

Abstract
Hematopoietic stem cell transplantation (HSCT) remains the only curative option for most patients with juvenile myelomonocytic leukemia (JMML). However, persistent disease and relapse rates after transplant range from 26% to 58%. We report the successful use of second HSCT after preparation with mitoxantrone and cytosine arabinoside (Ara-C) for patients with refractory or recurrent disease. Between 1993 and 2006, 5 children who underwent HSCT at our institution as initial therapy for JMML had persistent disease or relapsed. Pre-HSCT conditioning varied and donors were either HLA-matched siblings (n=2) or matched unrelated donors (n=3). After initial HSCT, they subsequently received high-dose Ara-C (3 g/m2 IV) every 12 hours on days −8 through −3 and mitoxantrone (10 mg/m2/d IV) on days −8, −7, −6 followed by second HSCT from their original donors. All 5 patients are alive at 88, 179, 199, 234, and 246 months with no evidence of JMML, no significant toxicity, and 100% donor chimera as determined by PCR short-tandem repeat analysis. Our experience supports second transplant utilizing high-dose Ara-C and mitoxantrone in children with JMML who do not respond or relapse after first transplant.
Key Words: JMML, chimerism, allogeneic transplant
Juvenile myelomonocytic leukemia (JMML) is a rare mixed myelodysplastic and myeloproliferative clonal disorder of early childhood representing about 1% to 2% of all pediatric leukemias.1–3 Most cases present before 6 years of age, with a median age of 2 years. Clinical presentation includes pallor, rash, hepatomegaly, splenomegaly, lymphadenopathy, and recurrent infections.2–5 JMML has an increased incidence in patients with neurofibromatosis and Noonan syndrome as well as genetic mutations in the RAS signaling pathway, PTPN11, CBL, and NF1 gene.6–10 Although most children demonstrate a normal karyotype, monosomy 7 and other chromosome 7 abnormalities are reported in 25% of cases.4
Diagnostic criteria as defined by the International JMML Working Group include peripheral blood monocytosis >1000/mm3, absence of the Philadelphia chromosome t(9:22) and the BCR-ABL1 fusion gene rearrangement, and bone marrow blasts <20%.11 Other findings include leukocytosis, anemia, thrombocytopenia, increased circulating hematopoietic precursors, hypersensitivity to GM-CSF in culture, and elevated fetal hemoglobin.11–13
The only curative option for most patients with JMML is the use of hematopoietic stem cell transplantation (HSCT), with the exception of some patients with a Noonan or Noonan-like phenotype or a germline mutation in the protein tyrosine phosphatase, nonreceptor type 11 (PTPN11), or the E3 ubiquitin ligase CBL who may have a self-limited course.10 Event-free survival is reported at 55% at 5 years and 40% at 10 years.11 Relapse-related and transplant-related mortality are the major reasons for therapeutic failure, with relapse occurring in approximately 35% of patients.13–16 Although second transplant for other recurrent malignancies has poor outcome, the only reported effective salvage therapy after recurrence of JMML is a second HSCT. A recent review confirms this assessment, while reinforcing the importance of decreasing the intensity of graft-versus-host disease (GVHD) prophylaxis with the second HSCT to optimize the graft-versus-leukemic (GVL) effect.17
We present 5 children diagnosed and treated for JMML at our institution between 1993 and 2006, all of whom had mixed chimerism after initial HSCT and either had persistent disease, evidence of progression, or relapse. All 5 patients received a second HSCT from the same HLA-matched sibling donor (2 patients) or the same HLA-matched unrelated donor (3 patients) after high-dose cytosine arabinoside (Ara-C) and mitoxantrone and all remain disease free with 100% donor chimerism.
PATIENTS AND METHODS
Between 1993 and 2006, 5 children with JMML received a bone marrow HSCT at The Nebraska Medical Center. Patient data at the time of diagnosis and before transplant are shown in Table 1. All patients underwent splenectomy and received cis-retinoic acid alone (n=2) or in combination with hydroxyurea (n=2), or Ara-C and fludarabine (n=1). All 5 patients demonstrated progressive disease despite these interventions. The median time from diagnosis to initial HSCT was 4.5 months (range, 4 to 7 mo). The median age at first HSCT was 13.5 months (range, 8 to 17 mo). Pretransplant conditioning regimens, donor type, and GVHD prophylaxis are summarized in Table 2. Two patients were transplanted with bone marrow from HLA-matched sibling donors, whereas the remaining 3 had bone marrow from matched unrelated donors. Preparative regimens for the first HSCT included VP-16, Cytoxan, and total body irradiation (1200 cGy) in 2 patients. Two patients received Busulfan, ATG, VP-16, and Cytoxan, whereas 1 patient received Cytoxan, ATG, and total body irradiation (1200 cGy). GVHD prophylaxis after first HSCT included Cyclosporin and Methotrexate in 4 patients and Cyclosporine alone in 1 patient. Supportive care after HCST included prophylaxis with diflucan, trimethoprim-sulfa, and acyclovir.
TABLE 1.
Patient Clinical and Laboratory Features at Time of Diagnosis

TABLE 2.
Patient Clinical Features at Time of Hematopoietic Stem Cell Transplant
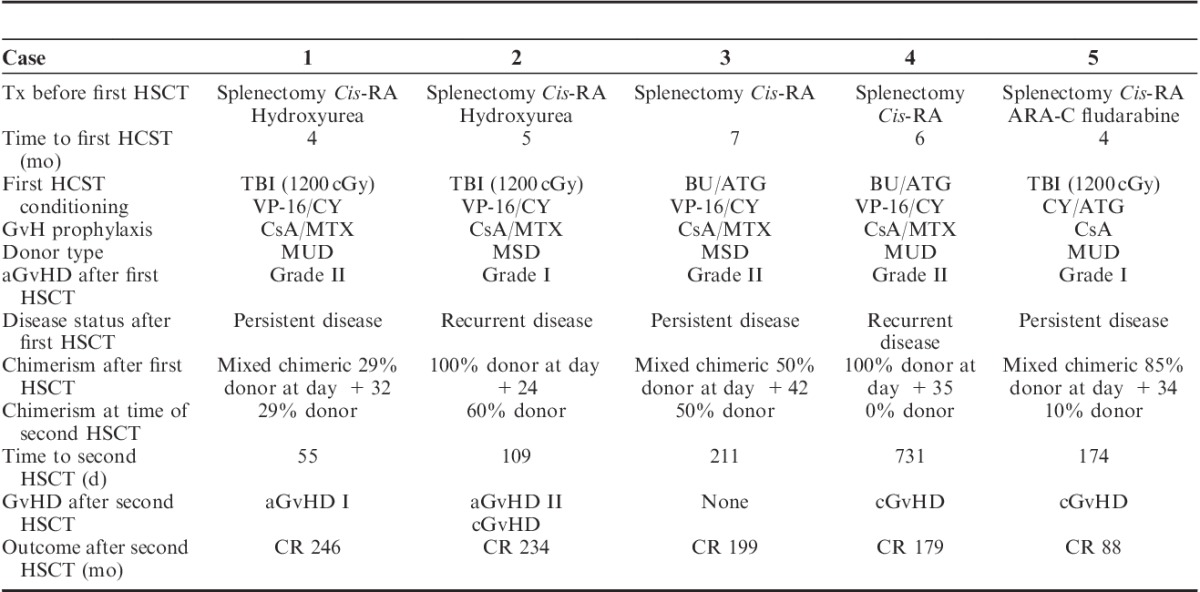
All 5 patients were engrafted with a median time to ANC>500 cells/μL for 3 consecutive days of 18 days. Acute GVHD after first HSCT was mild (grade I to II) in all patients, and was successfully managed with a short course of corticosteroids. Greater than 25% recipient chimerism was demonstrated in all patients before second SCT. Once mixed chimerism was detected, cyclosporine and methotrexate were discontinued in all patients. All patients also had evidence of persistent or recurrent disease verified by bone marrow aspiration completed between days +35 to +185.
Indications for second transplant varied among the 5 patients in addition to all demonstrating decreased donor chimerism after initial transplant. Chimerism studies obtained before and after second HSCT are detailed in Table 2. Time to second transplant for all patients ranged from 55 to 731 days. All 5 patients were prepared with Ara-C (3000 mg/m2 IV) every 12 hours on days −8 through −3, and mitoxantrone (10 mg/m2/d IV) on days −8, −7, −6 followed by 2 days of rest and second bone marrow HSCT from their original donors. No GVHD prophylaxis was given after the second transplant.
Cytogenetics and Chimeric Status
Standard G-banded analysis was performed on direct and 24-hour unstimulated bone marrow aspirates. The methods of chromosome preparation for cytogenetic analysis are described elsewhere.18,19 Peripheral blood specimens were obtained periodically posttransplant for studies of hematopoietic chimerism of either white blood cells or separated CD3(+) cells. Specimens were analyzed by polymerase chain reaction amplification for polymorphic markers (AmpFISTR Profiler PCR Amplification Kit; Applied Biosystems, Foster City, CA) composed of short-tandem repeats. Nine short-tandem repeat markers were used, including D3S1358, vWA, FGA, TH01, TPOX, CSF1PO, D5S818, D13S317, D7S820, and the sex marker Amelogenin.20
RESULTS
After second HSCT, median time to ANC>500 cells/μL for 3 consecutive days was 12 days (range, 9 to 28 d), last red cell transfusion was 45 days (range, 13 to 95 d), and last platelet transfusion was 63 days (range, 9 to 122 d). Two patients had mild acute GVHD (grade I to II), 3 patients developed chronic GVHD, and 1 patient had no evidence of GVHD after second HSCT. All 5 patients are alive and disease free at 88, 179, 199, 234, and 246 months after second HSCT with continued 100% donor chimerism (Table 2). The preparative regimen was well tolerated with no or minimal organ dysfunction and no significant infectious complications. All 5 patients have returned for follow-up within the last 12 months, and all are well with no evidence of cGVHD. Four of 5 patients demonstrate Karnofsky scores of 100% and 1 child (case 4) has a score of 80% secondary to persistent pulmonary hypertension that developed soon after first transplant and delayed the second transplant.
DISCUSSION
HSCT is a life-saving treatment modality for JMML. However, persistent disease and relapse rates after transplant range from 26% to 58%, along with significant transplant-related morbidity and mortality.11,21 Optimal pre-HSCT chemotherapy and preparative regimens are varied11,21,22 and experience with second HSCT is limited with 50% 3-year and 32% 5-year survival reported.10,16
Other investigators have suggested either the withdrawal of immunosuppressive therapy or donor lymphocyte infusion (DLI) as possible treatment modalities, given the lower relapse rates in patients who develop GVHD.23 However, in an evaluation of 21 patients with mixed chimerism after initial HSCT for JMML, only 1 patient was alive and in remission for >1500 days after receiving DLI.24 Moreover, all 21 patients in the study with mixed chimerism experienced a hematological relapse, further clarifying mixed chimerism status as a risk factor.24 A recent review of 16 patients with JMML who received HSCT by Inagaki et al25 confirmed these results, given that only 1 of 5 patients who received DLI achieved a longstanding remission.
For this reason, second HSCT is the treatment of choice at our institution for patients with increasing mixed chimerism and frank evidence of relapsed or progressive JMML. Previous reports have highlighted the importance of GVL effect in the successful control of JMML.26 We reasoned that if we could reduce the malignant cell burden by using myeolablative chemotherapy, GVL would be augmented. Given the efficacy of high-dose Ara-C and mitoxantrone for reinduction of relapsed myeloid leukemias we chose these agents as our cytoreductive regimen. As this regimen causes profound and prolonged myelosuppression, we provided a second stem cell infusion using the same donor in an effort to achieve earlier recovery of counts. In an effort to promote the GVL effect, we chose not to use GVHD prophylaxis after the second transplant. We felt this was reasonable as all patients only had mild, self-limited aGVHD after first HSCT from the same donor. We theorize that the elimination of GVHD prophylaxis after second transplant promoted a more robust GVL effect, thus eradicating residual disease.21 The omission of GVHD prophylaxis did not result in increased transplant morbidity, and only 2 of 5 patients developed grade I and II aGVHD and 3 of 5 mild self-limited chronic GVHD (Table 2). Furthermore, Ara-C and mitoxantrone were well tolerated by all 5 patients with minimal nonhematopoietic toxicity.
Considering that up to 35% to 58% of JMML patients who survive first transplant are reported to have persistent disease or relapse, second HSCT are frequently performed.11,25 In our 5 patients we were able to reharvest sibling donors for 2 patients (case 2 and 3) and harvested sufficient cells for 2 transplants from 3 unrelated donors. In our first case, a large collection fortuitously allowed us to store the additional harvested cells. In cases 4 and 5, we requested larger collections and stored enough cells for a second transplant. This was easily facilitated with small recipients and adult unrelated marrow donors. The potential need for a second transplant argues against utilizing unrelated cord blood donors and for obtaining enough stem cells for the 2 transplants when initially harvesting unrelated donors. A recent analysis by Locatelli and colleagues verifies this strategy, as they retrospectively analyzed 110 patients with JMML who received unrelated donor umbilical cord blood transplantation. Thirty-seven children (33%) relapsed at a median time of 2.6 months after transplant, and 28 of them died due to disease progression after a median of 6 months after umbilical cord blood transplantation.27
The recent report by Inagaki et al25 and the accompanying highlight by Locatelli and Lucarelli17 clarify some of the issues raised in the current report. It is clear that JMML responds differently to DLI and second HSCT than either acute leukemias or Philadelphia Chromosome positive (Ph+) CML. As in JMML, DLI infrequently results in eradication of the leukemic clone in recurrent acute myeloid or lymphoid leukemia and only in cases with minimal leukemic burden. DLI is efficacious in many cases of relapse in Ph+ CML.17 Second HSCT is rarely successful in either recurrent acute leukemias or Ph+ CML, but efficacious in JMML.
In conclusion, our findings support the strategy of second HSCT with the same donor combined with reduced or no GVHD prophylaxis to optimize the GVL effect in patients with persistent or recurrent JMML. Our experience further suggests utilizing a relatively nontoxic regimen of high-dose Ara-C and mitoxantrone as the preparative regimen in children with JMML.
Footnotes
All authors participated in patient care. S.A.P. collected patient data and wrote the manuscript. D.W.C. wrote the manuscript. A.C.G., B.G.G., J.L.H., P.I.W., and J.L.W. reviewed and approved the manuscript. W.G.S. performed cytogenetic analysis and reviewed and approved the manuscript. P.F.C. wrote, critically reviewed, and approved the manuscript.
The authors declare no conflict of interest.
REFERENCES
- 1.Gassas A, Doyle J, Dror Y, et al. Basic classification and a comprehensive examination of pediatric myeloproliferative syndromes.J Pediatr Hematol Oncol. 2005;27:192–196 [DOI] [PubMed] [Google Scholar]
- 2.Emanuel PD.Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia.Leukemia. 2008;22:1335–1342 [DOI] [PubMed] [Google Scholar]
- 3.Niemeyer CM, Kratz CP.Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options.Br J Haematol. 2008;140:610–624 [DOI] [PubMed] [Google Scholar]
- 4.Niemeyer CM, Locatelli F.Chronic Myeloproliferative Disorders. Childhood Luekemias. 2006:2nd edNew York:Cambridge University Press;571–598 [Google Scholar]
- 5.Arico M, Biondi A, Pui CH.Juvenile myelomonocytic leukemia.Blood. 1997;90:479–488 [PubMed] [Google Scholar]
- 6.Side LE, Emanuel PD, Taylor B, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1.Blood. 1998;92:267–272 [PubMed] [Google Scholar]
- 7.Bentires-Alj M, Paez JG, David FS, et al. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia.Cancer Res. 2004;64:8816–8820 [DOI] [PubMed] [Google Scholar]
- 8.Sheng XM, Kawamura M, Ohnishi H, et al. Mutations of the RAS genes in childhood acute myeloid leukemia, myelodysplastic syndrome and juvenile chronic myelocytic leukemia.Leuk Res. 1997;21:697–701 [DOI] [PubMed] [Google Scholar]
- 9.Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome.Nat Genet. 2006;38:331–336 [DOI] [PubMed] [Google Scholar]
- 10.Loh ML.Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukemia.Br J Haematol. 2011;152:677–687 [DOI] [PubMed] [Google Scholar]
- 11.Locatelli F, Nollke P, Zecca M, et al. HSCT in children with JMML: results of EWOG-MDS/EBMT trial.Blood. 2005;105:410–419 [DOI] [PubMed] [Google Scholar]
- 12.Niemeyer C, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases.Blood. 1997;89:3534–3534 [PubMed] [Google Scholar]
- 13.Chan RJ, Cooper T, Kratz CP, et al. Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium.Leuk Res. 2009;33:355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yusuf U, Frangoul HA, Gooley TA, et al. Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience.Bone Marrow Transplant. 2004;33:805–814 [DOI] [PubMed] [Google Scholar]
- 15.Smith FO, King R, Nelson G, et al. Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia.Br J Haematol. 2002;116:716–724 [DOI] [PubMed] [Google Scholar]
- 16.Yoshimi A, Mohamed M, Beirings M, et al. Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia.Leukemia. 2007;21:556–560 [DOI] [PubMed] [Google Scholar]
- 17.Locatelli F, Lucarelli B.Treatment of disease recurrence after allogeneic hematopoietic stem cell transplantation in children with juvenile myelomonocytic leukemia: a great challenge still to be won.Pediatr Blood Cancer. 2013;60:1–2 [DOI] [PubMed] [Google Scholar]
- 18.Dave BJ, Nelson M, Sanger WG.Lymphoma Cytogenetics.Clin Lab Med. 2011;31:725–761 [DOI] [PubMed] [Google Scholar]
- 19.Horsman DE, Conners JM, Pantzar T, et al. Analysis of secondary chromosomal alterations in 165 cases of follicular lymphoma with t(14:18).Genes Chromosomes Cancer. 2001;30:375–382 [DOI] [PubMed] [Google Scholar]
- 20.Rubocki RJ, Parsa JR, Hershfield MS, et al. Full hematopoietic engraftment after allogeneic bone marrow transplantation without cytoreduction in a child with severe combined immunodeficiency.Blood. 2001;97:809–811 [DOI] [PubMed] [Google Scholar]
- 21.Manabe A, Okamura J, Yumera-Yagi K, et al. Allogeneic hematopoietic stem cell transplantation for 27 children with juvenile myelomonocytic leukemia diagnosed based on the criteria of the International JMML Working Group.Leukemia. 2002;16:645–649 [DOI] [PubMed] [Google Scholar]
- 22.Bergstraesser E, Hasle H, Rogge T, et al. Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria.Pediatr Blood Cancer. 2007;49:629–633 [DOI] [PubMed] [Google Scholar]
- 23.Korthof ET, Snijder PP, de Graaff AA, et al. Allogeneic bone marrow transplantation for juvenile myelomonocytic leukemia: a single center experience of 23 patients.Bone Marrow Transplant. 2005;35:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshimi A, Bader P, Matthes-Martin S, et al. Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia.Leukemia. 2005;19:971–977 [DOI] [PubMed] [Google Scholar]
- 25.Inagaki J, Fukano R, Nishiwaka T, et al. Outcomes of Immunological Interventions for mixed chimerism following allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia.Pediatr Blood Cancer. 2013;60:116–120 [DOI] [PubMed] [Google Scholar]
- 26.Locatelli F, Niemeyer C, Angelucci E, et al. Allogeneic bone marrow transplantation for chronic myelomonocytic leukemia in childhood: a report from the European Working Group on Myelodysplastic Syndrome in Childhood.J Clin Oncol. 1997;15:566–573 [DOI] [PubMed] [Google Scholar]
- 27.Locatelli F, Crotta A, Ruggeri A, et al. Analysis of risk factors influencing outcomes after cord-blood-transplantation in children with juvenile myelomonocytic leukemia.Blood. 2001;97:809–811 [DOI] [PMC free article] [PubMed] [Google Scholar]