

Summary
The aryl hydrocarbon receptor (AHR) has garnered considerable attention as a modulator of CD4 lineage development and function. It also regulates antiviral CD8+ T cell responses, but via indirect mechanisms that have yet to be determined. Here, we show that during acute influenza virus infection, AHR activation skews dendritic cell (DC) subsets in the lung-draining lymph nodes, such that there are fewer conventional CD103+DCs and CD11b+DCs. Sorting DC subsets reveals AHR activation reduces immunostimulatory function of CD103+DCs in the MLN, and decreases their frequency in the lung. DNA binding domain (DBD) Ahr mutants demonstrate that alterations in DC subsets require the ligand-activated AHR to contain its inherent DBD. To evaluate the intrinsic role of AHR in DCs, conditional knockouts were created using Cre-LoxP technology, which reveal that AHR in CD11c+ cells plays a key role in controlling the acquisition of effector CD8+ T cells in the infected lung. However, AHR within other leukocyte lineages contributes to diminished naïve CD8+ T cell activation in the draining lymphoid nodes. These findings indicate DCs are among direct targets of AHR ligands in vivo, and AHR signaling modifies host responses to a common respiratory pathogen by affecting the complex interplay of multiple cell types.
Keywords: Dendritic cells, Influenza A virus, CD8 T cells, Antiviral immunity
Introduction
The aryl hydrocarbon receptor (AHR) is an environment sensing transcription factor that binds a wide variety of ligands, ranging from dioxins and related pollutants to metabolites derived from foods we consume [1, 2]. There is a substantial body of evidence supporting a role for AHR-mediated signaling in the development and function of the immune system. This is a rich literature, and will not be reviewed in its entirety herein. Briefly, AHR activation has a tremendous influence on conventional CD4+ T cell function and T cell-dependent B cell responses [3]. Several studies support a regulatory role for AHR agonists in autoimmune diseases [4–7], allergic inflammation [8], and inflammatory bowel diseases [9–11], via an influence on Th17 and CD4+ regulatory T (Treg) cells. When considering infectious diseases, several reports indicate that AHR plays a critical role, but that the nature of AHR's function varies with the type of pathogen and target organ [12–19]. For instance, AHR activation by exogenous ligands profoundly modulates host resistance, leading to either improved or worse outcomes following infection [12, 14, 18]. Conflicting results with AHR-deficient mice suggest that AHR likely plays pathway-specific roles in modulating host defense mechanisms [13–17, 19].
Using influenza virus as a model system to probe how AHR modulates host responses to infection, it has been shown that triggering AHR significantly reduces the proliferation and differentiation of virus specific CD8+ T cells via an indirect mechanism [15, 20]. Specifically, experiments with bone marrow chimeras and adoptive transfer of Ahr-null CD8+ T cells into congenic Ahr+/+ recipients revealed that AHR-regulated events within hematopoietic cells, but not directly within CD8+ T cells underlie this reduced response [15]. Yet, the consequences to the host are profound, as evidenced by enhanced morbidity and mortality [12, 21, 22]. The critical window for this effect is within the first 4 days of infection [20], suggesting AHR regulates accessory cell functions that control naïve CD8+ T cell activation, such as DCs. DCs are the major APCs that stimulate naïve CD8+ T cells in a primary infection. Consistent with this idea, AHR activation reduces the trafficking of DCs from the infected lung to the draining mediastinal lymph nodes (MLNs) [20]. Experimental evidence from other model systems further supports that DCs are affected by exposure to AHR agonists [6, 8, 20, 23–30].
DCs are heterogeneous cells, and the in vivo function of distinct DC subsets is not fully understood [31]. Likewise, the consequences of AHR signaling within phenotypically distinct DC subsets remain poorly explored. For instance, conventional DCs can be further divided into CD11b+ and CD103+ subsets. In the lung these represent two major DC subsets that migrate during respiratory antigen challenge, and present antigen to T cells in the lung-draining lymph nodes [32–43]. However, whether AHR activation modulates the proportion of these two specific DC subsets in vivo, and whether AHR-modulated events within DCs are directly responsible for the poorer response of CD8+ T cells during infection remain unknown. Thus, in the present study, we sought to determine whether AHR alters the frequency, trafficking, or functional capacity of CD11b+ and CD103+DC subsets during influenza virus infection. Moreover, we used the Cre/loxP system to create novel conditional knockout mice to delineate whether AHR within CD11c lineage cells directly contributes to poorer responsive capacity of CD8+ T cells during respiratory viral infection.
Results
AHR activation alters the profile of DC subsets in the MLN
To determine whether AHR activation affects DCs during influenza virus infection, we used the prototypical AHR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD or dioxin). While the identity and physiologic function of endogenous AHR ligands remain enigmatic, we are exposed to AHR-binding dioxins and polychlorinated biphenyls (PCBs) regularly, primarily through ingestion [1, 2, 44], and epidemiological reports show exposure to these pollutants are significantly and directly associated with more respiratory tract infections [45, 46]. Moreover, in a mouse model, a single low dose of TCDD sustains AHR activation throughout the response to influenza virus [47]. We first examined the effect of AHR activation on the proportion of DC subsets in the lung-draining MLNs. There are two different populations of CD11c+ cells in the MLN (Fig. 1A): CD11chiMHCIIlo cells (R1 gate) and CD11c+MHCIIhi cells (R2 gate). CD11chiMHCIIlo cells are described as either CD11c+ monocytes or as resident DCs [34, 48]; whereas CD11c+MHCIIhi cells are consistently thought to represent DCs, including those that have recently emigrated from peripheral tissues [34, 43]. Sorted CD11c+MHCIIhi cells (R2 gate) from virus-infected mice were able to activate naïve CD8+ T cells, but sorted cells from the R1 gate (CD11chiMHCIIlo cells) were unable to do so (data not shown). Therefore, we defined DCs as cells in the R2 gate. In the absence of infection, AHR activation with TCDD caused no discernable difference in the percent or number of total DCs in the MLN (data not shown). However, 3 days after infection AHR activation reduced the percentage and number of DCs in the MLN (Fig. 1B), with a significant reduction in both the CD11b+DCs and CD103+DC subsets (Fig. 1C). As an indicator of DC maturation we examined several cell surface molecules important for DC activation of naïve T cells [49, 50]. Although the percentage of CD11b+ and CD103+DCs expressing CD80, CD86 or CD40 did not change with AHR activation (data not shown), AHR activation significantly reduced MFI of CD40 on CD103+DCs, but not on CD11b+DCs, from infected mice (Fig.1D).
Figure 1.
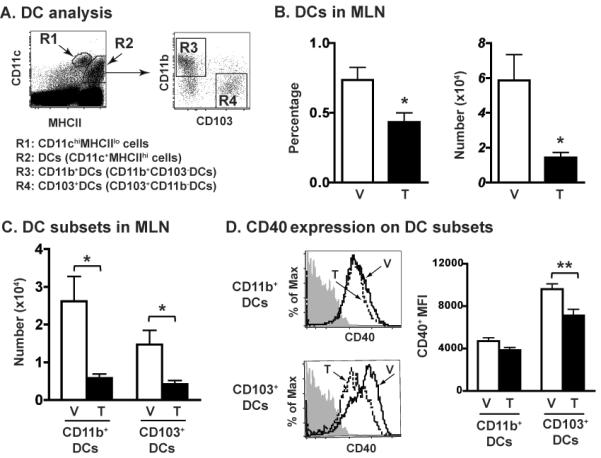
AHR activation reduces the number of CD11b+ and CD103+ DCs in the MLNs. Female B6 mice were dosed orally with 10 μg/kg of TCDD (TCDD-treated group, T) or peanut oil (vehicle-treated group, V) one day prior to influenza A virus infection (i.n., Mem/102, H3N2). MLN cells were collected 3 days later and stained with mAbs for flow cytometric analysis. (A) Doublet discrimination and live cell gating was used following strategies outlined in Supporting Information Fig. 4A. Two distinct populations of CD11c+ cells in the MLN: CD11chiMHCIIlo cells (R1 gate, monocytes) and CD11c+MHCIIhi cells (R2 gate, DCs). DCs are further analyzed to enumerate CD11b+DCs (R3 gate) and CD103+DCs (R4 gate). (B) The average percentage and number of DCs in the MLN. (C) The number of CD11b+DCs and CD103+DCs in the MLN. (D) Representative histograms of CD40 expression on the indicated DC subsets. Gray filled histograms depict the CD40 FMO controls; black lines indicate CD40 on cells from the vehicle group and dashed lines indicate CD40 from TCDD treatment group. The bar graph shows the average CD40 MFI in each DC subset from each treatment group. Data are shown as mean ± SEM (n = 7/group) from one experiment that is representative of 3 independent experiments. *p < 0.05, **p < 0.01, two-tailed unpaired Student's t-test.
To establish whether the reduced number of CD103+ and CD11b+DCs in the MLN reflects enhanced death of DCs, we determined whether AHR activation increases the frequency of apoptotic or dead DCs. We found no evidence to support this, as the percentage of Annexin-V single positive or Annexin-V,Live/Dead double positive DCs was not different in CD11c+ subsets from vehicle control or TCDD-treated mice before or after infection (Supporting Information Fig. 1A–D). We next determined whether the reduced number of CD103+ and CD11b+ DCs reflects that fewer are emigrating to the MLN from the infected lung by instilling (i.n.) CFSE to label cells in the lung [20, 51]. CFSE was instilled one day prior to infection, and CFSE+ DCs in the MLN were analyzed 3 days after infection. Importantly, there were no differences in the percentage, number, or fluorescence intensity of CFSE-labeled cells in lungs of mice treated with vehicle vs. TCDD, including phenotypically distinct CD11c+ subsets ([20], and data not shown). Consistent with prior reports, CD103+DCs are the major subset that has migrated from the lung at this point in time after infection [42, 43]. AHR activation significantly reduced the percentage of CD103+DCs that were CFSE+, but did not alter the percentage of CD11b+DCs that were CFSE+ (Fig. 2A). However, since AHR activation reduced the total number of CFSE+DCs in the MLN, there was a significant decrease in the number of both CFSE+CD11b+ and CFSE+CD103+ DC subsets (Fig. 2B). To account for the dynamic nature of the DC compartment in the lung, in separate experiments CFSE was administered (i.n.) 48 h after infection, and the frequency of CFSE-labeled DC subsets in the MLN was examined on day 3 of infection. Similarly, while no differences in CFSE-labeling of cells in the lung were observed, AHR activation reduced the number of CFSE+CD11b+ and CFSE+CD103+ DCs in the MLN (data not shown). Thus, AhR activation reduces DC number in the MLN following influenza virus infection, suggesting reduced emigration from lung.
Figure 2.
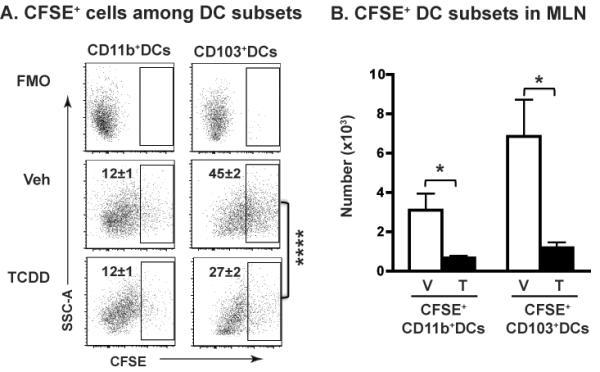
AHR activation reduces lung DC migration to the MLN. Mice were treated and infected as described in Fig. 1, except that they were given CFSE (i.n.) 18 h before infection. On day 3-post infection, MLNs were removed and processed for flow cytometry. CD11b+ and CD103+ DCs are defined as described in Fig. 1A, and the frequency of CFSE+ DCs was analyzed. CFSE+ cells were defined using MLN cells from mice that received media i.n. (FMO control). (A) Numbers on each gated region indicate the percentage of CFSE+ cells among CD11b+ or CD103+ DCs. (B) Bar graphs depict the average number of CFSE+CD11b+ and CFSE+CD103+ DCs in the MLN. Data are shown as mean ± SEM (n = 7/group) from one experiment that is representative of two independent experiments. *p < 0.05, ****p < 0.0001, two-tailed unpaired Student's t-test.
AHR activation impairs the ability of CD103+DCs to stimulate naïve virus-specific CD8+ T cells
To evaluate whether AHR activation alters the function of phenotypically defined DC subsets, we directly compared the activation of naïve, virus-specific CD8+ T cells by sorting CD11b+DCs and CD103+DCs from infected mice (± AHR activation; Fig. 3A). Three days after infection, sorted CD103+DCs from the MLN activate naïve F5 CD8+ T cells, as measured by the up-regulation of CD44, diminished CFSE staining, and secretion of IFNγ (Fig. 3B–D). In vivo AHR activation resulted in a 2-fold reduction in the ability of sorted CD103+DCs to drive the proliferation and differentiation of F5 CD8+ T cells (Fig. 3B–D), and this persisted across several different ratios of T cells to DCs. This is an important consideration because it has been suggested that partially matured DCs support proliferation only at higher T cell:DC ratios, but at lower ratios fail to support T cell proliferation and differentiation [52]. In contrast to CD103+DCs, CD11b+DCs, which were sorted from the same lymph nodes, were unable to activate naïve F5 CD8+ T cells at any T cell:DC ratio used, and AHR activation did not alter this (Fig. 3B and data not shown).
Figure 3.
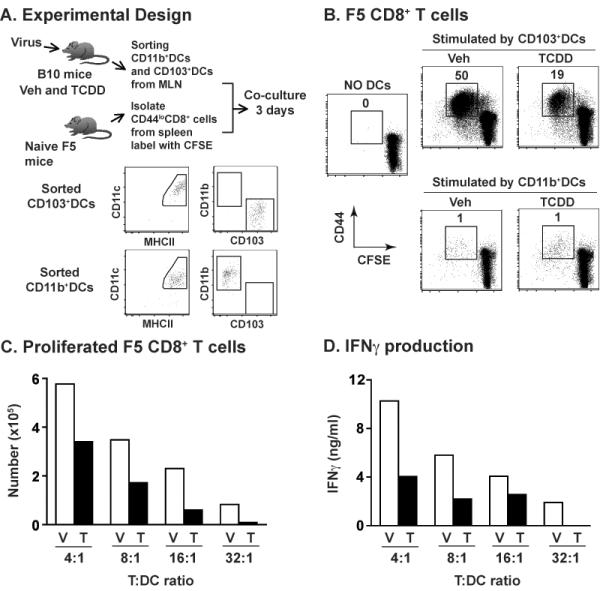
AHR activation reduces the ability of CD103+DCs to stimulate naïve virus-specific CD8+ T cells. (A) The overall approach: mice were treated and infected as described in Fig. 1. On day 3-post infection MLN cells were pooled from mice in the same group (≥30 mice/group) and stained with fluorochrome-conjugated mAbs. Using the same gating strategy as in Fig. 1A, DCs were sorted to obtain CD11b+DCs and CD103+DC (purity of sorted subsets ≥95%). Sorted DCs were serially diluted and used to stimulate CFSE-labeled naïve (CD44lo) F5 CD8+ T cells (2×105 cells/well), in a range from 4:1 to 32:1 T cells:DCs. Cells were collected after 3 days of ex vivo co-culture and stained for flow cytometric analysis. DCs in the co-culture were excluded and CD3+CD8+ cells were used to identify F5 CD8+ T cells (Supporting Information Fig. 4B). (B) Number on each dot plot indicates the percentage of activated (CFSEdecayCD44hi) F5 CD8+ T cells after culture with CD103+DCs (16:1 T:DC ratio) or CD11b+DCs (8:1 T:DC ratio). “NO DCs” shows CFSE-labeled naïve F5 CD8+ T cells cultured in the absence of antigen-bearing DCs. (C) Bar graphs show the number of activated F5 CD8+ T cells stimulated by CD103+DCs derived from MLN of vehicle (V) or TCDD (T) treated mice, and (D) IFNγ levels in corresponding co-culture supernatants. Other controls include the use of sorted CD11chiMHCIIlo cells from Mem/102 infected mice, and isolated CD11c+ cells from mice infected with HKx31, a strain that cannot be recognized by F5 transgenic CD8+ T cells. These cells failed to stimulate CD8+ T cell activation at any T cell:DC ratio used (not shown). Data shown are from one experiment that is representative of two independent experiments with the same results.
AHR activation reduces the number of DCs in the infected lung
Given that the primary source of DCs carrying influenza virus antigens to the MLN is the infected lung, we examined whether AHR activation with TCDD alters the profile of CD11c+ subsets in the lung. Similar to the MLN, there are two different populations of CD11c+ cells in the lung (Fig. 4A): CD11chiMHCIIlo cells (R1 gate) and CD11c+MHCIIhi cells (R2 gate). CD11chiMHCIIlo cells are a subset of lung monocytes/macrophages, whereas CD11c+MHCIIhi cells are DCs [37, 43, 48]. In uninfected mice, AHR activation did not alter the percentage or number of total DCs or CD11c+ macrophages in the lung (Fig. 4A–E). However, three days after infection there were 50% fewer lung DCs when AHR was activated (Fig. 4A,C,E). Consistent with prior reports, in the uninfected lung the proportion of CD11b+DCs is greater than CD103+DCs (e.g. 57% versus 16%, respectively), and the frequency of CD103+DCs decreases by day 3 after infection (Fig. 4F–H). Interestingly, AHR activation further reduced the percentage and number of CD103+DCs. In fact, AHR significantly diminished the number of lung CD103+DCs even in the absence of infection (Fig. 4J). In contrast, AHR activation did not change the frequency of CD11b+DCs in naïve lung, but did reduce the number of CD11b+DCs following infection on day 3 post infection (Fig. 4F,G,I). Similar to the MLN, AHR activation did not change the percentage of apoptotic CD11c+ subsets in the unchallenged or infected lung (Supporting Information Fig. 1E–H).
Figure 4.
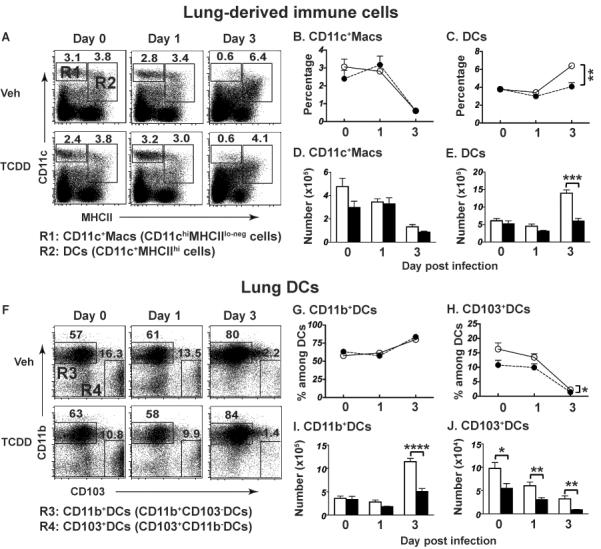
AHR activation modulates DCs in the lung. Female B6 mice were treated and infected as described in Fig. 1, and sacrificed on the indicated day relative to infection. Lung-derived immune cells were obtained using collagenase digestion and stained with fluorochrome-conjugated Abs. (A) Following gating to exclude doublets, dead and autofluorescent cells (Supporting Information Fig. 4C), CD45.2+CD11c+ cells were divided into two sub-populations: CD11chiMHCIIlo cells (R1 gate, CD11c+Macs) and CD11c+MHCIIhi cells (R2 gate, DCs). DCs were further analyzed as CD11b+DCs (R3 gate) and CD103+DCs (R4 gate). Numbers on dot plots show the average percentage of cells in the gated region. Panels B and C show the percentage of CD11c+Macs and DCs, D and E depict number of CD11c+Macs and DCs in the lung. Panels G and H show the percentage of CD11b+ and CD103+ cells among DCs, and I and J depict the number of CD11b+ and CD103+ DCs in the lung. Data are shown as mean ± SEM (n = 7–8/group) are from one experiment that is representative of at least two independent experiments with same results. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA followed by Tukey HSD.
It has been reported that AHR activation enhanced expression of the immunoregulatory genes Ido1 and Il10 in myeloid-derived cells [6, 24–26, 53]. Thus, we examined whether in vivo AHR activation up-regulated expression of these genes in sorted lung DCs and CD11c+ macrophages (Fig. 5). The AHR target gene cytochrome P4501A1 (Cyp1a1) is strongly induced in DCs and CD11c+ macrophages from TCDD treated mice (Fig. 5B,C). Ido1 was also significantly induced by AHR activation in sorted DCs, but Il10 mRNA levels were not significantly affected in this subpopulation of lung cells (Fig. 5B). In sorted CD11c+ macrophages we observed the opposite: Ido1 was not significantly induced by in vivo AHR activation, but Il10 gene expression was significantly elevated (Fig. 5C).
Figure 5.
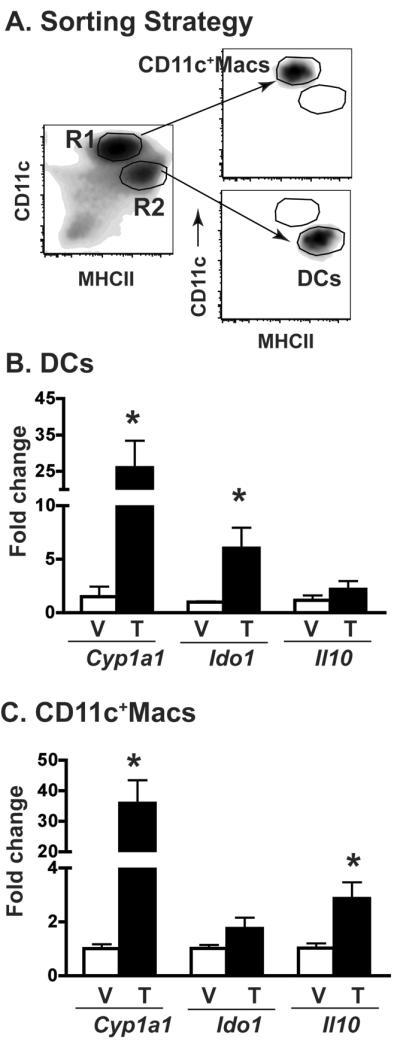
Cell type specific AHR-mediated changes in gene expression. Female B6 mice were treated and infected as described in Fig. 1. (A) Lung-derived CD11c+ cells were enriched using mouse CD11c microbeads and then sorted into CD11c+ Macs (R1 gate) and DCs (R2 gate); purity ≥99%. Total RNA was isolated from sorted cells, and Cyp1a1, Ido1, and Il-10 gene expression determined by quantitative real time PCR. (B,C) The average fold change (ΔΔCT) in gene expression compared to vehicle-treated mice and normalized to L13 is shown for sorted (B) DCs and (C) CD11c+Macs. V, indicates vehicle treatment group, T indicates TCDD treatment group. Data are shown as mean ± SEM (n=3 separate pools/group; where each pool consisted of cells from 10–15 mice). *p < 0.05, two-tailed unpaired Student's t-test. Data are representative of two independent experiments with same results.
AHR modulates DC phenotype via a mechanism that requires AHR's intrinsic DNA binding domain
Examination of Ahr−/− mice reveals mixed consequences to the immune system [54], and new information suggests AHR plays a key role in the development of certain innate immune cells [55–57]. With this in mind, we examined whether Ahr−/− mice exhibit discernable changes in the number of DCs, or an alteration in the proportion of DC subsets upon infection. Compared to age-matched wild-type mice, there were no differences in the number of DCs, CD103+DCs, CD11b+DCs, or CD11c+Macs in the MLN or lung of naïve or influenza virus infected Ahr−/− mice (Supporting Information Fig. 2 and data not shown). Moreover, in wild-type mice, all of these CD11c+ sub-populations express Ahr at similar levels (Supporting Information Fig. 3). Collectively, this provides a foundation for beginning to understand how the AHR modulates DC responses during infection.
In the canonical AHR signaling pathway, ligand binding triggers alterations in AHR conformation and chaperone binding, which initiates trafficking to the nucleus. Within the nucleus, activated AHR-ligand complexes bind aryl hydrocarbon response elements (AhRE) within AHR target genes, such as cyp1a1. Alternative pathways of AHR activation have been reported, in which AHR alters cell signaling or gene expression independently of AHR binding to these consensus DNA sequences [58–60]. Thus, we next determined whether AHR alters the proportion of DC subsets during infection via a pathway that requires AHR binding to DNA via its DNA binding domain (DBD). To accomplish this, we used Ahrdbd/dbd mice, which express a mutated AHR protein that lacks its DBD [61]. The Ahrdbd/dbd gene encodes a protein that binds ligand, translocates to the nucleus, but cannot bind consensus AhRE to induce expression of AHR target genes (e.g., cyp1a1). In contrast to effects observed in wild-type mice, TCDD treatment of Ahrdbd/dbd mice did not reduce the percentage or number of DCs (Fig. 6A), reduce the number of CD11b+ and CD103+DCs in the MLN (Fig. 6B), or alter the CD40 MFI on CD103+DCs (Fig. 6C). These results suggest that the cellular processes affected by AHR require that it likely acts in vivo directly as a transcription factor, rather than via crosstalk with other transcriptional regulators in the cytosol, or modulation of other transcriptional regulators such as NF-κB or the estrogen receptor.
Figure 6.
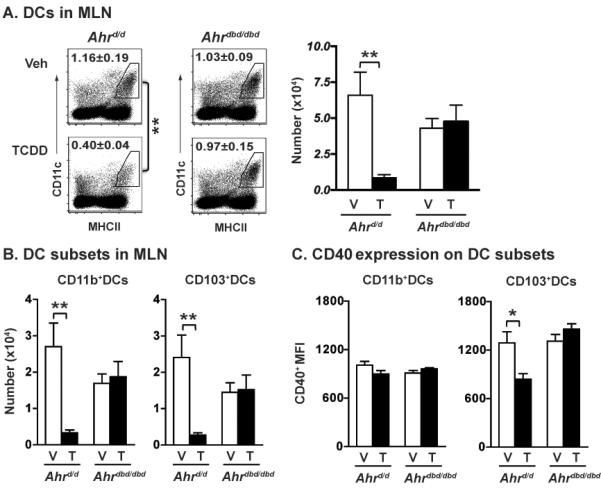
Alterations in DC subset distribution require the ligand-activated AHR have a functional DNA binding domain. Female B6 congenic (Ahrd/d) and Ahrdbd/dbd mutant mice received a single oral dose of either peanut oil or TCDD (100 μg/kg) one day before infection (i.n.). Three days later, MLN cells were collected, stained with mAbs, and data were analyzed as described in Fig. 1A. (A) The number on each gated region shows the average percentage of DCs in each group, and bar graphs depict the average number of DCs in the MLN. (B) The graphs show the number of CD11b+DCs (left) and CD103+DCs (right) in the MLN. (C) The bar graphs depict the CD40 MFI in each DC subset. Data are shown as mean ± SEM (n = 5–6/group) from one experiment representative of three performed. *p < 0.05, **p < 0.01 compared within genotype using one-way ANOVA with post hoc tests.
Conditional knockout reveals CD11c-lineage intrinsic and extrinsic role of AhR
To test whether DC intrinsic AHR signaling is upstream of the reduced CD8+ T cell response to influenza virus infection, we conditionally deleted Ahr by crossing Ahrfx/fx mice with mice that express Cre recombinase under control of the CD11c promoter (CD11ccre mice) [62–64]. To verify selective excision of Ahr from DCs, we sorted CD11c+MHCIIhi cells from CD11ccreAhrfx/fx and Ahrfx/fx mice, and used PCR probes that distinguish the excised and unexcised Ahr gene [65]. DCs from Ahrfx/fx mice have the unexcised Ahr gene, which encodes a functional AHR protein, whereas DCs from CD11ccreAhrfx/fx mice have a PCR product consistent with successful excision of Ahr (Fig. 7A). We next determined whether the absence of Ahr in CD11c-lineage cells affects the AHR-mediated suppression of the CD8+ T cell response to infection. In the lung, which is the ultimate site in which virus-specific CD8+ CTL kill infected cells and clear infection, the absence of Ahr in CD11c+ cells fully reverses the defective CD8+ T cell response observed upon TCDD treatment (Fig. 7B). That is, AHR activation in CD11ccreAhrfx/fx mice did not reduce the percent or number of nucleoprotein (NP)-specific CD8+ T cells and IFNγ+CD8+ T cells, compared to vehicle treated littermates. This is in direct contrast to the difference between vehicle- and TCDD-treated Ahrfx/fx mice, in which AHR activation significantly reduced in the percentage and number of influenza NP-specific CD8+ T cells and IFNγ+CD8+ T cells in the lung. These data reveal that expression of Ahr in CD11c lineage cells plays a key role in controlling the acquisition of effector, virus-specific CD8+ T cells in the infected lung.
Figure 7.

Excision of Ahr in CD11c+ cells restores TCDD-induced decrease in virus-specific CD8+ T cells in the lung. (A) Splenic CD11c+MHCII+ cells from Ahrfx/fx and CD11cCreAhrfx/fx mice were stained with Abs and sorted (FACS Aria). Genomic DNA from sorted cells was used to validate expression of the excised and unexcised Ahrfx gene by PCR. (B and C) Ahrfx/fx and CD11cCreAhrfx/fx mice were treated and infected as described in Fig. 6. On day 9 post infection, lung-derived immune cells (B) and MLN cells (C) were stained with MHCI tetramers (Db/NP366–374) or anti-IFNγ Ab combined with anti-CD8 Ab. Doublets and dead cells were gated out (Supporting Information Fig. 4D and 4E). Numbers on each gated region of representative dot plots show the percentage of Db/NP366–374+ or IFNγ+ CD8+ T cells in the lung and MLN. Bar graphs depict the number of NP-specific CD8+ T cells and IFNγ+ CD8+ T cells in each compartment. V, indicates vehicle treatment group, T indicates TCDD treatment group. Data are shown as mean ±SEM (n=7–8/group) from one of two independent experiments with same results. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with V group of same genotype; # p < 0.05 compared with Ahrfx/fx group treated with TCDD; one-way ANOVA follow by post-hoc test (Tukey HSD).
However, AHR's role in modulating CD8+ T cell responses to infection likely involves AHR-mediated events in other immune cell lineages as well. Evidence for additional cellular players derives from the observation that conditional deletion of Ahr from CD11c lineage cells only partially modified the effect on CD8+ T cell responses in the MLN. In contrast to the lung, AHR activation in CD11ccreAhrfx/fx mice did not fully restore the reduced frequency of virus-specific CD8+ T cells and IFNγ+CD8+ T cells in the MLN (Fig. 7C). However, whereas the response of CD8+ T cells in TCDD-treated CD11ccreAhrfx/fx mice was suppressed compared to the vehicle-treated controls, the magnitude of suppression was not as severe as it is in TCDD-treated Ahrfx/fx mice. Instead, it was in between that of vehicle-treated infected mice (of either genotype), and the severely blunted response observed in TCDD-treated Ahrfx/fx mice. Thus, conditional deletion of Ahr in CD11c+ cells partially prevented repression of CD8+ T cell response in the MLN. These data suggest that in the draining lymph nodes, which are considered the principal site for the activation of naïve, virus-specific CD8+ T cells a combination of CD11c+ and other cell types contributes to the substantially reduced CD8+ T cell response to infection following AHR activation.
Discussion
The AHR modulates immune cell development and function, although details regarding precisely how it does this are only beginning to emerge. Much recent focus on AHR has centered on its control of CD4+ T cell differentiation, the development of autoimmune diseases, and immune homeostasis in the gastrointestinal (GI) tract [4, 5, 7, 17, 19, 66]. We present here another aspect of immune regulation by the AHR: DC responses during acute viral infection, and the ensuing impact on CD8+ T cell activation. During respiratory virus infection, AHR activation results in fewer mature, immunostimulatory CD103+DCs from the lung getting to the MLN to activate naïve virus-specific CD8+ T cells and generate a robust CTL response to infection. Moreover, AHR signaling in DCs directly contributes to this poorer response. This extends prior reports showing effects of AHR ligands on DCs [6, 23–25, 27–30, 67] in several ways, and suggests that when thinking about host responses to common respiratory infections, such as influenza virus, AHR-regulated pathways that influence the function of DCs are overlooked but potentially critical factors influencing disease outcome.
The study presented here shows that TCDD-induced AHR signaling altered the response of DC subsets upon infection. We found no evidence that this is explained by AHR-mediated elevation in DC apoptosis, which is in contrast to other reports in which in vitro treatment of bone marrow derived DCs with TCDD increased death [67], but is consistent with prior reports that AHR ligands perturb in vivo DC homeostasis [68]. Another possible explanation for the reduced adaptive immune response to infection is that AHR activation impairs viral replication. In other words, what appears to be an attenuated response reflects the fact that the virus is not able to replicate successfully. However, while morbidity and mortality are increased, AHR activation does not significantly change pulmonary viral load or the kinetics at which virus in cleared from the lung [21, 22, 69]. Instead, we found that AHR signaling altered the response of CD103+ and CD11b+ DC subsets upon infection, as measured by reduction in trafficking to and frequency in the lung-draining lymph nodes during infection. Given that CD103+DCs are better able to take up influenza virus, directly present antigen in MHCI, and may be better at cross-presenting antigen to naïve CD8+ T cells [33, 36, 38, 40], AHR activation may diminish the ability of CD103+DCs to prime naïve virus specific CD8+ T cells. Thus, AHR may specifically regulate genes and signaling pathways involved in these processes. Further evidence of selectivity in AHR-mediated effects on DC subsets comes from the observation that, in the absence of infection, triggering the AHR reduced only the frequency of CD103+DCs in the lung. This is interesting because in the lung CD11b+DCs are thought to reside primarily in the parenchyma, whereas CD103+DCs are predominantly found just below the airway epithelium, possibly extending processes up into airspaces [35]. Thus, AHR-mediated changes in DC function may be due in part to events that begin in the lung, and affect signaling events in CD103+DCs that are important early on after infection.
Indeed, the time after infection in which DC subsets are examined is an important consideration. We examined the function of CD11b+DCs and CD103+DCs 3 days after infection, whereas Ballesteros-Tato et. al. [32] measured DC function 7 days after infection. In that study, they found CD11b+DCs played the dominant immunostimulatory role [32]. However, if the AHR is not activated until 4 days after infection, then it no longer attenuates the response of CD8+ T cells [20], suggesting that the fundamental AHR sensitive events are prior to day 4 of infection. Thus, the contribution of CD11b+DCs during primary influenza virus infection may not overlap with pathways that are regulated by the AHR earlier on during infection. Additionally, it is possible that the principal DC subset that stimulates naïve CD8+ T cells in the draining lymph nodes changes over time.
Depending on context, DCs can act in an immunostimulatory or regulatory capacity. Immunostimulatory DCs drive naïve T cell activation and promote adaptive immunity. Regulatory DCs do not sustain T cell activation because they make factors that act directly on T cells to reduce or block activation or promote the expansion of Treg cells [50, 70]. The mechanisms that drive DCs to become `immunostimulatory' or `regulatory' are not fully understood, but involve integration of multiple signals [31, 50]. Several studies suggest that AHR influences these processes, such that upon AHR activation, regulatory DC functions dominate over stimulatory ones [6, 24–26, 28, 71]. However, the specific DC subsets and particular DC functions modified by AHR ligands remain to be fully defined. Our findings suggest that in the context of infection with influenza virus, AHR skews the profile of DC subsets such that there are fewer immunostimulatory CD103+DCs. Yet, the effects of AHR are probably not limited to simply reducing the number and function of CD103+DCs in the draining lymph nodes. AHR may simultaneously influence distinct CD11c+ cell subpopulations, albeit in a different manner. For instance, AHR ligands, including TCDD, induce Ido1 and Il10 in monocytes and DCs derived from mice and humans [6, 24–26, 53]. In this study, we found that Ido1 was more strongly induced in sorted DCs, whereas AHR-mediated elevation in Il10 expression was more pronounced in sorted CD11c+ macrophages. The idea that AHR selectively induces gene expression in distinct subsets of CD11c+ cells suggests that AHR's down-regulatory action derives from the confluence of AHR-mediated alterations in regulatory pathways in multiple cell types, including DCs and monocytes/macrophages.
The appreciation that AHR modulates immune responses not by controlling the function of a single cell type, but by affecting the integrated functions of multiple cell types is supported by findings using conditional knockouts [72]. While the reduced CTL response in the lung was fully reversed by ablating Ahr in CD11c lineage cells, suppression of CD8+ T cell activation in lymph nodes was only partially affected. In the lung, AHR-mediated events in DCs and CD11c+ macrophages could contribute to negative regulation of the response to infection. As this is teased out further, it will be important to bear in mind that AHR may differentially alter the function of these two subsets of CD11c+ cells, which could act together or separately to influence host responses to infection. In contrast, CD8+ T cell suppression in the MLN also requires AHR in other cell types. One candidate is plasmacytoid DCs (pDCs) because they contribute to antiviral host defenses, but also exhibit regulatory functions, including Ido1 and downstream production of regulatory tryptophan metabolites [73, 74]. Given that pDCs express lower levels of CD11c, it is uncertain whether loxP flanked genes are fully ablated in CD11clo cells [62, 63]; thus they may retain AHR and contribute to poorer CD8+ T cell response in the MLN in the CD11ccreAhrfx/fx mice. In the context of the conditional knockouts it is unclear whether pDCs are sufficient to explain the partial effect observed in the MLN. Another likely candidate is CD4+ Treg cells. AHR activation by several ligands, including TCDD, induces Treg cells in a variety of disease models [4, 5, 18, 66, 75]. It remains less clearly defined precisely how AHR does this; and it may use different mechanisms to enhance the number of Treg cells depending on the type of antigen, tissue localization, and even AHR ligand involved. A transient increase in Treg cells has been reported after influenza virus infection, although their role has yet to be defined [76]. Yet, they are a logical candidate population because reciprocal interactions between Treg cells and DCs have been reported. For example, Treg cells can inhibit stable contacts between T cells and DCs, and improperly matured DCs can induce Treg cells [77–80]. Moreover, during respiratory syncytial virus infection, Treg cells modulate CD8+ T cell responses and tissue inflammation [81]. Therefore, AHR may reduce the activation of naïve CD8+ T cells in the lymph nodes by reducing the response of conventional, immunostimulatory DCs, and by increasing down-regulatory immune functions, perhaps via Treg-DC cross-talk and/or Treg-CD8 interactions.
In summary, the work presented here expands our understanding of how the AHR regulates immune responses. In addition to influencing CD4 lineage cells, progression of autoimmune diseases, and GI tract homeostasis, AHR is an important and potent regulator of DC responses during acute respiratory viral infection. Whether this reflects a particularly unique role of AHR in the context of viral infections, or pertains more generally to AHR regulation of factors that control naïve CD8+ T cell activation remains to be determined. The large descriptive database demonstrating that exposure to a variety of AHR ligands profoundly affects Th1, Th2, Th17, and CD8+ T cell responses in numerous different models, which include replicating, non-replicating, pathogenic and non-pathogenic antigens, speaks to a more global pathway regulated by AHR. Moreover, when considering the mechanism by which AHR ligands modulate the immune system, it is likely that AHR controls events in more than one cell type, which act together to alter host immune responses. This concept suggests that further understanding of AHR immunobiology needs to be undertaken using in vivo model systems, such that the complex impact of AHR signaling on cell-cell interactions and cellular trafficking can become fully appreciated.
Materials and methods
Mice and treatments
C57BL/6 (B6), C57BL/10 (B10), and B6.Cg-Tg (Itgax-cre)1-1Reiz/J (CD11cCre) mice were purchased from the National Cancer Institute (Frederick, MD) or from The Jackson Laboratory (Bar Harbor, ME). Dr. Christopher Bradfield (University of Wisconsin, Madison, WI) provided breeding stock of B6 Ahr−/−, Ahrdbd/dbd and Ahrfx/fx mice, and Dr. Demetrius Moskophidis (Medical College of Georgia, Augusta, GA) and Dr. Dimitris Kioussis (National Institute for Medical Research, London, UK) provided F5 TCR transgenic mice (F5 mice). Colonies are maintained at URMC. Ahrdbd/dbd and Ahr−/− mice were backcrossed onto a B6 genetic background possessing the Ahrd allele. B6.Ahrd/d mice, maintained at URMC, were used as controls for Ahrdbd/dbd mutant and Ahr−/− mice. Total DNA from ear tissue was extracted using QIAamp DNA Mini Kit (QIAGEN, Valencia, CA), and genotyping was performed by PCR using Ahr sense and antisense primers OL941 and OL942 (Integrated DNA Technologies (IDT), San Diego, CA) as described previously [61]. Conditional knockout CD11ccreAhrfx/fx mice were generated in our laboratory by crossing male CD11ccre mice with female Ahrfx/fx mice. Genotyping was performed by PCR using ear punch DNA, as described above, with CD11c Cre transgene (5'-ACTTGGCAGCTGTCTCCAAG-3'; 5'-GCGAACATCTTCAGGTTCTG-3', IDT) and Ahrfx/fx sense and antisense primers (OL4062, OL4064 and OL4088, IDT), as described previously [82]. The F5 mice were derived using a TCR isolated from a cytotoxic T cell clone that recognizes amino acids 366-374 of the nucleoprotein (NP366-374) of influenza virus strain A/Memphis/102/72 in the context of H-2Db [83], and were maintained and phenotyped as described previously [20]. All mice were housed in pathogen-free micro-isolator cages, kept on a 12-h light/dark cycle, and provided food and water ad libitum.
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; ≥99% purity, Cambridge Isotope Laboratories, Woburn, MA) was diluted in peanut oil. For all experiments, seven to eight week old mice received a single oral treatment of TCDD by gavage one day before infection. Control mice received the peanut oil vehicle in the same manner. The LD50 of TCDD in B6 mice (Ahrb/b) is 114 μg/kg [84]. Mice on the B6 and B10 genetic background are Ahrb/b and were administered 10 μg TCDD per kg of body weight. Lineages expressing Ahrd/d encode a protein with 10-fold lower binding affinity for TCDD, and thus require a 10-fold higher dose of TCDD than the Ahrb/b allele mice in order to elicit similar effects [85]. Therefore B6.Ahrd/d, Ahrdbd/dbd, Ahrfx/fx and CD11ccreAhrfx/fx mice, which express Ahrd/d allele, were treated with 100 μg/kg of TCDD. Importantly, B6.Ahrd/d mice dosed with 100 μg/kg TCDD present the same magnitude defect in their CD8+ T cell response to influenza virus infection as B6.Ahrb/b mice given 10 μg/kg, indicating they are sensitive to this immunomodulatory effect [20, 72].
Mice were anesthetized by i.p. injection of Avertin (2,2,2-tribromoethanol; Aldrich, Milwaukee, WI) and inoculated i.n. with 25 μl sterile PBS containing 1 × 107 PFU of influenza virus strain A/Memphis/102/72 (Mem/102; H3N2). In some experiments, CFSE (Invitrogen, Grand Island, NY, 25 mM in DMSO) was diluted in sterile endotoxin-free PBS to a concentration of 8 mM. One day prior to infection, mice were anesthetized and given 50 μl of diluted CFSE (i.n.). The University of Rochester Institutional Animal Care and Use and Institutional Biosafety Committees reviewed and approved all procedures involving laboratory animals and infectious agents. The University of Rochester is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), and handling of vertebrate animals is conducted following guidelines set for by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Collection and preparation of immune cells
To obtain lung-derived immune cells, pulmonary vessels were perfused with 5 ml of 0.6 mM EDTA/PBS, and lungs were digested with collagenase, as previously described [86]. Single cell suspensions of MLN or spleen were prepared as previously described [12]. Erythrocytes were removed using an ammonium chloride lysing solution, and then cell suspensions were washed, passed through a cell strainer, and kept on ice until labeling, further cell isolation, or use in an assay. The number of viable cells in each sample was determined using TC10 Automated cell Counter (Bio-Rad, Hercules, CA).
Flow cytometry
Cells were incubated with anti-mouse CD16/CD32, and stained with previously determined optimal concentrations of the following fluorochrome-conjugated mAbs: CD3 (145-2C11), CD8 (53.67), CD11b (M1/70), CD11c (N418), CD40 (1C10), CD44 (IM7), CD45.2 (104), CD62L (MEL-14), CD103 (M290), IFNγ (XMG1.2), MHC class II (M5/114.15.2), and Vβ11 (RR3–15); purchased from eBioscience, BD Biosciences or BioLegend. Virus-specific CD8+ T cells were further identified using MHC class I tetramers corresponding to a major Mem/102 influenza A virus epitope (Db/NP366-374), as described [15]. Intracellular IFNγ staining was performed as described previously [86]. To measure apoptosis, Annexin V (eBioscience) and Live/Dead Fixable Dead Cell Stain kit (eBioscience) were used, as per the manufacturers' recommendations. Data acquisition was performed using LSR-II cytometers (BD Biosciences). A combination of fluorescence minus one (FMO) controls and isotype-matched fluorochrome-labeled antibodies were used to define gating parameters. Doublet discrimination and viable cell gating was used according to gating strategies presented in Supporting Information Fig. 4. Data analyses were performed using FlowJo software (Tree Star, Ashland, OR).
Cellular isolation and sorting
Naïve F5 CD8+ T cells were negatively isolated from spleens of 7–8 week old untreated F5 mice using a MagCellect Mouse Naïve CD8 T Cell Isolation Kit (R&D System, Minneapolis, MD). The purity of naïve F5 CD8+ T cells (CD8+Vβ11+CD44loCD62Lhi) was >95%. Cells from lung or MLN of infected mice (±AHR activation) were pooled and CD11c+ cells were enriched using Mouse CD11c Microbeads Kit (Miltenyi Biotec, Auburn, CA). Enriched cells were stained with fluorochrome-conjugated mAbs for sorting (FACS Aria). All sorted DCs were resuspended in RPMI1640 media containing 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM nonessential amino acids, 50 μM 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Ex vivo DC function assay
MLN DC subsets from either vehicle control or TCDD treated infected mice were isolated as described above, and used to stimulate naïve F5 CD8+ T cells ex vivo [20]. Briefly, isolated naïve F5 CD8+ T cells labeled with 2 μM CFSE were co-cultured in 96-well plates with serially diluted, sorted DC subpopulations. After 3 days in culture, cells were collected and stained with antibodies to CD3, CD8, and CD44 for flow cytometric analysis. For data analysis, CD11c+ and MHCII+ cells were excluded, and F5 cells were identified as CD3+CD8+ cells. Activated F5 CD8+ T cells were defined based on up-regulation of CD44 and loss of CFSE staining (CFSEdecayCD44hi). Culture supernatants were collected and the concentration of IFNγ was measured using an ELISA (BD Biosciences).
Real-time PCR analysis
Total RNA was isolated from sorted lung CD11c+Macs, DCs, CD11b+DCs, CD103+DCs, and liver tissue using RNeasy Mini Kit (QIAGEN), and quantified using a NanoDrop (Thermo Scientific, Wilmington, DE). RT was performed using SuperScript II (Invitrogen). For gene-specific amplification, the following primers were used: mouse Cyp1a1 (5'TTTGGAGCTGGGTTTGACAC3', 5'CTGCCAATCACTGTGTCTA3'; IDT), Il-10 primer-probe mixture (Applied Biosystems), Ido1 primer-probe set (QIAGEN) or Ahr primers (QIAGEN). Real time-RTPCR was performed using a Bio-Rad iCycler MyiQ2 with IQ SYBR Green Supermix (Bio-Rad) or primer-specific probes. L13 was used as and internal control gene (5'CTACAGTGAGATACCACACCAAG3'; 5'TGGACTTGTTTCGCCTCCTC3', IDT). Changes in the expression of a particular gene, compared to the same (sorted) cell type from vehicle-treated mice and normalized to the housekeeping gene L13, were calculated using the 2−ΔΔCT method [87].
Statistical analyses
Statistical analyses were performed using JMP 9.0.0 (SAS Institute, Cary, NC) or Prism 6 (GraphPad Software, La Jolla, CA). Differences between means of multiple independent variables were compared between each genotype, time, and/or treatment group using one-way ANOVA followed by post-hoc tests (Tukey HSD). Differences between vehicle and TCDD treatment groups within the same genotype and at a single point in time were analyzed using a Student's t-test. Differences in mean values were considered significant when p < 0.05, with p values indicated in legends for significant differences.
Supplementary Material
Acknowledgements
We gratefully acknowledge the generosity of Dr. Christopher Bradfield for his willingness to share the unique mouse strains created by his research group, and Drs. Deborah Fowell, Michael R. Elliott, and Nancy Kerkvliet for thoughtful discussion of our data and helpful comments during the preparation of this manuscript. We also thank Dr. Timothy P. Bushnell and the Flow Cytometry Core at the University of Rochester for their assistance with cell sorting. The following research, training, and center grants from the US National Institutes of Health supported this work: RC2-ES018750, R01-ES017250, R01-HL097141, T32-ES07026, and P30-ES01247.
Abbreviations
- AHR
Aryl hydrocarbon receptor
- AhRE
aryl hydrocarbon response element
- cyp1a1
cytochrome P4501A1
- DBD
DNA binding domain
- MLN
mediastinal lymph nodes
- NP
nucleoprotein
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Denison M, Nagy S. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Phramacol. Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence BP, Kerkvliet NI. Immune modulation by TCDD and related polyhalogenated aromatic hydrocarbones. In: Luebke R, House R, Kimber I, editors. Immunotoxicology and Immumopharmacology. 3 Edn CRC Press, Taylor & Francis Group; Boca Raton, FL: 2006. pp. 239–258. [Google Scholar]
- 4.Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, Pham D, Mourich D. Activaiton of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–547. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 6.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah A-M, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veldhoen M, Hirota K, Christensen JL, O'Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp Med. 2008;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawrence BP, Denison M, Novak H, Vorderstrasse BA, Harrer N, Neruda W, Reichel C, Woisetschlager M. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low molecular weight compound. Blood. 2008;112:1158–1165. doi: 10.1182/blood-2007-08-109645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takamura T, Harama D, Fukumoto S, Nakamura Y, Shimokawa N, Ishimaru K, Ikegami S, Makino S, Kitamura M, Nakao M. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol Cell Biol. 2011 doi: 10.1038/icb.2010.165. doi:10.1038/icb.2010.165: 10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson J, Shepherd DM. Aryl hydrocarbon receptor activaiton by TCDD reduces inflammation associated with Crohn's disease. Toxicol Sci. 2011;120:68–78. doi: 10.1093/toxsci/kfq360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chmill S, Kadow S, Winter M, Weighardt H, Esser C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin impairs stable establishment of oral tolerance in mice. Toxicol Sci. 2010;118 doi: 10.1093/toxsci/kfq232. [DOI] [PubMed] [Google Scholar]
- 12.Warren TK, Mitchell KA, Lawrence BP. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin suppresses the cell-mediated and humoral immune response to influenza A virus without affecting cytolytic activity in the lung. Toxicol Sci. 2000;56:114–123. doi: 10.1093/toxsci/56.1.114. [DOI] [PubMed] [Google Scholar]
- 13.Teske S, Bohn AA, Regal JF, Neumiller JJ, Lawrence BP. Exploring mechanisms that underlie aryl hydrocarbon receptor-mediated increases in pulmonary neutrophilia and diminished host resistance to influenza A virus. Am J Physiol Lung Cell Mol Physiol. 2005;289:111–124. doi: 10.1152/ajplung.00318.2004. [DOI] [PubMed] [Google Scholar]
- 14.Vorderstrasse BA, Lawrence BP. Protection against lethal challenge with S. pneumoniae is conferred by aryl hydrocarbon receptor activation, but is not associated with enhanced inflammation. Infect Immun. 2006;74:5679–5686. doi: 10.1128/IAI.00837-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence BP, Roberts AD, Neumiller JJ, Cundiff JA, Woodland DL. Aryl hydrocarbon receptor activation impairs the priming but not the recall of influenza virus-specific CD8+ T cells in the lung. J. Immunol. 2006;177:5819–5858. doi: 10.4049/jimmunol.177.9.5819. [DOI] [PubMed] [Google Scholar]
- 16.Shi LZ, Faith NG, Nakayama Y, Suresh M, Steinberg H, Czupyrynski C. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J. Immunol. 2007;179:6952–6962. doi: 10.4049/jimmunol.179.10.6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiss E, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Sciencexpress. 2011 doi: 10.1126/science.1214914. 10.1126.science.1214914. [DOI] [PubMed] [Google Scholar]
- 18.Veiga-Parga T, Suryawanshi A, Rouse Controlling viral immuno-inflammatory lesions by modulating aryl hydrocarbon receptor signaling. PLoS Pathog. 2011 Dec;7(12):e1002427. doi: 10.1371/journal.ppat.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Cella M, McDonald K, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry R, Colonna M. AHR drives the development of ILC22 and post-natal lymphoid tissues through Notch-dependent and independent pathways. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin GB, Moore AJ, Head JL, Neumiller JJ, Lawrence BP. Aryl hydrocarbon receptor activation reduces dendritic cell function during influenza virus infection. Toxicol Sci. 2010;116:514–522. doi: 10.1093/toxsci/kfq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burleson G, Lebrec H, Yang Y, Ibanes J, Pennington K, Birnbaum L. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on influenza virus host resistance in mice. Fund. App. Toxicol. 1996;29:40–47. doi: 10.1006/faat.1996.0004. [DOI] [PubMed] [Google Scholar]
- 22.Luebke R, Copeland C, Bishop L, Daniels M, Gilmour M. Mortality in dioxin-exposed mice infected with influenza: Mitochondrial toxicity (Reye's like syndrome) versus enhanced inflammation as the mode of action. Toxicol. Sci. 2002;69:109–116. doi: 10.1093/toxsci/69.1.109. [DOI] [PubMed] [Google Scholar]
- 23.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, Roncarolo MG. Activation of the aryl hydrocarbon receptor promotes allograft specific tolerance through direct- and DC-mediated effects on regulatory T cells. Blood. 2008;112:1214–1222. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 24.Simones T, Shepherd DM. Consequences of AhR activation in steady-state dendritic cells. Toxicol Sci. 2011;119:293–307. doi: 10.1093/toxsci/kfq354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol Appl Pharmacol. 2010;246:18–28. doi: 10.1016/j.taap.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction betweeen kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Hwang J-A, Sung H-N, Jeon C-H, Gill B-C, Youn H-J, Park J-H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin modulates functional differentiation of mouse bone marrow derived dendritic cells: Downregulation of RelB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Tox. Letters. 2007;173:31–40. doi: 10.1016/j.toxlet.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Singh N, Nagarkatti M, Nagarkatti P. Primary peripheral T cells become susceptible to 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated apoptosis in vitro upon activation and in the presence of dendritic cells. Mol. Pharmacol. 2008;73:1722–1735. doi: 10.1124/mol.107.043406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschlager M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J. Immunol. 2009;183:66–74. doi: 10.4049/jimmunol.0802997. [DOI] [PubMed] [Google Scholar]
- 30.Vorderstrasse B, Kerkvliet N. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine dendritic cells and their expression of accessory molecules. Toxicol. Appl. Pharmacol. 2001;171:117–125. doi: 10.1006/taap.2000.9119. [DOI] [PubMed] [Google Scholar]
- 31.Ueno H, Klechevsky E, Morita R, Aspord C, Cao T, Matsui T, Di Pucchio T, Connolly J, Fay JW, Pascual V, Palucka AK, Banchereau J. Dendritic cell subsets in health and disease. Immunol. Rev. 2007;219:118–142. doi: 10.1111/j.1600-065X.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 32.Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ T cell responses to influenza. Nat Immunol. 2010;11:216–224. doi: 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao X, Kim TS, Braciale TJ. Differential response of respiratory DC subsets to influenza virus infection. J. Virol. 2008;82:4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vermaelen K, Carro-Muino I, Lambrecht B, Pauwels R. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 2001;193:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung SS, Fu SM, Rose CE, Gaskin F, Ju ST, Beaty SR. A major lung CD103 (alphE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 36.Ho AWS, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, Lew FC, Wong KL, Hanson BJ, Macary PA, Kemeny DM. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHCI for presentation to CD8+ T cells. J. Immunol. 2011;187:6011–6021. doi: 10.4049/jimmunol.1100987. [DOI] [PubMed] [Google Scholar]
- 37.Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown B, Schmolke M, Miller J, Leboeuf M, Murphy K, Garcia-Sastre A, Merad M. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin Invest. 2012;122:4037–4047. doi: 10.1172/JCI60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.del Rio M-L, Rodriquez-Barbosa J-I, Kremmer E, Forster R. CD103− and CD103+ brochial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ an CD8+ T cells. J. Immunol. 2007;178:6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 39.McGill J, Van Rooijen N, Legge KL. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lung. J. Exp Med. 2008;205:1635–1646. doi: 10.1084/jem.20080314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukens M, Kruijsen D, Coenjaerts F, Kimpen J, van Bleek G. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J. Virol. 2009;83:7235–7243. doi: 10.1128/JVI.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim T, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One. 2009;4:e4204. doi: 10.1371/journal.pone.0004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beauchamp NM, Busick RY, Alexander-Miller M. Functional divergence among CD103+ dendritic cell subpopulations following pulmonary poxvirus infection. J. Virol. 2010;84:10191–10199. doi: 10.1128/JVI.00892-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kandasamy M, Ying P, Ho A, Sumatoh H, Schlitzer A, Hughes T, Kemeny D, Morgan B, Ginhoux F, Sivasankar B. Complement mediated signaling on pulmonary CD103+ dendritic cells is critical for their migratory function in response to influenza infection. PLoS Pathog. 2013 Jan;9(1):e1003115. doi: 10.1371/journal.ppat.1003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schecter A, Cramer P, Boggess K, Stanley J, Papke O, Olson J, Silver A, Schmitz M. Intake of dioxins and related compounds from food in the US population. J. Toxicol. Environ. Health, Part A. 2001;63:1–18. doi: 10.1080/152873901750128326. [DOI] [PubMed] [Google Scholar]
- 45.Stolevik S, Nygaard U, Namork E, Haugen M, Kvalem H, Meltzer H, Alexander J, van Delft J, Van Loveren H, Lovik M, Granum B. Prenatal exposure to polychlorinated biphenyls and dioxins is associated iwth increased risk of wheeze and infections in infants. Food Chem Toxicol. 2011;49:1843–1848. doi: 10.1016/j.fct.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Dallaire F, Dewailly E, Muckle G, Vizena C, Jacobson S, Jacobson J, Ayotte P. Acute infections and environmental exposure to organochlorines in Inuit infants from Nunavik. Environ Health Perspect. 2004;112:1359–1364. doi: 10.1289/ehp.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler J, Martin K, Resseguie E, Lawrence BP. Differential consequences of two distinct AhR ligands on innate and adaptive immune responses to influenza A virus. Toxicolog. Sci. 2013 doi: 10.1093/toxsci/kft255. doi: 10.1093/toxsci/kft255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol. 2012;189:946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 49.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 50.Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: The signal integration model of dendritic cell activation. Trends Immunol. 2007;28:227–233. doi: 10.1016/j.it.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Legge K, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 52.Hopken U, Lehmann I, Droese J, Lipp M, Schuler T, Rehm A. The ratio between dendritic cells and T cells determines the outcome of their encounter: proliferation versus deletion. Eur J. Immunol. 2005;35:2851–2863. doi: 10.1002/eji.200526298. [DOI] [PubMed] [Google Scholar]
- 53.Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Comm. 2008;375:331–335. doi: 10.1016/j.bbrc.2008.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawrence BP, Vorderstrasse BA. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin Immunopathol. 2013;35:615–626. doi: 10.1007/s00281-013-0395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh KP, Casado FL, Opanashuk LA, Gasiewicz TA. The aryl hydrocarbon receptor has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations. Biochem Pharmacol. 2009;77:577–587. doi: 10.1016/j.bcp.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimura A, Naka T, Nohara K, Jujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105 doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadow S, Jux B, Zahner S, Wingerath B, Chmill S, Clausen BE, Hengstler J, Esser C. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cell in the murine epidermis. J. Immunol. 2011;187:3104–3110. doi: 10.4049/jimmunol.1100912. [DOI] [PubMed] [Google Scholar]
- 58.Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. Rel B, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–2955. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baglole CJ, Maggirwar S, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J. Biol. Chem. 2008;283:28944–28957. doi: 10.1074/jbc.M800685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanos R, Patel R, Murray I, Smith P, Patterson A, Perdew GH. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology. 2012;55:1994–2004. doi: 10.1002/hep.25571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bunger M, Glover E, Moran SM, Walisser J, Lahvis G, Hsu EL, Bradfield CA. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA binding domain of the aryl hydrocarbon receptor. Toxicol Sci. 2008;106:83–92. doi: 10.1093/toxsci/kfn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caton M, Smith-Raska M, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J. Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Melillo J, Song L, Bhagat G, Blazquez A, Plumlee C, Lee C, Berin C, Reizis B, Schindler C. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol. 2010;184:2638–2645. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kudo M, Melton A, Chen C, Engler M, Huang K, Ren X, Wang Y, Bernstein X, Li J, Atabai K, Huang X, Sheppard D. IL-17A produced by abT cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Medicine. 2012;18:547–555. doi: 10.1038/nm.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walisser J, Glover E, Pande K, Liss A, Bradfield C. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci U S A. 2005;102:17858–17863. doi: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Funatake C, Marshall N, Steppan LB, Mourich D, Kerkvliet NI. Cutting Edge: Activation of the aryl hydrocarbon receptor (AhR) by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) generates a population of CD4+CD25+ cells with characteristics of regulatory T cells. J. Immunol. 2005;175:4184–4188. doi: 10.4049/jimmunol.175.7.4184. [DOI] [PubMed] [Google Scholar]
- 67.Ruby C, Funatake C, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) directly enhances the maturation and apoptosis of dendritic cells in vitro. J. Immunotox. 2004;1:159–166. doi: 10.1080/15476910490920968. [DOI] [PubMed] [Google Scholar]
- 68.Bankoti J, Burnett A, Navarro S, Miller A, Rase B, Shepherd DM. Effects of TCDD on the fate of naive dendritic cells. Toxicolog. Sci. 2010;115 doi: 10.1093/toxsci/kfq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neff-LaFord HD, Vorderstrasse BA, Lawrence BP. Fewer CTL, not enhanced NK cells, are sufficient for viral clearance from the lungs of immunocompromised mice. Cell. Immunol. 2003;226:54–64. doi: 10.1016/j.cellimm.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 2007;7:610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 71.Nguyen N, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:19961–19966. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wheeler J, Martin K, Lawrence BP. Novel cellular targets of AhR underlie alterations in neutrophilic inflammation and iNOS expression during influenza virus infection. J. Immunol. 2013;190:659–668. doi: 10.4049/jimmunol.1201341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson B, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3-dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boasso A, Royle C, Doumazos S, Aquino V, Baisin M, Piacentini L, Tavano B, Fuchs D, Mazzotta F, Lo Caputo S, Shearer G, Clerici G, Clerici M, Graham D. Overactivation of plasmacytoid dendritic cells inhibits antiviral T-cell responses: a model for HIV immunopathogenesis. Blood. 2011;118:5152–5162. doi: 10.1182/blood-2011-03-344218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schulz V, Smit JJ, Willemsen K, Fletcher D, Hassing I, Bleumink R, Boon L, van den Berg M, van Duursen M, Pieters R. Activation of the aryl hydrocarbon receptor suppresses sensitization in a mouse peanut allergy model. Toxicol Sci. 2011;123:491–500. doi: 10.1093/toxsci/kfr175. [DOI] [PubMed] [Google Scholar]
- 76.Betts RJ, Prabhu N, Ho AWS, Hutchinson PE, Rotzschke O, Macary PA, Kemeny DM. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J. Virol. 2012;86:2817–2825. doi: 10.1128/JVI.05685-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krathwohl M, Schacker T, Anderson J. Abnormal presence of semimature dendritic cells that induce regulatory T cells in HIV-infected subjects. J. Infect. Dis. 2006;193:494–504. doi: 10.1086/499597. [DOI] [PubMed] [Google Scholar]
- 78.Tadokoro C, Shakhar G, Shen S, Ding Y, Lino A, Maraver A, Lafaille J, Dustin M. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med. 2006;203:505–511. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Darrasse-Jeze G, Seroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao K, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp Med. 2009;206:1853–1862. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Catani L, Sollazzo D, Trabanelli S, Curti A, Evangelisti C, Polveraelli N, Palandri F, Baccarani M, Vianelli N, Lemoli R. Decreased expression of indoleamine 2,3-dioxygenase 1 in dendritic cells contributes to impaired regulatory T cell development in immune thrombocytopenia. Ann Hematol. 2012 doi: 10.1007/s00277-012-1556-5. doi:10.1007/s00277-012-1556-5. [DOI] [PubMed] [Google Scholar]
- 81.Fulton RB, Meyerholz DK, Varga S. FoxP3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J. Immunol. 2010;185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walisser J, Bunger M, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A. 2004;101:16677–16682. doi: 10.1073/pnas.0404379101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mamalaki C, Elliott J, NOrton T, Yannoutsos N, Townsend A, Chandler P, Simpson E, Kioussis D. Positive and negative selection in transgenic mice expression a T cell receptor specific for influenza nucleoprotein and endogenous superantigen. Develop. Immunol. 1993;3:159–174. doi: 10.1155/1993/98015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vos J, Moore J. Suppression of cellular immunity in rats and mice by maternal treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Int. Arch. Allergy, Appl. Immunol. 1974;47:777–794. doi: 10.1159/000231268. [DOI] [PubMed] [Google Scholar]
- 85.Poland A, Palen D, Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- 86.Neff-LaFord HD, Teske S, Bushnell TP, Lawrence BP. Aryl hydrocarbon receptor activation during influenza virus infection unveils a novel pathway of IFN© production by phagocytic cells. J. Immunol. 2007;179:247–255. doi: 10.4049/jimmunol.179.1.247. [DOI] [PubMed] [Google Scholar]
- 87.Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.