

Abstract
A major barrier to understanding the mechanism of nitric oxide reductases (NORs) is the lack of selective probe of NO binding to the non-heme FeB center. By replacing the heme in a biosynthetic model of NORs (L29H/F43H/V68E Mb), that structurally and functionally mimics NORs, with isostructural ZnPP, we report herein a study where the electronic structure and functional properties of the FeB-nitrosyl complex has been probed selectively. This approach allowed us to observe the first S=3/2 non-heme {FeNO}7 complex in a protein system. Such feats are not achievable in native NORs as these are complex membrane proteins containing multiple hemes. Detailed spectroscopic and computational studies show that the electronic state of the {FeNO}7 complex is best described as a HS ferrous iron (S=2) antiferromagnetically coupled to NO radical (S=1/2) [Fe2+-NO•]. The radical nature of the FeB-bound NO would facilitate N-N bond formation by radical coupling with the heme-bound NO. This finding, therefore, supports the proposed trans mechanism of NO reduction by NORs.
Keywords: Metalloproteins, Nitric Oxide Reductases, Non-heme Iron-Nitrosyl, QM/MM, Reaction Mechanism, Spectroscopy
Nitric oxide reductases (NORs) catalyze two electron reduction of nitric oxide to nitrous oxide (2NO + 2H2+ + 2e− ➔ N2O + H2O), a critical step in the biological denitrification process.[1] Additionally, many pathogenic non-denitrifying bacteria use NORs to inactivate NO produced by the host’s immune system.[2] Thus, understanding the nitrosyl complexes of the enzyme and their role in the NO reduction mechanism is not only of biochemical significance, but also has broad implications for the global nitrogen cycle. Bacterial NORs are integral membrane proteins that contain a binuclear active site, consisting of a high spin (HS) heme b3 and a non-heme iron (FeB) center. Three mechanisms of NO reduction to form N2O have been proposed: the cis-heme b3, cis FeB and trans mechanisms (Scheme S1).[1a, 3] Important to differentiating these mechanisms is the study of nitrosyl complexes of either the heme or the FeB center. Spectroscopic features for the heme nitrosyl complexes in NORs have been probed; however, those of the FeB nitrosyl complexes have been elusive because its spectroscopic signatures are often masked by those of the heme nitrosyl (Keq for NO binding to ferrous hemes is ~1010–1012 M−1).[4] Therefore, this issue has become one of the most critical barriers to our current understanding of the electronic structure and reaction mechanism of NORs. Because NORs are large membrane proteins, their heme cofactor(s) cannot be successfully replaced to probe the effect of NO binding to the FeB site selectively.
Several studies have elucidated the electronic and spectroscopic properties of heme-nitrosyl complexes.[5] In addition, synthetic models of NORs have been reported.[6] To complement the studies of native NORs and synthetic models, we have taken advantage of small, easy-to-prepare and well-characterized proteins such as myoglobin to obtain biosynthetic models of NORs.[7] Using such an approach, we have successfully engineered an FeB site in the distal pocket of sperm whale myoglobin (swMb). These designed proteins, FeBMb1 (L29H, F43H, H64, V68E swMb),[7a] and FeBMb2 (I107E FeBMb1)[7c] not only reproduced the active site structure of NORs, such as the cytochrome c dependent NOR (cNOR),[8] but also displayed NOR activity.
One of the major advantages of using the biosynthetic model proteins is that the heme can be readily replaced with isostructural Zn-protoporphyrin IX (ZnPP), thus allowing us to spectroscopically probe the effect of NO binding to the FeB site exclusively.
Here, we report the preparation of Fe(II)-ZnPPFeBMb1, where the Fe(II) is in the FeB center and the heme in FeBMb1 is substituted with ZnPP, as confirmed by X-ray crystallography. Spectroscopic studies of Fe(II)-ZnPPFeBMb1 and its NO complex by UV-vis absorption, electron paramagnetic resonance (EPR) and Mössbauer spectroscopic methods, in conjunction with DFT analysis, have allowed us to gain insight into the NOR reaction mechanism. This study is the first report where the FeB-nitrosyl complex of NOR or its models have been probed exclusively in a protein system, and the results support the trans mechanism of the NOR reaction.
The FeBMb1 was prepared as reported previously.[7a] After extracting its heme using a slightly modified method from the literature (see the SI),[10] the protein was reconstituted with ZnPP (Figure 1). The UV-vis spectrum of ZnPP-reconstituted protein, ZnPPFeBMb1, has absorption peaks at 427 nm, 553 nm, and 595 nm (Figure 2). These spectral features are different from those of ZnPP alone (Figure S1), or from FeBMb1 that contains heme (Figure 2), suggesting that the non-native cofactor ZnPP was successfully incorporated into FeBMb1. To obtain further evidence of successful incorporation of the ZnPP into FeBMb1, an X-ray structure of ZnPPFeBMb1, refined to 1.5 Å resolution, was obtained (Figure S2) which shows successful incorporation of the non-native cofactor.
Figure 1.
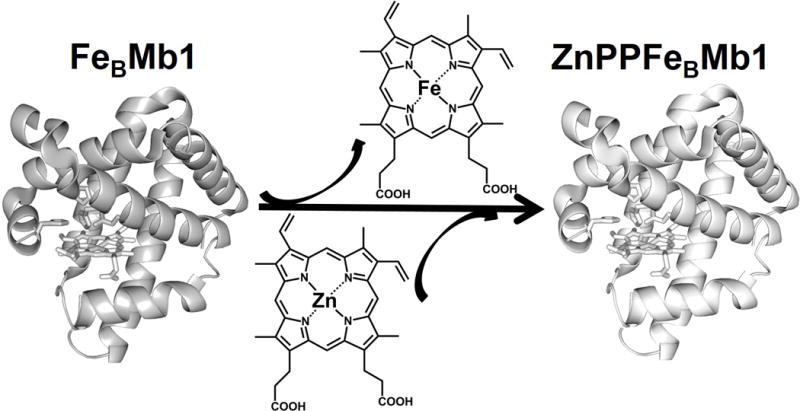
Schematic representation of the replacement of heme from FeBMb1 with ZnPP, yielding ZnPPFeBMb1. Figures were generated in PyMol[9] using PDB codes 3K9Z[7a], and 4MXL, respectively.
Figure 2.
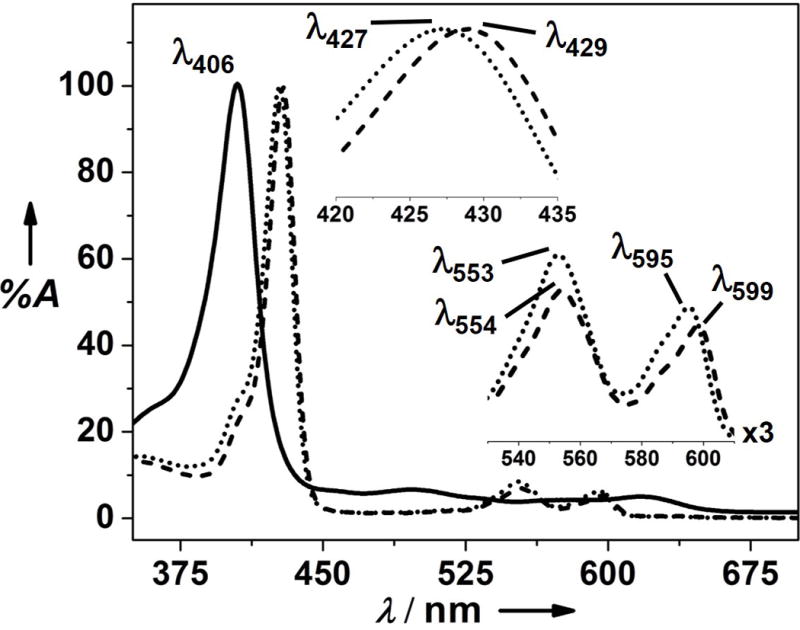
UV-vis spectra of FeBMb1 (solid line), ZnPPFeBMb1 (dotted line), and ZnPPFeBMb1 in the presence of 1.0 equivalent (eq.) Fe(II) (dashed line) in 50 mM Bis-Tris buffer pH 7.3. Peak positions in the Soret and visible region of ZnPPFeBMb1 and Fe(II)-ZnPPFeBMb1 are shown as insets.
Having established that FeBMb1 can be reconstituted with ZnPP, Fe(II) binding to the FeB site of ZnPPFeBMb1 was probed by UV-vis spectroscopy. As shown in Figure 2, addition of 1.0 eq. FeCl2 to 7 μM ZnPPFeBMb1 in 50 mM Bis-Tris pH 7.3, resulted in a red shift of the Soret band from 427 nm to 429 nm and concomitant changes in the visible region of the spectra, with isosbestic points at 432 nm, 562 nm, and 598 nm (Figure S3). As the electronic environment around the ZnPP framework is being perturbed upon Fe(II) binding at the distal pocket, only slight changes in the wavelength maxima are expected to be observed.[7a, 7c, 7d, 11] By following the spectral changes upon Fe(II) addition (Figure S3), a dissociation constant (Kd) of 7.2 ± 0.4 μM for Fe(II) binding to the FeB site of ZnPPFeBMb1 was obtained. Interestingly, compared to the Kd of 21.5 ± 0.5 μM for the same FeBMb1 but with heme in the active site (Figure S4), these data suggest that substitution of heme with ZnPP into FeBMb1 increases Fe(II) binding affinity at the FeB site by ~3-fold.
The Fe(II) binding was further investigated by crystallography. A 1.52 Å resolution X-ray structure of the FeCl2-soaked crystal of ZnPPFeBMb1 clearly shows that a metal is present at the FeB site. Anomalous X-ray scattering data of the crystals collected above Fe K-edge at 7.2 keV unambiguously assigned this metal to be Fe (Figure 3). Furthermore, anomalous data collected below the Fe K-edge at 7.0 keV did not show the presence of any anomalous electron density at the FeB site (Figure S5), ruling out the possibility of any other metal ion at the FeB site. Structural overlays of Fe(II)-ZnPPFeBMb1 with the active sites of Fe(II)-FeBMb1[7a] and cNOR[8] demonstrated that these structures were strikingly similar to each other (Figure 4). Therefore, the FeB site of Fe(II)-ZnPPFeBMb1 is a structural analog of the non-heme site of cNOR and FeBMb1. The close structural similarity between Fe(II)-ZnPPFeBMb1 and Fe(II)-FeBMb1 also suggests that the ~3-fold higher affinity of Fe(II) in the former was likely influenced by changes in the electronic environment around the metal binding site caused by replacement of iron protoporphyrin IX with ZnPP, and not due to changes in the overall structure of the FeB site.
Figure 3.
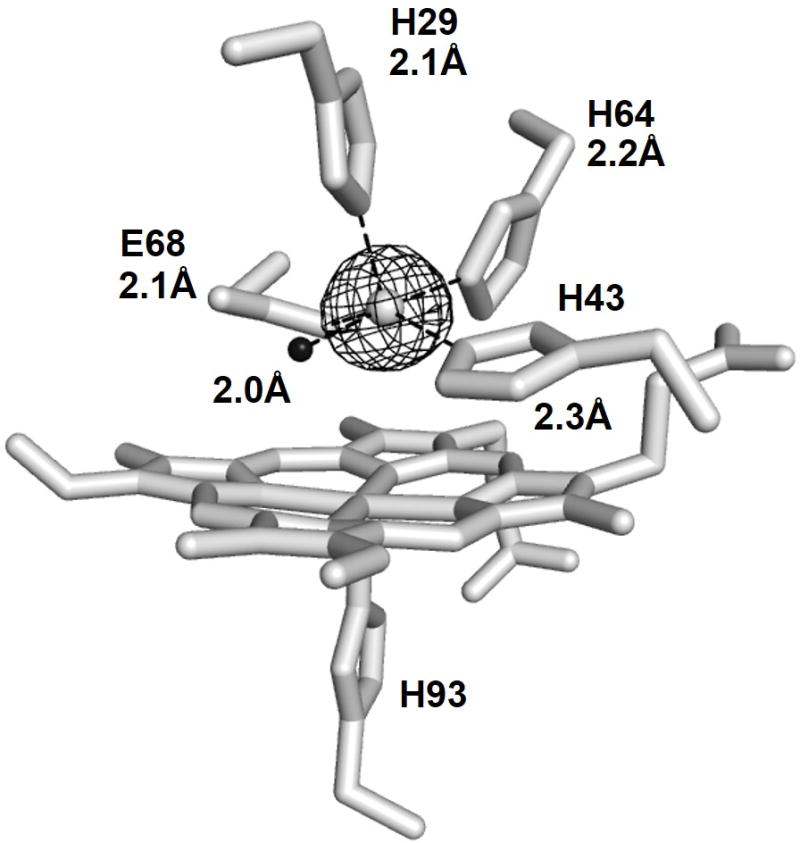
1.52 Å X-ray structure of Fe(II)-ZnPPFeBMb1. ZnPP, and the coordinating residues are shown as sticks. Fe is shown as gray sphere, the water molecule is shown as black sphere. The anomalous map generated in PHENIX[12] from the data collected at the Fe K-edge at 7.2 keV is drawn at 7σ and shown as gray mesh. Rfac=0.18, Rf=0.22. Figure was generated using PyMol.[9] PDB ID 4MXK.
Figure 4.
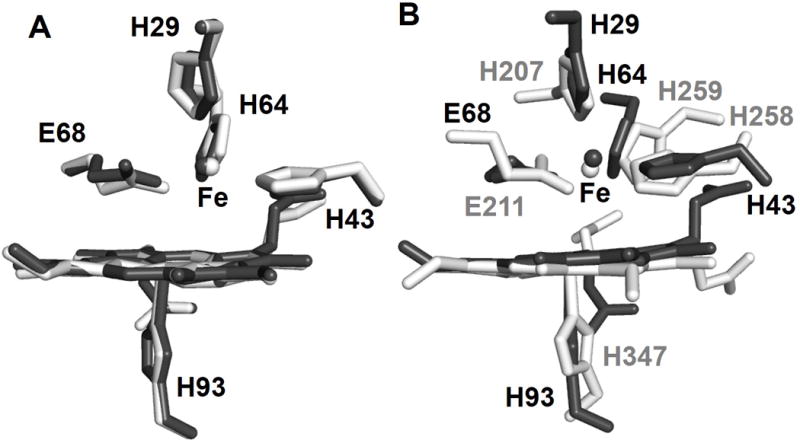
Overlay of Fe(II)-ZnPPFeBMb1 (PDB ID: 4MXK, dark gray) with Fe(II)-FeBMb1 (PDB ID: 3K9Z, light gray) (A), and cNOR (PDB ID: 3O0R, light gray) (B). Figures were generated using PyMol.
We next studied NO binding properties of Fe(II)-ZnPPFeBMb1 by EPR spectroscopy. The sample containing Fe(II)-ZnPPFeBMb1 in the absence of NO had no detectable EPR signal (Figure S6). With addition of up to 5 eq. of NO, the major peak observed was a radical-like signal at g ~ 2.01, with no other significant spectral features (Figure S7). However, in the presence of 10 eq. of NO, an EPR feature at g ~ 4.04 appeared, indicating the formation of a new species. Similar features were also present in the presence of 20 eq. of NO. Encouraged by the results, we collected EPR spectrum under the condition to maximize the signal of this new species (T = 5K, power = 20dB). The resulting spectrum shows two well-resolved doublets 1x,1y, and 2x,2y at g = 4.36, 3.58, and 4.13, 3.73, respectively (Figure 5, Table 1). No other features were observed at lower field (Figure S8). The EPR spectrum was simulated as a {FeNO}7 ferrous-nitrosyl complex with S=3/2 ground state, according to Enemark-Feltham nomenclature.[13] The sharp outer doublet 1x,1y, accounting for 46% of the spin, is more rhombic with an E/D = 0.063 than the major (54%) broad inner doublet 2x,2y which has an E/D of 0.040. The corresponding g‖ components are not well resolved in the region of g ~ 1.97. The two different components can be ascribed to either different orientations of NO bound to the FeB site, or slight changes in the orientation of Fe(II) ligands upon NO binding. Multiple Fe-NO symmetries with different rhombicities are commonly observed in heme nitrosyls.[5g, 14] To test whether different components of the FeB-NO complex observed here are a result of hyperfine interaction with 14N nucleus, EPR simulations were performed taking into consideration this interaction. Figure S9 rules out this possibility, as a satisfactory fit was not obtained. Detailed characterization of the saturation behavior of the radical-like signal at g ~ 2.01 shows that it is not a “free” radical instead it is most likely a radical associated with a metal ion (see the SI, Figures S10–12, Tables S1–2).
Figure 5.
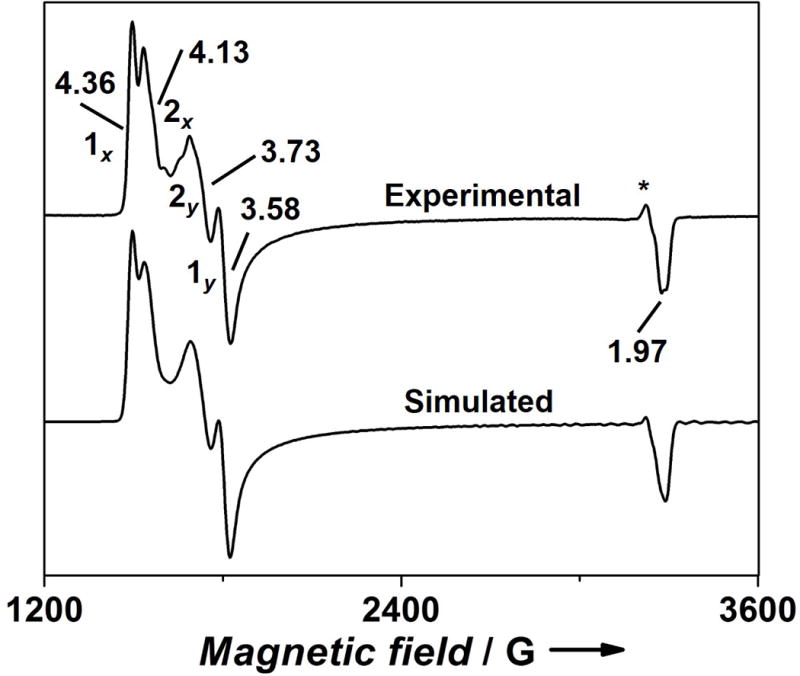
X-band EPR spectrum of a sample containing 0.7 mM ZnPPFeBMb1 in the presence of 1.0 eq. FeCl2 and 20 eq. of NO in 50 mM Bis-Tris buffer pH 7.3, and the simulated spectrum. Excess NO was used to ensure saturation of the FeB site. Experimental parameters: T = 5 K, microwave frequency = 9.053 GHz, microwave power = 20 dB, modulation amplitude = 4 G. g values and the components are labeled. *A radical type peak (<1%).
Table 1.
Parameters extracted from fitting of the EPR spectrum in Figure 5.
The ferrous nitrosyl complex is present in two rhombic geometries with E/D = 0.063, and 0.040, respectively. D = 7.2 cm−1 as obtained from the Mössbauer measurements (Table S3) was used to simulate the EPR spectrum.
Freeze-quench EPR studies of the reaction of reduced NOR from Ps. Aeruginosa[15] with NO-saturated buffer showed a peak at g = 4 within 0.5 ms of mixing, corresponding to ~30% population of the FeB-nitrosyl {FeNO}7 complex with S=3/2 ground state. Similar EPR features have been reported for the reaction of NO with various non-heme iron proteins,[16] as well as model complexes.[6b, 17] ICP-MS analysis and spin quantification against Fe(III)-EDTA gave 61% of total S=3/2 species in Fe(II)-ZnPPFeBMb1-NO. These results, therefore, suggest that the S=3/2 FeB-NO complex can be formed in heme substituted Fe(II)-ZnPPFeBMb1. Control experiments containing ZnPPFeBMb1 and NO in the absence of Fe(II) in the FeB site did not show any EPR feature (Figure S13), confirming that the aforementioned EPR feature of S=3/2 ferrous-nitrosyl species can be observed only when NO is present with the FeB site reconstituted by Fe(II).
To understand the electronic and spin state of the FeB-nitrosyl species further, we carried out field-dependent Mössbauer measurements at 4.2K. In the absence of NO, a sample containing 3.5 mM 57Fe(II)-ZnPPFeBMb1 shows a single quadrupole doublet at 0.01 Tesla (T) (Figure S14). In the presence of magnetic field (1–9 T) the doublet splits into multiplets, resulting from magnetic hyperfine interaction with the electron spin. A quadrupole splitting (ΔEQ) = 2.85 ± 0.01 mm/s, and isomer shift δFe = 1.13 ± 0.01 mm/s are obtained from fits indicative of a high spin ferrous (S = 2) species (Tables 2 and S3).[18] Similar parameters have been reported for non-heme ferrous sites in proteins.[16a] In the presence of 20 eq. NO, the Mössbauer spectrum (Figure 6) of 57Fe(II)-ZnPPFeBMb1-NO at low field (0.01T) shows two doublets (solid and dashed gray lines), and a magnetically split component (short gray dots). The solid gray line in the 0.01T field spectrum represents 35% of unreacted Fe(II) species. To simplify the high-field spectra, this unreacted component was subtracted, and the splitting pattern of the 65% iron-nitrosyl component is shown in Figure 6. Simultaneous least-squares fits to the three applied field spectra gave Mössbauer parameters (ΔEQ) = −1.70± 0.01 mm/s, and δFe = 0.69 ± 0.01 mm/s, indicative of S=3/2 six-coordinate {FeNO}7 species (Tables 2 and S3), found in other proteins as well as small molecule models.[16a, 16d, 17d, 19]
Table 2.
Experimental (bold), and calculated (parentheses) Mössbauer parameters of 57Fe-ZnPPFeBMb1 in the absence and presence of NO. Experimental conditions: T=4.2K, field=0.01T, 1T, 5T, 9T.
| Sample | δFe (mm/s) | ΔEQ (mm/s) | S | Fe Spin Population | NO Spin population |
|---|---|---|---|---|---|
| 57Fe-ZnPPFeBMb1 | 1.13 (1.13)a | 2.85 (3.15)a | 2 | 3.75b | N/A |
| 57Fe-ZnPPFeBMb1-NO | 0.69 (0.70)a | −1.70 (−2.01)a | 3/2 | 3.66b | −0.90b |
Mössbauer parameters were calculated from active site model, and
spin populations were calculated from QM/MM calculations.
Figure 6.
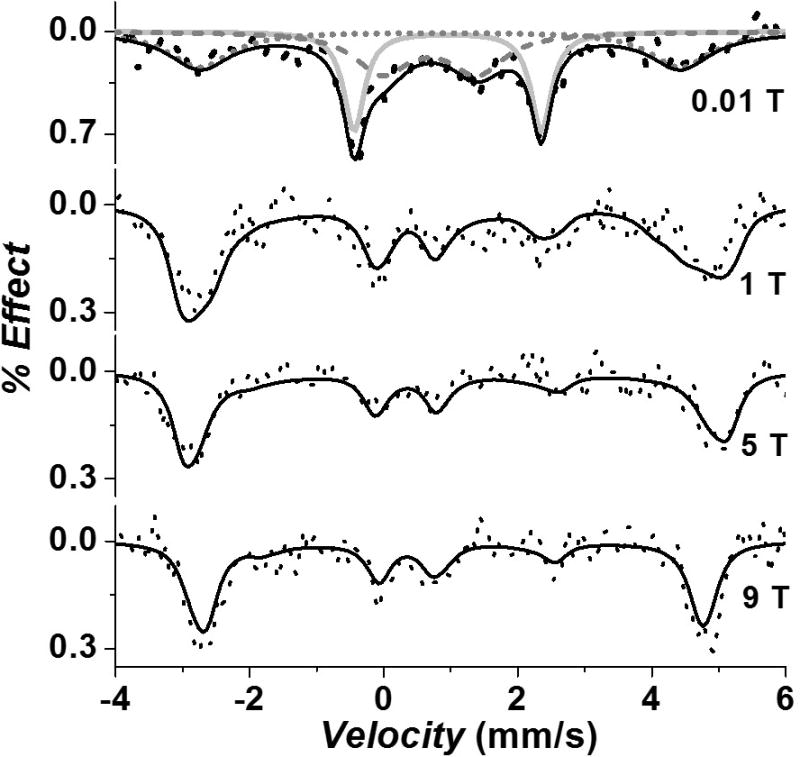
Field-dependent Mössbauer spectra of a sample containing 7 mM 57Fe(II)-ZnPPFeBMb1, and 20 eq. of NO in 50 mM Bis-Tris pH 7.3, collected at 4.2K. Black dots: experimental data; black lines: sum of fit; gray solid line: ferrous S=2 starting material; gray dashes: S=3/2 {FeNO}7 component in intermediate relaxation; gray short dots: magnetically split S=3/2 {FeNO}7 component. Only the splitting pattern of 65% S=3/2 {FeNO}7 species is shown in high field. The S=3/2 species at all fields was simulated using a slow relaxation model. The S=2 species in low-field was simulated as Lorentzian. (See Figure S15 for a color representation of this figure).
Some of the S=3/2 species (gray dashed line) is in intermediate relaxation at low field (0.01T), which is most likely due to the proximity to the small population of the metal-associated radical species detected by EPR (vide supra, SI). In applied field, this S=3/2 component is in the slow relaxation limit and is therefore indistinguishable to the other S=3/2 center (short gray dots, 0.01T) that is already in the slow relaxation regime. Simulations to the high-field spectra show excellent matches to the positions and intensity of all the spectral features, thus supporting that there is only a single type of S=3/2 species. These conclusions are supported by low-field Mössbauer data obtained under lower NO equivalents at higher temperatures (Figure S16, Table S4).
The electronic state of the S=3/2 {FeNO}7 species in both proteins and model complexes has been most frequently described as a result of antiferromagnetic coupling of HS ferric (S=5/2) to NO− (S=1) [Fe3+-NO−][16f, 17b, 17d] or as antiferromagnetic coupling of HS ferrous (S=2) to NO radical (S=1/2) [Fe2+-NO•].[16a, 19c, 20] To obtain further insight into the electronic structure of the {FeNO}7 moiety, we calculated the Mulliken spin populations (Table 2) at the Fe and NO centers of the structures obtained from quantum mechanical/molecular mechanical (QM/MM) calculations as well as partially optimized active site structures (Figures S17–18). In these structures, NO occupies the vacant axial coordination site as the sixth ligand to FeB. The Mulliken spin population on the FeB center remains virtually the same in the absence and presence of NO, at 3.75 and 3.66, respectively, indicating four unpaired electrons, while the spin population on NO is found to be −0.90, indicating one unpaired electron. These results unambiguously support the assignment of HS ferrous center (S=2) antiferromagnetically coupled to NO radical (S=1/2) [Fe2+-NO•]. Furthermore, we computed the Mössbauer parameters at the FeB site in the absence and presence of NO for partially optimized active site structures (see the SI for details). As can be seen from Table 2, both the calculated ΔEQ and δFe values agree well with the experimental data.
In conclusion, we have succeeded in probing the NO binding properties of the FeB site in a biosynthetic model protein of NOR by replacing the high affinity heme with isostructural ZnPP in the protein ZnPPFeBMb1. Such feats are not easily achievable in native NORs, as these are complex membrane proteins with multiple heme cofactors. Structural overlays with cNOR clearly show that the FeB site in Fe(II)-ZnPPFeBMb1 is a structural mimic of the FeB site in the native enzyme. EPR spectral studies show that NO binding to the FeB site yields rhombic signals corresponding to a {FeNO}7 S=3/2 ferrous-nitrosyl complex. Mössbauer, and QM/MM studies confirmed the electronic properties of the {FeNO}7 complex in Fe(II)-ZnPPFeBMb1-NO as HS ferrous (S=2) antiferromagnetically coupled with NO radical (S=1/2) [Fe2+-NO•]. This exciting finding indicates that the radical nature of the NO would facilitate the radical coupling of the second heme-bound NO to promote N-N bond formation, supporting the proposed trans mechanism of NO reduction by NORs.
Experimental Section
Details of protein expression and purification, reconstitution of FeBMb1 with ZnPP, experimental details of determination of molar absorptivity, synthesis of 57FeCl2, Fe(II) titration, EPR, Mössbauer sample preparation and data analysis, X-ray crystallography, ICP, and QM/MM calculations are provided in the supporting information.
Supplementary Material
Acknowledgments
This work is supported by the US National Institute of Health (5T32-GM070421 to JR, GM056207 to SHS, GM085774 to YZ, and GM062211 to YL) and National Science Foundation (CHE-1026369 to JTS). We thank Dr. Sebastian Stoian for helpful discussions and Prof. Eric Oldfield for access to their glove bag in a cold room.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Saumen Chakraborty, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
Julian Reed, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
Matthew Ross, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
Mark J. Nilges, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA
Igor D. Petrik, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA
Soumya Ghosh, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
Sharon Hammes-Schiffer, Email: shs3@illinois.edu, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
J. Timothy Sage, Department of Physics, Northeastern university, Boston, MA, USA.
Yong Zhang, Email: yong.zhang@stevens.edu, Department of Chemistry, Chemical Biology and Biomedical Engineering, Stevens Institute of Technology, Hoboken, NJ, USA.
Charles E. Schulz, Email: cschulz@knox.edu, Department of Physics, Knox College, Galesburg, IL, USA
Yi Lu, Email: yi-lu@illinois.edu, Department of Chemistry and Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
References
- 1.a) Shiro Y, Sugimoto H, Tosha T, Nagano S, Hino T. Phil Trans R Soc B. 2012;367:1195–1203. doi: 10.1098/rstb.2011.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schopfer MP, Wang J, Karlin KD. Inorg Chem. 2010;49:6267–6282. doi: 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 2.a) Laver JR, Stevanin TM, Messenger SL, Lunn AD, Lee ME, Moir JW, Poole RK, Read RC. FASEB J. 2010;24:286–295. doi: 10.1096/fj.08-128330. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stevanin TM, Moir JW, Read RC. Infect Immun. 2005;73:3322–3329. doi: 10.1128/IAI.73.6.3322-3329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Möenne-Loccoz P. Nat Prod Rep. 2007;24:610–620. doi: 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu N, Campbell ALO, Powell DR, Khandogin J, Richter-Addo GB. J Am Chem Soc. 2009;131:2460–2461. doi: 10.1021/ja809781r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodrich LE, Paulat F, Praneeth VKK, Lehnert N. Inorg Chem. 2010;49:6293–6316. doi: 10.1021/ic902304a. [DOI] [PubMed] [Google Scholar]
- 5.a) Cheng L, Richter-Addo GB. In: The Porphyrin Handbook Kadish, Vol 4, Chapter 33. Kadish KM, Smith KM, Guilard R, editors. Academic Press; New York: 2000. pp. 219–291. [Google Scholar]; b) Coyle CM, Vogel KM, Rush TS, Kozlowski PM, Williams R, Spiro TG, Dou Y, Ikeda-Saito M, Olson JS, Zgierski MZ. Biochemistry. 2003;42:4896–4903. doi: 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]; c) Lehnert N, Berto TC, Galinato MGI, Goodrich LE. In: The Handbook of Porphyrin Science, Vol 14, Chapter 63. Kadish KM, Smith KM, Guilard R, editors. World Scientific; Singapore: 2011. pp. 1–247. [Google Scholar]; d) Silvernail NJ, Barabanschikov A, Sage JT, Noll BC, Scheidt WR. J Am Chem Soc. 2009;131:2131–2140. doi: 10.1021/ja8055613. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wyllie GR, Scheidt WR. Chem Rev. 2002;102:1067–1090. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]; f) Praneeth V, Neese F, Lehnert N. Inorg Chem. 2005;44:2570–2572. doi: 10.1021/ic050144k. [DOI] [PubMed] [Google Scholar]; g) Radoul M, Sundararajan M, Potapov A, Riplinger C, Neese F, Goldfarb D. PCCP. 2010;12:7276–7289. doi: 10.1039/c000652a. [DOI] [PubMed] [Google Scholar]
- 6.a) Collman JP, Yang Y, Dey A, Decréau RA, Ghosh S, Ohta T, Solomon EI. Proc Natl Acad Sci, USA. 2008;105:15660–15665. doi: 10.1073/pnas.0808606105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Berto TC, Hoffman MB, Murata Y, Landenberger KB, Alp EE, Zhao J, Lehnert N. J Am Chem Soc. 2011;133:16714–16717. doi: 10.1021/ja111693f. [DOI] [PubMed] [Google Scholar]
- 7.a) Yeung N, Lin YW, Gao YG, Zhao X, Russell BS, Lei L, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079–1082. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lin YW, Yeung N, Gao YG, Miner KD, Tian S, Robinson H, Lu Y. Proc Natl Acad Sci, USA. 2010;107:8581–8586. doi: 10.1073/pnas.1000526107. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lin YW, Yeung N, Gao YG, Miner KD, Lei L, Robinson H, Lu Y. J Am Chem Soc. 2010;132:9970–9972. doi: 10.1021/ja103516n. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lu Y, Chakraborty S, Miner KD, Wilson TD, Mukherjee A, Yu Y, Liu J, Marshall NM. In: Comprehensive Inorganic Chemistry II. Reedijk J, Poeppelmeier K, editors. Elsevier; Amsterdam: 2013. pp. 565–593. [Google Scholar]; f) Hayashi T, Miner KD, Yeung N, Lin YW, Lu Y, Möenne-Loccoz P. Biochemistry. 2011;50:5939–5947. doi: 10.1021/bi200409a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S, Shiro Y. Science. 2010;330:1666–1670. doi: 10.1126/science.1195591. [DOI] [PubMed] [Google Scholar]
- 9.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, California, USA: 2005. http://www.pymol.org. [Google Scholar]
- 10.a) Teale FWJ. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]; b) Koshiyama T, Shirai M, Hikage T, Tabe H, Tanaka K, Kitagawa S, Ueno T. Angew Chem Int Ed. 2011;50:4849–4852. doi: 10.1002/anie.201008004. [DOI] [PubMed] [Google Scholar]; c) Liang ZX, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. J Am Chem Soc. 2002;124:6849–6859. doi: 10.1021/ja0127032. [DOI] [PubMed] [Google Scholar]; d) Wang N, Zhao X, Lu Y. J Am Chem Soc. 2005;127:16541–16547. doi: 10.1021/ja052659g. [DOI] [PubMed] [Google Scholar]
- 11.a) Sigman JA, Kwok BC, Gengenbach A, Lu Y. J Am Chem Soc. 1999;121:8949–8950. [Google Scholar]; b) Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 12.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr Sect D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enemark J, Feltham R. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 14.Schmidt P, Kappl R, Hüttermann J. Appl Magn Reson. 2001;21:423–440. [Google Scholar]
- 15.Kumita H, Matsuura K, Hino T, Takahashi S, Hori H, Fukumori Y, Morishima I, Shiro Y. J Biol Chem. 2004;279:55247–55254. doi: 10.1074/jbc.M409996200. [DOI] [PubMed] [Google Scholar]
- 16.a) Arciero DM, Lipscornb JD, Huynh BH, Kent TA, Münck E. J Biol Chem. 1983;258:14981–14991. [PubMed] [Google Scholar]; b) D’Autréaux B, Tucker NP, Dixon R, Spiro S. Nature. 2005;437:769–772. doi: 10.1038/nature03953. [DOI] [PubMed] [Google Scholar]; c) Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]; d) Orville AM, Chen VJ, Kriauciunas A, Harpel MR, Fox BG, Münck E, Lipscomb JD. Biochemistry. 1992;31:4602–4612. doi: 10.1021/bi00134a010. [DOI] [PubMed] [Google Scholar]; e) Rodriguez JH, Xia YM, Debrunner PG. J Am Chem Soc. 1999;121:7846–7863. [Google Scholar]; f) Zhang Y, Pavlosky MA, Brown CA, Westre TE, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1992;114:9191–9192. [Google Scholar]; g) Nocek JM, Kurtz DM, Jr, Sage JT, Xia YM, Debrunner P, Shiemke AK, Sanders-Loehr J, Loehr TM. Biochemistry. 1988;27:1014–1024. doi: 10.1021/bi00403a026. [DOI] [PubMed] [Google Scholar]
- 17.a) Wells F, McCann S, Wickman H, Kessel S, Hendrickson D, Feltham R. Inorg Chem. 1982;21:2306–2311. [Google Scholar]; b) Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1995;117:715–732. [Google Scholar]; c) Li M, Bonnet D, Bill E, Neese F, Weyhermüller T, Blum N, Sellmann D, Wieghardt K. Inorg Chem. 2002;41:3444–3456. doi: 10.1021/ic011243a. [DOI] [PubMed] [Google Scholar]; d) Hauser C, Glaser T, Bill E, Weyhermüller T, Wieghardt K. J Am Chem Soc. 2000;122:4352–4365. [Google Scholar]; e) Berto TC, Speelman AL, Zheng S, Lehnert N. Coord Chem Rev. 2013;257:244–259. [Google Scholar]
- 18.a) Debrunner PG. In: Spectroscopic Approaches to Biomolecular Conformation. Urry DW, editor. A.M.A. Press; Chicago: 1970. pp. 209–262. [Google Scholar]; b) Greenwood NN, Gibb TC. Mossbauer Spectroscopy. Chapman and Hall; London: 1971. [Google Scholar]
- 19.a) Bill E, Bernhardt FH, Trautwein AX, Winkler H. Eur J Biochem. 1985;147:177–182. doi: 10.1111/j.1432-1033.1985.tb08734.x. [DOI] [PubMed] [Google Scholar]; b) Haskin CJ, Ravi N, Lynch JB, Münck E, Que L., Jr Biochemistry. 1995;34:11090–11098. doi: 10.1021/bi00035a014. [DOI] [PubMed] [Google Scholar]; c) Zhang Y, Oldfield E. J Am Chem Soc. 2004;126:9494–9495. doi: 10.1021/ja0401242. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Oldfield E. J Phys Chem A. 2003;107:4147–4150. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.