

Abstract
The molybdopterin (MPT) is a complex molecule, made out of three distinctly different components. A retrosynthetic analysis provides a possible route for its synthesis that utilizes the coupling of a diamine with an osone analog. A regioselective condensation of the diamine with an osone affords the dephospho MPT, which has been characterized by NMR and IR spectroscopies, as well as high-resolution mass spectrometry.
Introduction
The molybdopterin (MPT, 1) lies at the heart of more than fifty enzymes that catalyse important chemical transformations in all forms of life. 1-3 These reactions play important roles in global cycling of C, S, N, As and Se to prodrug metabolism. In humans, failures to biosynthesize MPT cofactor due to genetic predispositions lead to severe physiological disorders and even death.4 The molybdopterin itself is a metal chelating ligand, that is known to bind molybdenum as well as tungsten in biology. When coordinated to molybdenum, it is called molybdenum cofactor.5 During the biosynthesis molybdenum cofactor, MPT binds to copper, which subsequently is replaced by molybdebun.4 Structurally MPT is comprised of three distinct components,6 and they are: a pterin moiety, a pyran ring fused to the pyrazine ring of the pterin and an ene-dithiolate unit that binds to metal ions.7 The pyran ring, however, opens up at times through C7—O bond scission resulting in a 6-substituted pterin with an alcohol functionality connected to the dithiolene moeity. 8 In prokaryotic enzymes, the phosphate moiety is often modified by a dinucleotide.
The MPT molecule is redox active, and in the reduced state it is unstable in the absence of a protein environment. The MPT molecule was originally characterized by degradative studies, and it was proposed to coordinate molybdenum via its dithiolene moiety.6, 9 Subsequently, both the open and the closed forms of the MPT have been confirmed by protein crystallography.10 Despite the biochemical interest, complete synthesis of this MPT remains a challenge that has compromised in-depth analyses of it properties, although progress has been made through years of efforts.11-15 In recent years progress has been made particularly towards the syntheses of the precursor of the MPT cofactor14, 16 and Mo coordinated MPT.13 A general strategy to prepare the MPT is to functionalize a pterin molecule at the C-6 position to introduce a dithiolene functionality leading to a fully protected pterin moiety.11 For past several years we have been engaged in developing robust synthetic methodologies for making molecules directed towards understanding the effect of different components of the cofactors with17 or without metal, 18, 19 including the topological20, 21 and hydrogen bonding22 aspects. Herein we report our approach in meeting the synthetic challenge by constructing the pterin unit while carrying the dithiolene moiety. This approach originates from a retrosynthetic analysis of the cofactor (Scheme 1) and provides a versatile route for unprotected pterin functionality.
Scheme 1.
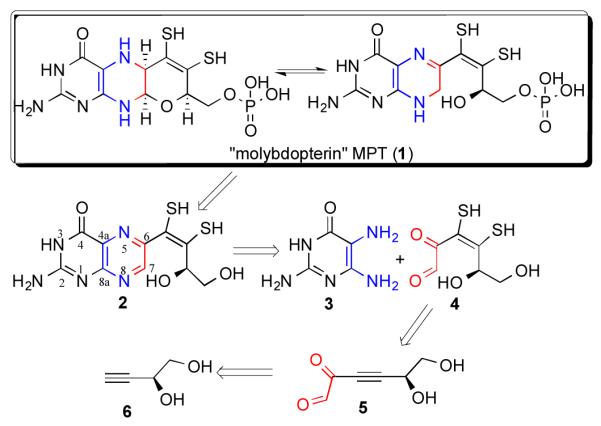
The retrosynthetic analysis of molybdopterin cofactor
Results and Discussion
The retrosynthetic analysis, shown in Scheme 1, of the MPT provides a conceptual frame work for this investigation. The fragile N8-C7-O moiety of the MPT can be oxidatively cleaved under acidic conditions,11, 16 yielding a fully oxidized pyrazine ring, concomitantly opening the pyran ring by breaking the C7-O bond as in compound 2. It is possible to reconstruct the N8-C7-O moiety by nucleophilic addition of the alcohol to an N-acetylated pyrazine ring, which can be formed by treating the pterin with chloroformate reagents e.g., benzyl chloroformate, 9H-fluoren-9-ylmethyl chloroformate (FMOC-Cl) that can subsequently be reduced by NaBH3CN.11 It has been proposed that both the open and the closed forms exist in equilibrium, and the open form can be oxidized to fully oxidized pterin as in 2.8, 23 The retrosynthetic analysis also shows compound 2 as the core MPT, without the phosphate group, as has the dithiolene and pterin functionalities. We envision that the pyrazine ring of the oxidized molybdopterin can be constructed via the condensation of an α-keto aldehyde (4), with 2,5,6-triamino-3,4-dihydropyrimidin-4-one (3). The retrosynthetic analysis highlights 4 as a key precursor to MPT, which is an ‘osone’ harboring a dithiolene moiety. A protected form of 4 could be achieved from an activated acetylene derivative (5) to which a dithiolene moiety can be introduced. 24, 25 In 5, the adjacent keto group activates the acetylene. Compound 5 could be prepared from an acetylene e.g., compound 6, through a nucleophilic addition reaction via an acetylide formation.17
The synthesis of dithiolene protected dephospho MPT has been realized by using dibromoalkene26 (7) as a key building block (Scheme 2). Compound 7 is expected to afford the same stereochemistry as the MPT, whose absolute configuration has been crystallographically described.10 Compound 7 was prepared in 68% yield, from 2,3-O-isopropylidene-D-glyceraldehyde27 with modification of the Corey-Fuchs reaction.28, 29 Compound 7 was reacted with nBuLi resulting in first a lithiated intermediate, 6a, from which compound 5a was prepared via an electrophilic substitution reaction with ethyl 2,2-diethoxyacetate. From this one-pot reaction, 5a was isolated by radial chromatography in a 31% yield. Structurally, compounds 6a and 5a are very similar to 6 and 5, respectively, with the exception of alcohol and aldehyde groups being protected. Compound 5a contains an acetylene moiety next to an electron withdrawing keto group which makes it suitable for the introduction of a protected dithiolene unit24, 25 with phenyl-1,3-dithiolane-2-thione30 resulting in compound 4a in a 58% yield. Compound 4a is a protected form of precursor 4 and contains a protected aldehyde moiety as an acetal, a keto group, a protected dithiolene, and protected alcohol moieties.
Scheme 2.
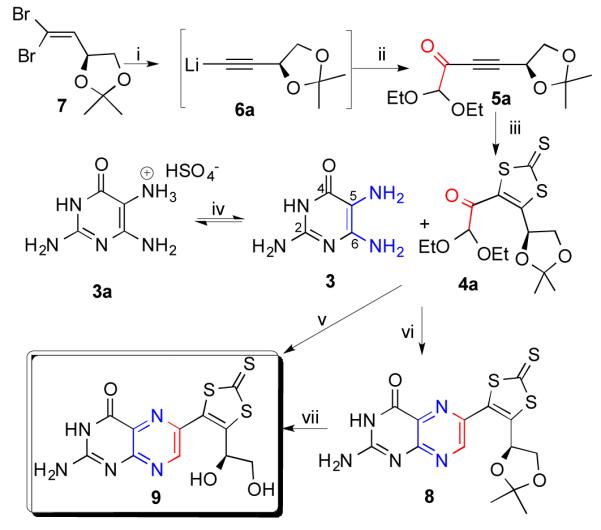
(i) nBuLi, Et2O at −78 °C, room temperature; (ii) ethyl 2,2-diethoxyacetate at −78 °C, room temperature (31%); (iii) phenyl-1,3-dithiolane-2-thione at 130 °C (58%); (iv) Na2SO3, DMSO; (v) 150 °C, Na2SO3, DMSO (24%); (vi) 120 °C, 2-mercaptoethanol, DMSO (31%) (vii) CF3COOH, DMF at 60 °C (82%).
The reaction of the compound containing a keto group next to the acetal with diamines follows that of the Isay-Gabriel-Coleman condensation31, 32 for synthesizing pteridines.33 The regioselective potential of this reaction is realized by exploiting the higher reactivity of the keto group relative to the acetal towards condensation with a diamine. This type of condensation reactions often lead to stereoselective formation of the undesired 7-isomer, although, a microwave assisted synthesis has afforded the 6-substituted pterin preferentially.34 Earlier work on regiospecific synthesis of biopterin and anapterin involved condensation of the same pyrimidine with a sugar derivative.35, 36 A similar methodology has been applied in synthesizing a pyranopterin.37 In the present case, 4a was successfully connected with 3a in the presence of sodium sulfite as an additive. In the condensation reaction with the amine functionality in 3, the keto group in 4a serves as an electrophile. The free 5-NH2 in 3 is the most nucleophilic among the three amine moieties38, and is protonated forming a salt in 3a. To obtain the desired regioisomer, the 5-NH2 group in 3a was deprotonated with a base (i.e., Na2SO3) forming 3 which then reacts with 4a resulting in 8 in the desired regioisomer. No appreciable amount of the 7-isomer was detected in the 1H NMR spectra of the reaction mixtures suggesting the regioselective nature of the condensation reaction (supplementary information). Additive assisted regioselective syntheses of pterins, like the one reported in here, have been reported before.39 Yellow-orange 8 has been isolated in 31% yields from reactions conducted at 120 °C in the presence of 2-mercaptoethanol. At a slightly elevated temperature, the ketal protection of 8 can be removed in acidic conditions e.g., with trifluroacetic acid, resulting in the target compound, 9 in 82% yields. The ketal protection in 8 can be removed at room temperature, however, the reaction proceeds slowly.
Compound 9 can also be prepared directly by reacting 3 and 4a in a 24% yield, optimally at 150 °C. During the reaction, the formation and decay of 8 can be observed by 1H NMR spectroscopy (supplementary information). Here also only the desired isomer (i.e., 9) is obtained in one step reaction in a similar overall yield. Condensation reactions of 3 with 4a, but with unprotected acetal and ketal functionality, resulted in both the 6- and 7-isomers, and this route was not pursued further. Similar condensation reaction of 3 and 4a bearing only the acetal protection, results in intractable products. Therefore, the protection of the aldehyde and the alcohol is important for regioselective condensationin this case.
Compound 9 has been characterized by IR, 1H and 13C NMR spectroscopies as well as high-resolution mass spectrometry (supplementary information). The 1H NMR spectra of 9 are shown in Figure 1. Consistent with the literature11, 40 a peak at 8.97 ppm corresponding to one proton is assigned to the proton at C7 in the pterin ring. Resonances due to the NH and NH2 groups appear at 11.87 and 7.31 ppm, respectively, and the signals disappear in the presence of D2O. The two alcohol protons appear at 6.43 and 5.26 ppm, respectively for the secondary and primary alcohols, and both alcohol protons are exchanged in D2O. The CH and CH2 protons appear at 5.11 (q), 3.67 (ddd) and 3.59 (ddd) ppm respectively. The latter two signals in equivalency of the two CH2 protons. In the presence of D2O these reduced splitting is observed.
Figure 1.
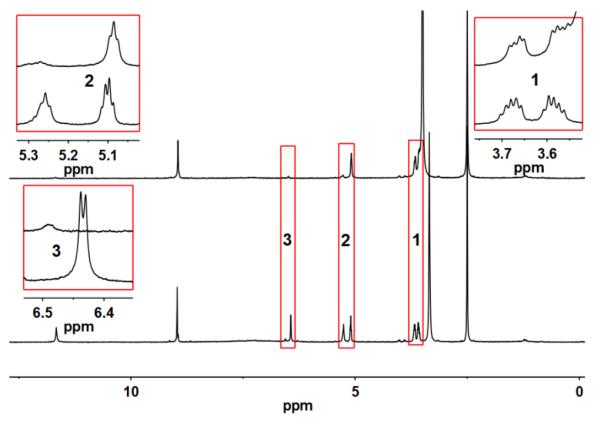
Room temperature 1H NMR of 9: bottom, in DMSO-d6; top, same solution as bottom with added D2O.
The 13C NMR spectroscopy has been used in characterizing the synthesized compounds. The trithiocarbonate carbon resonates almost at same place, in 4a, 8 and 9, at ~210 ppm. In addition to the thiocarbonyl group, a major difference between 5a and 4a is the conversion of acetylene to ene-dithiolate moiety. Two new peaks at ~167 and 129 ppm appeared in 4a at the expense of peaks at 111 and 93 ppm present in 5a. When the pterin heterocycle is attached to the dithiolene moiety, the situation becomes complicated due to the presence of several electron deficient carbon centres in the ring, making unambiguous assignment from 1D spectrum difficult. The protection of the alcohol group present in 8 is absent in 9 and consequently peaks at ~25 ppm and ~110 ppm observed in the spectrum of 8 are absent in 9 (Figure 2).
Figure 2.
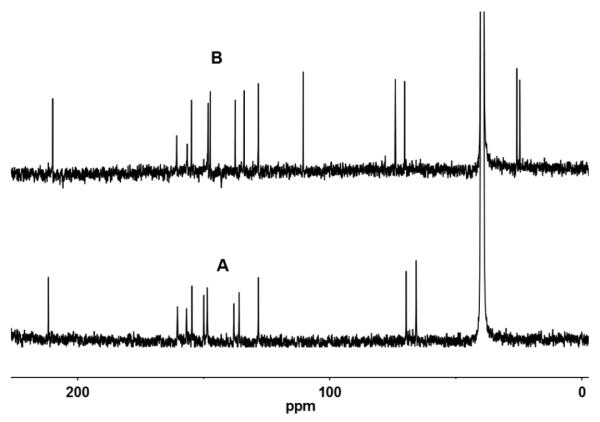
Room temperature 13C NMR spectra of (A) 9 and (B) 8 taken in DMSO-d6.
Conclusion
In conclusion, we disclose here a new approach to synthesize the core structure of the MPT, compound 9, in a regioselective condensation reaction.31, 32 The condensation reaction of oxo-aldehyde (4a) with the triaminohydroxy pyrimidine to obtain the desired 6-substituted regioisomer. The pterin functionality bears no protection, the only protection is in the dithiolene unit, which can be modified to introduce metal ions following the procedures developed by others8, 25 and us17. Molecule 9 has all the components of the dephospho MPT and can serve as a spectroscopic benchmark for future studies. Results from our on going investigations to introduce metal ion, and reduction of the pterin moiety as well as introduction of a phosphate group will be reported in due course.
Experimental
General
Starting materials were purchased either from Aldrich Chemical Company or Acros Chemical Company and were used without further purification. Silica gel for column chromatography was purchased from Sorbent Technologies, and that for radial chromatography was purchased from EMD Chemicals. Ether was dried either by distillation over sodium-benzophenone or using a column (LC Technology). DMSO and DMF were dried by passing using solvent purification systems (Pure Solv, Innovative Technology) and LC Technology, respectively. All other solvents were used as received. NMR spectra were collected on a Bruker Avance 400 or a Bruker Avance 500 spectrometer operating at 400 MHz and 500 MHz for 1H spectra, respectively. IR spectra were recorded using a Nicolet 380 FT spectrometer. High-resolution mass spectra were recorded on a Agilent 6200 time of flight LC MS system using a nano ESI and APCI-TOF interface.
Synthesis of 4-(2,2-dimethyl-1,3-dioxolan-4-yl)-1,1-diethoxybut-3-yn-2-one (5a)
1.7 g (5.7 mmol) of 4-(2,2-dibromovinyl)-2,2-dimethyl-1,3-dioxolane (7) was dissolved in 8.0 mL of dry diethyl ether and kept under N2 for the duration of the reaction. The solution was then cooled to −78 °C before 4.16 mL (10.4 mmol) of 2.5 M n-butyllithium in hexanes was added. The reaction continued for 1 hour at −78 °C, then warmed to room temperature for an additional hour. The solution was then cooled to −78 °C before 2.0 g (11.3 mmol) of ethyl 2,2-diethoxyacetate was added. The reaction mixture was stirred for 10 min at −78 °C, then for another 30 minutes at ambient temperature before it was transferred to a flask containing 30 mL of an ice-cold aqueous saturated solution of ammonium chloride and stirred vigorously. The organic layer was extracted with ethyl acetate (3 × 20 mL), and dried with anhydrous sodium sulfate and filtered, and the solvent was removed under vacuum. Crude product was purified by radial chromatography (4 mm silica gel, 60PF254) using a mixture of 9:1 hexanes and ethyl acetate yielding pure product as an oil. Yield, 31.5 % (0.463 g, 1.8 mmol). 1H NMR in CDCl3 (δ, ppm): 4.89 (1H, dd, J=3.6, 4.4 Hz, CH), 4.74 (1H, s, acetal), 4.21 (1H, dd, J=5.6, 9.2 Hz, CH2), 4.07 (1H, dd, J= 5.2, 8.0 Hz, CH2), 3.75-3.67 (2H, m, acetal), 3.66-3.59 (2H, m, acetal), 1.51 (3H, s, CH3, ketal), 1.40 (3H, s, CH3, ketal), 1.27 (6H, t, J=7.2 Hz, CH3, acetal). 13C NMR in CDCl3 (δ, ppm): 182.58, 111.40, 92.90, 82.28, 69.44, 65.36, 63.25, 26.17, 25.96, 15.24. IR (neat, cm−1): 2974, 2943, 2876, 2214, 1687, 1376, 1151, 1067, 837. HR ESIMS+ with acetonitrile as the mobile phase, (m/z): 257.1379 [C13H20O5 (M+H)+, 257.1384], 279.1200 [C13H20O5 (M+Na)+, 279.1203], 295.0938 [C13H20O5 (M+K)+, 295.0942].
Synthesis of 1-(5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-thioxo-1,3-dithiol-4-yl)-2,2-diethoxyethan-1-one (4a)
0.34 g (1.32 mmol) of 4-(2,2-dimethyl-1,3-dioxolan-4-yl)-1,1-diethoxybut-3-yn-2-one (5a) and 3.0 g (14.12 mmol) of 4-phenyl-1,3-dithiolane-2-thione were dissolved in 5 mL of dichloromethane placed in a 100 mL round bottom flask. Addition of dichloromethane makes the reaction mixture more homogeneous, but it was removed under low pressure, before the reaction mixture was heated to 130 °C for 60 min under N2. The reaction mixture was cooled to room temperature yielding a yellow-brown solid, which was purified by column chromatography on silica gel (65-250 mesh, 60A) using dichloromethane-n-hexanes (1:1) followed by dichloromethane which elutes the target compound as the second fraction. The solvent was evaporated under vacuum as a yellow liquid, which solidifies upon cooling. Yield, 58% (0.28 g, 0.768 mmol). 1H NMR in CDCl3 (δ, ppm): 5.55 (1H, dd, J=7.2, 7.2 Hz, CH), 4.75 (1H, s, acetal), 4.60 (1H, dd, J=8.8, 9.2 Hz, CH2), 3.86 (1H, dd, J= 8.8, 9.2 Hz, CH2), 3.79-3.76 (2H, m, acetal), 3.65-3.56 (2H, m, acetal), 1.53 (3H, s, CH3, ketal), 1.38 (3H, t, CH3, ketal), 1.29 (3H, t, J=6.8 Hz, CH3, acetal), 1.28 (3H, t, J=6.8 Hz, CH3, acetal). 13C NMR in CDCl3 (δ, ppm): 211.91, 184.83, 167.04, 129.82, 111.65, 102.04, 75.67, 70.75, 64.13, 26.04, 24.60, 15.17. IR (neat, cm−1): 2978, 2925, 2884, 1679, 1053, 863. HR ESIMS+ with acetonitrile as the mobile phase, (m/z): 365.0547 [C13H20O5S3 (M+H)+, 365.0546], 387.0365 [C13H20O5S3 (M+Na)+, 387.0386], 403.0127 [C13H20O5S3 (M+K)+, 403.0103]
Synthesis of 2-amino-6-(5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-thioxo-1,3-dithiol-4-yl)pteridin-4(3H)-one (8)
100 mg (0.27 mmol) of 1-(5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-thioxo-1,3-dithiol-4-yl)-2,2-diethoxyethan-1-one (4a), 180 mg (0.75 mmol) of 4-hydroxy-2,5,6-triaminopyrimidine sulfate, 330 mg (1.30 mmol) of Na2SO3.7H2O and 1.1 g (~50 drops) of 2-mercaptoethanol, were taken in 10 mL dry DMSO. The mixture was warmed to 120 °C for 30 min and then cooled to room temperature. The reaction mixture turned dark brown, which was diluted with 10 mL DCM. The reaction mixture was purified by radial chromatography (4 mm silica, 60PF254) using a mixture of 1:2 DMF:DCM, only a yellow fraction was collected, and the organic solvent was evaporated under vacuum. The yellow solid was dissolved in 3 mL DMF, then diluted with 3 mL DCM. This mixture was purified again by radial chromatography using the same solvent mixture. The organic solvent was evaporated under reduced pressure. The dark orange solid was washed with water (3 × 5 mL), and then dried under vacuum yielding an orange solid. Yield, 33 mg (0.083 mmol, 31%). 1H-NMR in DMSO (δ, ppm): 11.66 (1H, s, =NH) 8.75 (1H, s, H-C7), 7.22 (2H, s, −NH2), 5.56(1H, dd, J=7.2, 6.0 Hz, CH), 4.88 (1H, dd, J=7.2, 9.2 Hz, CH), 4.01 (1H, dd, J=6.0, 9.2 Hz, CH), 1.48 (6H, s, CH3), 1.34 (6H, s, CH3). 13C NMR in CDCl3 (δ, ppm): 209.87, 160.69, 156.53, 154.80, 148.24, 147.38, 137.48, 133.93, 128.32, 110.49, 73.99, 70.26, 25.78, 24.60. IR (neat, cm−1): 3129, 2982, 2757, 1691, 1679, 1606, 1368, 1049, 812, 502. HR ESIMS+ with acetonitrile as the mobile phase, (m/z): 396.0251 [C14H13N5O3S3 (M+H)+, 396.0253], 418.0067 [C14H13N5O3S3 (M+Na)+, 418.0072], 433.9822 [C14H13N5O3S3 (M+K)+, 433.9812]
Synthesis of 2-amino-6-(5-(1,2-dihydroxyethyl)-2-thioxo-1,3-dithiol-4-yl)pteridin-4(3H)-one (9)
This compound was synthesized in two methods
Method a
100 mg (0.27 mmol) of 1-(5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-thioxo-1,3-dithiol-4-yl)-2,2-diethoxyethan-1-one (4a), 262 mg (1.09 mmol) of 4-hydroxy-2,5,6-triaminopyrimidine sulfate, and 552 mg (2.19 mmol) of Na2SO3.7H2O were suspended in 3 mL DMSO. The mixture was warmed for 35 min at 150 °C. The mixture was extracted with DMF (6 × 3 mL), and the supernatant was separated after centrifugation. The DMF-DMSO solution was evaporated under vacuum, the ensuing solid was dissolved in 3 mL DMF, and purified by radial chromatography (2 mm silica, 60PF254,) using DMF:DCM 1:2 then DMF as eluent. The second yellow fraction was collected, evaporated yielding orange solid. The solid was recrystallized from hot water (15 mL), and dried under vacuum. Yield, 23 mg (0.064 mmol, 24%).
Method b
20 mg (0.050 mmol) of 2-amino-6-(5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-thioxo-1,3-dithiol-4-yl)pteridin-4(3H)-one (8) was dissolved in 3 mL of dry DMF, and to this reaction mixture 1 mL of trifluoroacetic acid was added. The mixture was stirred for 3 h at 60 °C. The solvent was evaporated under low pressure at 60 °C. The mixture was recrystallized from boiling water. The yellow precipitate was separated by centrifugation, then dried under reduced pressure. Yield, 14.7 mg (0.041 mmol, 81%).
Characterization
1H-NMR in DMSO (δ, ppm): 11.87 (1H, s, =NH) 8.97 (1H, s, H-C7), 7.31 (2H, s, −NH2), 6.43 (1H, J=5.0, Hz, −OH), 5.26 (1H, t, J= 5.0 Hz, −OH), 5.11 (1H, q, J=5.0 Hz, CH), 3.67 (1H, ddd, J= 5.0, 5.0, 5.0 Hz, CH), 3.59 (1H, ddd, J= 5.0, 5.0, 5.0 Hz, CH).13C NMR in CDCl3 (δ, ppm): 211.62, 160.40, 156.80, 154.65, 149.95, 148.58, 138.01, 135.94, 128.32, 69.69, 65.71. IR (neat, cm−1): 3313, 3129, 2925, 1617, 1466, 1339, 1067, 1037, 988, 812, 506. HR ESIMS+ with acetonitrile as the mobile phase, (m/z): 355.9973 [C11H9N5O3S3 (M+H)+, 355.9940], 377.9793 [C11H9N5O3S3 (M+Na)+, 377.9759], 393.9553 [C11H9N5O3S3 (M+K)+, 393.9499]
Supplementary Material
Acknowledgements
We thank Professors Fraser F. Fleming and Patrick T. Flaherty for numerous stimulating discussions. Funding for this research from the National Institutes of Health (GM 061555) is gratefully acknowledged. Support from the National Science Foundation DBI 0821401 for mass spectrometry, and CHE 0614785 for NMR spectrometer are gratefully acknowledged.
Footnotes
Department of Chemistry and Biochemistry, Duquesne University, Pittsburgh, Pennsylvania 15282.
Electronic Supplementary Information (ESI) available: concentration profiles of reaction mixtures. See DOI: 10.1039/b000000x/
Notes and references
- 1.Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 2.Sparacino-Watkins C, Stolz JF, Basu P. Chem. Soc. Rev. 2014;43:676–706. doi: 10.1039/c3cs60249d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hille R, Hall J, Basu P. Chem. Rev. 2014 doi: 10.1021/cr400443z. Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz G, Mendel RR, Ribbe MW. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 5.Fischer B, Enemark JH, Basu P. J. Inorg. Biochem. 1998;72:13–21. doi: 10.1016/s0162-0134(98)10054-5. [DOI] [PubMed] [Google Scholar]
- 6.Johnson JL, Hainline BE, Rajagopalan KV, Arison BH. J. Biol. Chem. 1984;259:5414–5422. [PubMed] [Google Scholar]
- 7.Basu P, Burgmayer SJN. Coord. Chem. Rev. 2011;255:1016–1038. doi: 10.1016/j.ccr.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hine FJ, Taylor AJ, Garner CD. Coord. Chem. Rev. 2010;254:1570–1579. [Google Scholar]
- 9.Johnson JL, Rajagopalan KV. Proc. Natl. Acad. Sci. U. S. A. 1982;79:6856–6860. doi: 10.1073/pnas.79.22.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romao MJ. Dalton Trans. 2009:4053–4068. doi: 10.1039/b821108f. [DOI] [PubMed] [Google Scholar]
- 11.Bradshaw B, Dinsmore A, Ajana W, Collison D, Garner CD, Joule JA. J. Chem.Soc., Perkin Trans. 1. 2001:3239–3244. [Google Scholar]
- 12.Taylor EC, Doetzer R. J. Org. Chem. 1991;56:1816–1822. [Google Scholar]
- 13.Williams BR, Fu Y, Yap GPA, Burgmayer SJN. J. Am. Chem. Soc. 2012;134:19584–19587. doi: 10.1021/ja310018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA. J. Med. Chem. 2013;56:1730–1738. doi: 10.1021/jm301855r. [DOI] [PubMed] [Google Scholar]
- 15.Pilato RS, Eriksen KA, Greaney MA, Stiefel EI, Goswami S, Kilpatrick L, Spiro TG, Taylor EC, Rheingold AL. J. Am. Chem. Soc. 1991;113:9372–9374. [Google Scholar]
- 16.Hilken S, Kaletta F, Heinsch A, Neudoerfl J-M, Berkessel A. Eur. J. Org. Chem. 2014 Ahead of Print. [Google Scholar]
- 17.Marbella L, Serli-Mitasev B, Basu P. Angewandte Chemie, International Edition. 2009;48:3996–3998. doi: 10.1002/anie.200806297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pimkov IV, Nigam A, Venna K, Fleming FF, Solntsev PV, Nemykin VN, Basu P. J. Heterocycl. Chem. 2013;50:879–886. doi: 10.1002/jhet.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sengar RS, Nemykin VN, Basu P. New J. Chem. 2003;27:1115–1123. [Google Scholar]
- 20.Basu P, Nemykin VN, Sengar RS. Inorg. Chem. 2003;42:7489–7501. doi: 10.1021/ic034821r. [DOI] [PubMed] [Google Scholar]
- 21.Sengar RS, Basu P. Inorg. Chim. Acta. 2007;360:2092–2099. doi: 10.1016/j.ica.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sengar RS, Miller JJ, Basu P. Dalton Trans. 2008:2569–2577. doi: 10.1039/b714386a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer B, Enemark JH, Basu P. J Inorg Biochem. 1998;72:13–21. doi: 10.1016/s0162-0134(98)10054-5. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor BR, Jones FN. J. Org. Chem. 1970;35:2002–2005. [Google Scholar]
- 25.Bradshaw B, Dinsmore A, Collison D, Garner CD, Joule JA. J. Chem. Soc., Perkin Trans. 1. 2001:3232–3238. [Google Scholar]
- 26.Gung BW, Kumi G. J. Org. Chem. 2003;68:5956–5960. doi: 10.1021/jo0344900. [DOI] [PubMed] [Google Scholar]
- 27.Schmid CR, Bryant JD, Dowlatzedah M, Phillips JL, Prather DE, Schantz RD, Sear NL, Vianco CS. J. Org. Chem. 1991;56:4056–4058. [Google Scholar]
- 28.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972:3769–3772. [Google Scholar]
- 29.Habrant D, Rauhala V, Koskinen AMP. Chem Soc Rev. 39:2007–2017. doi: 10.1039/b915418c. [DOI] [PubMed] [Google Scholar]
- 30.Culvenor CCJ, Davies W, Pausacker KH. Journal of the Chemical Society. 1946:1050–1052. [Google Scholar]
- 31.Isay O. Berichte der deutschen Chemischen Gesellschaft. 1906;39:250–265. [Google Scholar]
- 32.Gabriel S, Coleman J. Chem. Ber. 1901;34:1234–1257. [Google Scholar]
- 33.Angier RB. J. Org. Chem. 1963;28:1398–1401. [Google Scholar]
- 34.Goswami S, Adak AK. Tetrahedron Letters. 2002;43:8371–8373. [Google Scholar]
- 35.Schircks B, Bieri JH, Viscontini M. Helv. Chim. Acta. 1985;68:1639–1643. doi: 10.1002/hlca.19760590128. [DOI] [PubMed] [Google Scholar]
- 36.Viscontini M, Bosshard R. de Gruyter. 1990. pp. 73–76.
- 37.Soyka R, Pfleiderer W, Prewo R. Helv. Chim. Acta. 1990;73:808–826. [Google Scholar]
- 38.Murata S, Ichinose H, Urano F. Top. Heterocycl. Chem. 2007;8:127–171. [Google Scholar]
- 39.Ivery MTG, Gready JE. Biol. Chem. Hoppe-Seyler. 1992;373:1125–1137. doi: 10.1515/bchm3.1992.373.2.1125. [DOI] [PubMed] [Google Scholar]
- 40.Hanaya T, Baba H, Toyota H, Yamamoto H. Tetrahedron. 2009;65:7989–7997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.