived from the GWA meta-analyses results of the TAG Consortium we predicted alcohol- and cannabis use in an independent sample from the Netherlands Twin Register. The risk scores for cigarettes per day explained .4–.5% of the variance in glasses alcohol per week and .6–.9% of the variance in cannabis initiation. The polygenic risk scores for age at onset of smoking predicted about 1.1–1.5% of the variance in age at regular alcohol use. The risk scores for smoking initiation and smoking cessation were not significantly associated with alcohol- and cannabis use.
When complex phenotypes, like addiction phenotypes, display a polygenic genetic architecture it is unlikely that GWA studies lead to straightforward results that can be replicated in independent samples. The TAG meta-analyses showed that even with large samples sizes no genome-wide significant results were obtained for smoking initiation and age at smoking initiation. For CPD a very strong association was observed for the SNPs in the cluster of Nicotinic Receptor Genes on chromosome 15 (15q25.1) (38). This CPD phenotype was also responsible for significant results in the present study. Interestingly, the significant associations we observed were not driven by the top SNPs on chromosome 15. The association between the polygenic risk score for CPD and glasses alcohol or cannabis initiation was not significant when a smaller number of SNPs was selected, for example: Pt=.01 (data not shown). A recent study composed a polygenic risk score based on 4 of the top-SNPs from the 15q25.1 region and 2 SNPs from another region (19q13.2). This score was unrelated with smoking initiation, but the individuals with a high score were more likely to convert to heavy, persistent smoking (43). Another study incorporated a SNP score of 92 top-SNPs (based on meta-analyses (23)) in a developmental model of CPD. The SNP score was associated with CPD, but not to the frequency of alcohol use at different ages (44). Our results suggested that, besides the top-SNPs from the meta-analyses, a large number of SNPs with all small individual effect sizes contribute to substance (ab)use.
The correlations between the 4 polygenic risk scores for smoking were moderate to low to non-significant. The highest correlation was found between the polygenic scores for CPD and former smoking and it was negative, suggesting that being an ex-smoker is associated with a high number of cigarettes per day. This can be explained by the fact that former smokers reported on the maximum number of cigarettes smoked per day while smokers report on their current number of cigarettes per day. The moderate correlations between the 4 polygenic risk scores might be the result of a lot of error variance resulting from random, non-generalizable, non-linear and/or interactive genetic effects. Previous twin studies have suggested some overlap between smoking-related variables, varying from only a small proportion of shared genetic variance (45,46) between age at first cigarette and smoking variables, to a higher genetic overlap between smoking persistence and initiation (47). A study of the Netherlands Twin Register showed two separate dimensions for smoking initiation and nicotine dependence, but those dimensions were not independent (5).
In the present study, the explained variance in the regression analyses varied from 0.4% for glasses alcohol per week up to 1.5% for age at regular drinking. Other studies reported explained variances varying from 0,1 to 3%.(32,48–50). Even when taking all available SNPs into account, the explained variance is lower than the heritability estimates from twin studies (26,30). An explanation for this ‘missing heritability’ problem is that the mutations causing variation in a trait are not in perfect linkage disequilibrium with any of the measured SNPs and therefore part of the genetic variance is undetected by the SNPs. The causal variants are expected to have lower minor allele frequencies (<0.1) because they are more likely to be subject to some form of natural selection that leads to variants negatively associated with reproductive fitness (51).
Low reported values of R2 might not directly reflect the degree of missing heritability; but could also reflect the effect of sampling variation on the variance explained by an estimated score (50). Because the individual SNP effects are very small they are estimated with much error. Although we can obtain an unbiased estimate of a SNP effect, a prediction of a phenotype using the estimated SNP effect suffers from the sampling variance with which the effect is estimated. The crudeness of the measures of substance use in the present study might have limited the explained variance. The worse the estimate of the effect size of the variant in the discovery sample, the worse the variance will be explained by the predictor in the validation sample (49,50).
Simulations showed that large sample sizes of the discovery sample are necessary (50). A strength of the present study is that summary-level results of the TAG meta-analyses were used as discovery sample. The TAG meta-analyses is currently the largest GWA meta-analyses for smoking behavior. The chances of success of polygenic risk score analyses depend primarily on the size of the discovery set. If the sample size is too small, the risk profiles will be based on random noise and are not expected to explain variance in the target set (31,50,52).
For traits with a moderate heritability (h2 .40) the required sample sizes of the discovery samples are about twice as high as for a trait with a high heritability (h2 .80) (50). Simulations showed that even with high-heritability traits the sample size of for example TAG is still rather low. Besides sample size other factors, like proportion of SNPs having effect on the trait, are of importance (50). We have used 4 smoking dimensions from the TAG meta-analyses and the heritability of these phenotypes varied. The fact that a significant association with alcohol- and cannabis use was found for the polygenic risk scores of CPD and AOS but not for smoking initiation and smoking cessation might (partly) be explained by differences in heritability. In samples of the NTR the heritability was 75% for Nicotine Dependence, 51% for CPD, 60% (males) and 39% (females) for age at first cigarette and 36%–44% for smoking initiation (5,53,54). CPD might mirror a more ‘severe’ phenotypes that reflect addictive behavior (like nicotine dependence).
The present results support the idea of a shared genetic background between smoking and use of alcohol and cannabis. In conclusion, our data point to a genetic architecture of many common variants with very small individual effect sizes, influencing both smoking behavior and alcohol- and cannabis use. This analysis provides the first evidence that aggregated genetic risk factors are shared between substances. The finding that genetic variants have cross-substance effects is an important step towards understanding the common co-occurrence of the use of different substances. Our findings suggest that besides ‘substance-specific’ genes, we’ll also have to search for ‘general substance-use’ genes.
Supplementary Material
Acknowledgments
This study was supported by the European Research Council (ERC Starting Grant 284167 PI Vink, ERC Advanced Grant 230374 PI Boomsma,), Netherlands Organization for Scientific Research (NWO: MagW/ZonMW grants 904-61-090, 985-10-002, 904-61-193, 480-04-004, 400-05-717, Addiction-31160008 Middelgroot-911-09-032, Spinozapremie 56-464-14192), Center for Medical Systems Biology (CSMB, NWO Genomics), NBIC/BioAssist/RK(2008.024), Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL, 184.021.007), the VU University’s Institute for Health and Care Research (EMGO+), Neuroscience Campus Amsterdam (NCA), the European Science Foundation (ESF, EU/QLRT-2001-01254), the European Community’s Seventh Framework Program (FP7/2007–2013), ENGAGE (HEALTH-F4-2007-201413), Rutgers University Cell and DNA Repository (NIMH U24 MH068457-06), the Avera Institute, Sioux Falls, South Dakota (USA), the National Institutes of Health (NIH, R01D0042157-01A, NIH DA-18673, DA-026119, PI Neale), the Genetic Association Information Network (GAIN) of the Foundation for the US National Institutes of Health, and the US National Institutes of Mental Health (NIMH, MH081802, 1RC2MH089951-01 PI Sullivan, 1RC2 MH089995- 01 PI Hudizak).
Footnotes
Declaration of interests: None.
References
- 1.John U, Meyer C, Rumpf H-J, Schumann A, Thyrian JR, Hapke U. Strength of the relationship between tobacco smoking, nicotine dependence and the severity of alcohol dependence syndrome criteria in a population-based sample. Alcohol and Alcoholism. 2003;38:606–612. doi: 10.1093/alcalc/agg122. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal A, Budney AJ, Lynskey MT. The co-occurring use and misuse of cannabis and tobacco: a review. Addiction. 2012;107:1221–1233. doi: 10.1111/j.1360-0443.2012.03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li MD, Cheng R, Ma JZ, Swan GE. A meta-analysis of estimated and environmental effects on smoking behavior in male and female adult twins. Addiction. 2003;98:23–31. doi: 10.1046/j.1360-0443.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan PF, Kendler KS. The genetic epidemiology of smoking. Nicotine Tobacco Res. 1999;1:S51–S57. doi: 10.1080/14622299050011811. [DOI] [PubMed] [Google Scholar]
- 5.Vink JM, Willemsen G, Boomsma DI. Heritability of smoking initiation and nicotine dependence. Behav Genet. 2005;35:397–406. doi: 10.1007/s10519-004-1327-8. [DOI] [PubMed] [Google Scholar]
- 6.van Beek JH, Kendler KS, de Moor MH, Geels LM, Bartels M, Vink JM, et al. Stable genetic effects on symptoms of alcohol abuse and dependence from adolescence into early adulthood. Behav Genet. 2012;42:40–56. doi: 10.1007/s10519-011-9488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Moor MH, Vink JM, van Beek JH, Geels LM, Bartels M, de Geus EJ, et al. Heritability of problem drinking and the genetic overlap with personality in a general population sample. Front Genet. 2011;2:76. doi: 10.3389/fgene.2011.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heath AC, Martin N, Lynskey MT, Todorov AA, Madden PAF. Estimating two-stage models for genetic influences on alcohol, tobacco or drug use initiation and dependence vulnerability in twin and family data. Twin Research. 2002;5:113–124. doi: 10.1375/1369052022983. [DOI] [PubMed] [Google Scholar]
- 9.Vink JM, Wolters LM, Neale MC, Boomsma DI. Heritability of cannabis initiation in Dutch adult twins. Addict Behav. 2010;35:172–174. doi: 10.1016/j.addbeh.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Distel MA, Vink JM, Bartels M, van Beijsterveldt CE, Neale MC, Boomsma DI. Age moderates non-genetic influences on the initiation of cannabis use: a twin-sibling study in Dutch adolescents and young adults. Addiction. 2011;106:1658–1666. doi: 10.1111/j.1360-0443.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verweij KJ, Zietsch BP, Lynskey MT, Medland SE, Neale MC, Martin NG, et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta-analysis of twin studies. Addiction. 2010;105:417–430. doi: 10.1111/j.1360-0443.2009.02831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kendler KS, Neale MC, Sullivan P, Corey LA, Gardner CO, Prescott CA. A population-based twin study in women of smoking initiation and nicotine dependence. Psychological Medicine. 1999;29:299–308. doi: 10.1017/s0033291798008022. [DOI] [PubMed] [Google Scholar]
- 13.Kendler KS, Schmitt E, Aggen SH, Prescott CA. Genetic and Environmental Influences on Alcohol, Caffeine, Cannabis, and Nicotine Use From Early Adolescence to Middle Adulthood. Arch Gen Psychiatry. 2008;65:674–682. doi: 10.1001/archpsyc.65.6.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koopmans JR, Doornen van LJP, Boomsma DI. Association between alcohol use and smoking in adolescent and young adult twins: a bivariate genetic analysis. Alcoholism: Clinical and Experimental Research. 1997;21:537–546. [PubMed] [Google Scholar]
- 15.Young S, Rhee SH, Stallings M, Corley R, Hewitt J. Genetic and Environmental Vulnerabilities Underlying Adolescent Substance Use and Problem Use: General or Specific? Behavior Genetics. 2006;36:603–615. doi: 10.1007/s10519-006-9066-7. [DOI] [PubMed] [Google Scholar]
- 16.Madden PAF, Heath AC. Shared genetic vulnerability in alcohol and cigarette use and dependence. Alcoholism. 2002;26:1919–1921. doi: 10.1097/01.ALC.0000040960.15151.30. [DOI] [PubMed] [Google Scholar]
- 17.Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE. Molecular genetics of nicotine dependence and abstinence: whole genome association using 520,000 SNPs. BMC genetics. 2007:8. doi: 10.1186/1471-2156-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu QR, Drgon T, Johnson C, Walther D, Hess J, Uhl GR. Addiction Molecular Genetics: 639,401 SNP whole genome association identifies many “cell adhesion” genes. American Journal of Medical Genetics Part B (Neuropsychiatric Genetics) 2006;141B:918–925. doi: 10.1002/ajmg.b.30436. [DOI] [PubMed] [Google Scholar]
- 19.Bierut LJ, Madden PAF, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Human Molecular Genetics. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vink JM, Smit AB, de Geus EJ, Sullivan P, Willemsen G, Hottenga JJ, et al. Genome-wide association study of smoking initiation and current smoking. Am J Hum Genet. 2009;84:367–379. doi: 10.1016/j.ajhg.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amos CI, Spitz MR, Cinciripini P. Chipping away at the genetics of smoking behavior. Nat Genet. 2010;42:366–368. doi: 10.1038/ng0510-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–440. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42:448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consortium TaG. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agrawal A, Lynskey MT. Candidate genes for cannabis use disorders: findings, challenges and directions. Addiction. 2009;104:518–532. doi: 10.1111/j.1360-0443.2009.02504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verweij KJH, Vinkhuyzen AAE, Benyamin B, Lynskey MT, Quaye L, Agrawal A, et al. The genetic aetiology of cannabis use initiation: a meta-analysis of genome-wide association studies and a SNP-based heritability estimation. Addiction Biology. 2012:n/a–n/a. doi: 10.1111/j.1369-1600.2012.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agrawal A, Lynskey MT, Hinrichs A, Grucza R, Saccone SF, Krueger R, et al. A genome-wide association study of DSM-IV cannabis dependence. Addiction Biology. 2011;16:514–518. doi: 10.1111/j.1369-1600.2010.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rietschel M, Treutlein J. The genetics of alcohol dependence. Annals of the New York Academy of Sciences. 2013;1282:39–70. doi: 10.1111/j.1749-6632.2012.06794.x. [DOI] [PubMed] [Google Scholar]
- 29.Symons M, Weng J, Diehl E, Heo E, Kleiber M, Singh S. Delineation of the Role of Nicotinic Acetylcholine Receptor Genes in Alcohol Preference in Mice. Behavior Genetics. 2010;40:660–671. doi: 10.1007/s10519-010-9366-9. [DOI] [PubMed] [Google Scholar]
- 30.Lubke GH, Hottenga JJ, Walters R, Laurin C, de Geus EJ, Willemsen G, et al. Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biol Psychiatry. 2012;72:707–709. doi: 10.1016/j.biopsych.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaun MP, Naomi RW, Jennifer LS, Peter MV, Michael COD, Patrick FS, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demirkan A, Penninx BWJH, Hek K, Wray NR, Amin N, Aulchenko YS, et al. Genetic risk profiles for depression and anxiety in adult and elderly cohorts. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luciano M, Huffman JE, Arias-Vásquez A, Vinkhuyzen AAE, Middeldorp CM, Giegling I, et al. Genome-wide association uncovers shared genetic effects among personality traits and mood states. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2012;159B:684–695. doi: 10.1002/ajmg.b.32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Middeldorp CM, Moor MHMd, McGrath LM, Gordon SD, Blackwood DH, Costa PT, et al. The genetic association between personality and major depression or bipolar disorder. A polygenic score analysis using genome-wide association data. Translational Psychiatry. 2011:1. doi: 10.1038/tp.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium C.-D. G. o. t. P. G. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. The Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vorstman JAS, Anney RJL, Derks EM, Gallagher L, Gill M, de Jonge MV, et al. No evidence that common genetic risk variation is shared between schizophrenia and autism. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2013;162:55–60. doi: 10.1002/ajmg.b.32121. [DOI] [PubMed] [Google Scholar]
- 37.van Scheltinga AFT, Bakker SC, van Haren NEM, Derks EM, Buizer-Voskamp JE, Cahn W, et al. Schizophrenia genetic variants are not associated with intelligence. Psychological Medicine. 2013:1–8. doi: 10.1017/S0033291713000196. FirstView. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tobacco-and-Genetic-Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boomsma DI, de Geus EJ, Vink JM, Stubbe JH, Distel MA, Hottenga JJ, et al. Netherlands Twin Register: from twins to twin families. Twin Res Hum Genet. 2006;9:849–857. doi: 10.1375/183242706779462426. [DOI] [PubMed] [Google Scholar]
- 40.Willemsen G, Vink JM, Abdellaoui A, den Braber A, van Beek JHDA, Draisma HHM, et al. The Adult Netherlands Twin Register: Twenty-Five Years of Survey and Biological Data Collection. Twin Research and Human Genetics. 2013;16:271–281. doi: 10.1017/thg.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willemsen G, de Geus EJC, Bartels M, van Beijsterveldt CEM, Brooks AI, Estourgie-van Burk GF, et al. The Netherlands Twin Register Biobank:A Resource for Genetic Epidemiological Studies. Twin Res Hum Genet. 2010;13:231–245. doi: 10.1375/twin.13.3.231. [DOI] [PubMed] [Google Scholar]
- 42.Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Human Molecular Genetics. 2009;18:3525–3531. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- 43.Belsky DwMTEBTB, et al. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: Evidence from a 4-decade longitudinal study. JAMA Psychiatry. 2013;70:534–542. doi: 10.1001/jamapsychiatry.2013.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vrieze S, McGue M, Iacono W. The interplay of genes and adolescent development in substance use disorders: leveraging findings from GWAS meta-analyses to test developmental hypotheses about nicotine consumption. Human genetics. 2012;131:791–801. doi: 10.1007/s00439-012-1167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardie TL, Moss HB, Lynch KG. Genetic correlations between smoking initiation and smoking behaviors in a twin sample. Addictive Behaviors. 2006;31:2030–2037. doi: 10.1016/j.addbeh.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 46.Broms U, Silventoinen K, Madden PAF, Heath AC, Kaprio J. Genetic architecture of smoking behavior: a study of Finnish adult twins. Hum Genet. 2006;9:64–72. doi: 10.1375/183242706776403046. [DOI] [PubMed] [Google Scholar]
- 47.Madden PAF, Heath AC, Pedersen NL, Kaprio J, Koskenvuo MJ, Martin NG. The genetics of smoking persistence in men and women: a multicultural study. Behavior Genetics. 1999;29:423–444. doi: 10.1023/a:1021674804714. [DOI] [PubMed] [Google Scholar]
- 48.International-Schizophrenia-Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatry. 2011;16:996–1005. doi: 10.1038/mp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudbridge F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genet. 2013;9:e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visscher PM, Yang J, Goddard ME. A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al. (2010) Twin Res Hum Genet. 2010;13:517–523. doi: 10.1375/twin.13.6.517. [DOI] [PubMed] [Google Scholar]
- 52.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Research. 2007;17:1520–1528. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vink JM, Beem AL, Posthuma D, Neale MC, Willemsen G, Kendler KS, et al. Linkage analysis of smoking initiation and quantity in Dutch sibling pairs. Pharmacogenomics J. 2004;4:274–282. doi: 10.1038/sj.tpj.6500255. [DOI] [PubMed] [Google Scholar]
- 54.Vink JM, Posthuma D, Neale MC, Eline Slagboom P, Boomsma DI. Genome-wide linkage scan to identify loci for age at first cigarette in Dutch sibling pairs. Behav Genet. 2006;36:100–111. doi: 10.1007/s10519-005-9012-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.