

Abstract
We introduce a Python-based program that utilizes the large database of 13C and 15N chemical shifts in the Biological Magnetic Resonance Bank to rapidly predict the amino acid type and secondary structure from correlated chemical shifts. The program, called PACSYlite Unified Query (PLUQ), is designed to help assign peaks obtained from 2D 13C–13C, 15N–13C, or 3D 15N–13C–13C magic-angle-spinning correlation spectra. We show secondary-structure specific 2D 13C–13C correlation maps of all twenty amino acids, constructed from a chemical shift database of 262,209 residues. The maps reveal interesting conformation-dependent chemical shift distributions and facilitate searching of correlation peaks during amino-acid type assignment. Based on these correlations, PLUQ outputs the most likely amino acid types and the associated secondary structures from inputs of experimental chemical shifts. We test the assignment accuracy using four high-quality protein structures. Based on only the Cα and Cβ chemical shifts, the highest-ranked PLUQ assignments were 40–60 % correct in both the amino-acid type and the secondary structure. For three input chemical shifts (CO–Cα–Cβ or N–Cα–Cβ), the first-ranked assignments were correct for 60 % of the residues, while within the top three predictions, the correct assignments were found for 80 % of the residues. PLUQ and the chemical shift maps are expected to be useful at the first stage of sequential assignment, for combination with automated sequential assignment programs, and for highly disordered proteins for which secondary structure analysis is the main goal of structure determination.
Keywords: Chemical shift correlation, Amino-acid type assignment, PLUQ, Secondary structure, Protein resonance assignment
Introduction
Assignment of 13C and 15N chemical shifts is the main bottleneck in structure determination of extensively labeled proteins by solid-state NMR (SSNMR) (Hong 2006; Hong et al. 2012; McDermott 2009). Backbone chemical shifts are the foundation for obtaining secondary structure information (Shen et al. 2009; Wishart and Sykes 1994; Wishart et al. 1991), while both backbone and sidechain chemical shifts are necessary for distance extraction. Recent progress in ab initio structural modeling methods (Rohl et al. 2004) has made chemical shift assignment even more important for small proteins. Now chemical shifts alone, without distance constraints, can be sufficient for generating high-resolution structural models (Shen et al. 2008).
Two types of automated assignment programs have been available in solution NMR for some time. The first deals with the assignment of nuclear Overhauser effect (NOE) cross peaks after sequence-specific assignment has been made (Herrmann et al. 2002; Linge et al. 2003; Vranken et al. 2005), while the second type produces sequential resonance assignment (Eghbalnia et al. 2005; Zawadzka-Kazimierczuk et al. 2012; Zimmerman et al. 1997). The latter programs require narrow linewidths, which are available mostly for small proteins in solution. Under these conditions, automated chemical-shift assignment is generally quite successful, with accuracies of ~90 %. However, for larger proteins in solution and most proteins in the solid state, which have large linewidths, the spectra are almost always assigned manually. In solid-state NMR, manual assignment usually begins with amino-acid type assignment of 2D 13C–13C correlation spectra, followed by sequential assignment of several 3D 15N–13C–13C correlation spectra. While experienced NMR spectroscopists generally have a knowledge base of the typical chemical shifts of each amino acid type, it remains common for spectroscopists to refer to tabulated amino acid chemical shifts (Ulrich et al. 2008; Wang and Jardetzky 2002; Ye et al. 1993) to make the initial amino-acid type assignment. Such chemical shift tables are inconvenient to use and provide limited information about the range of chemical shifts compatible with a given secondary structure. In principle, the best way to utilize the large knowledge base of protein 13C and 15N chemical shifts is to plot the chemical shifts in correlation maps analogous to experimental multidimensional spectra. A large number of chemical shifts have been deposited into the Biological Magnetic Resonance Databank (BMRB) (Ulrich et al. 2008), but it has been difficult to access these chemical shifts in their entirety. Very recently, a relational database management system, PACSY (Lee et al. 2012), was introduced to link the chemical shifts in BMRB (Markley et al. 2008; Ulrich et al. 2008) to structural information in the Protein Data Bank (PDB). The PACSY database can be queried using the Structured Query Language (SQL) or the PACSY Analyzer program. However, no tools are yet available for visualizing the chemical shifts in the database or for analyzing the query results.
In this work, we show 2D 13C–13C correlation maps of all 20 amino acids constructed using PACSY, to facilitate amino-acid type assignment of 2D and 3D solid-state NMR spectra. We describe interesting spectral features and conformation dependence of chemical shifts obtained from these correlation maps. Based on the information in these correlation maps, we introduce a Python/SQL program, PLUQ (PACSYlite Unified Query), which uses input 13C and 15N chemical shifts to predict residue types along with their secondary structures. The predicted amino-acid type and secondary structures are ranked according to their likelihood in the database. We describe the implementation of PLUQ and the accuracy of its assignment using four well-studied proteins.
Methods
PACSYMakerQT (Lee et al. 2012) was used to build a PACSY database on December 12, 2012. From this parent database, a smaller database called PACSYlite was derived using SQL. PACSYlite contains 13C and 15N chemical shifts of 4,406 unique proteins and 262,209 residues and is devoid of atomic coordinates. All 13C chemical shifts are referenced to DSS. PACSYlite also contains the STRIDE (Heinig and Frishman 2004) assigned secondary structure for each residue, using the first structural model in the deposited protein structure ensemble in the PDB. We chose only the first structure model in the ensemble to keep the derived database size manageable, and because the first structure is normally the lowest-energy structure. The database structure of PACSYlite is identical to that of PACSY, so that PLUQ can be used to query the full database, if desired, by altering only two lines of the Python code. The PACYlite database is stored in SQLite3, and occupies ~192 MB of disk space. It can be accessed through all modern operating systems without the need to rebuild the database. A list of the number of residues for each amino acid and secondary structure type in PACSY-lite is given in Table 1.
Table 1.
Number of residues in each secondary structure for the 20 amino acids used in the PACSYlite database
| Res | Helix | Sheet | Coil | Total |
|---|---|---|---|---|
| Ala | 8,462 | 3,064 | 7,123 | 18,649 |
| Cys | 1,224 | 1,297 | 2,187 | 4,708 |
| Asp | 4,441 | 1,893 | 9,005 | 15,339 |
| Glu | 8,862 | 3,590 | 7,824 | 20,276 |
| Phe | 3,358 | 3,387 | 2,879 | 9,624 |
| Gly | 2,209 | 2,314 | 14,470 | 18,993 |
| His | 1,844 | 1,253 | 2,915 | 6,012 |
| Ile | 4,914 | 5,323 | 3,712 | 13,949 |
| Lys | 6,992 | 3,607 | 8,119 | 18,718 |
| Leu | 9,832 | 5,765 | 7,150 | 22,747 |
| Met | 2,293 | 1,136 | 2,307 | 5,736 |
| Asn | 2,913 | 1,353 | 6,452 | 10,718 |
| Pro | 1,366 | 1,011 | 8,795 | 11,172 |
| Gln | 4,765 | 1,984 | 4,283 | 11,032 |
| Arg | 4,987 | 2,900 | 5,323 | 13,210 |
| Ser | 4,246 | 3,000 | 10,085 | 17,331 |
| Thr | 3,367 | 4,084 | 6,709 | 14,160 |
| Val | 5,287 | 7,668 | 5,367 | 18,322 |
| Trp | 1,077 | 1,112 | 1,023 | 3,212 |
| Tyr | 2,590 | 2,925 | 2,786 | 8,301 |
Total Number of Proteins = 4,406, Total Number of Residues = 262,209
To create 2D 13C–13C correlation maps, we used a MATLAB script to generate a chemical shift matrix for each amino acid. The script reads comma-separated-values files dumped from the PACSYlite database. A matrix with 0.1-ppm bins was populated with the chemical shift data by creating correlations for all atoms within three bonds. To find the correlations within three bonds, we represented the carbons and nitrogens of each amino acid in a bond distance matrix whose elements are the numbers of bonds between the two atoms of the specific column and row. If a matrix element (or bond number) is smaller than three, then a correlation peak is made between the corresponding chemical shifts.
The number of times a certain combination of chemical shifts in a 0.1 × 0.1 ppm bin is found in the database gives the intensity of each bin. Since these discrete intensity distributions are difficult to represent as contour plots, we broadened the delta-function peaks by Gaussian convolution to give peaks with a 0.3-ppm full width at half maximum. For each chemical shift map, eight equally spaced contour levels were plotted. The lowest contour level, which varies for different amino acids, was chosen to include at least two data points per 0.1-ppm bin, while the highest contour level was set to be 50 % of the maximum intensity of the most populated secondary structure category of the amino acid (Table 1). The chemical shift distribution of each secondary structure was separately broadened, color-coded, then superimposed in the 2D plot.
The PLUQ program queries the PACSYlite database using the SQLite3 Python module. The program searches for residues that contain the chemical shifts of interest within a default search range of ±0.25 ppm. If more than two chemical shifts are inputted, independent sub-queries are run for each pair of chemical shifts. The same residue type and secondary structure for the different sub-queries are grouped together, and the numbers of times they are found in the database are multiplied. By default, only atoms separated by no more than two bonds are considered in the assignment, but this bond number can be changed with an option to reflect experimental constraints. For example, 2D correlation spectra measured using through-bond J-couplings should use a bond number of 1.
Since some amino acids such as Glu and Ser have similar chemical shifts for multiple sites, and occasional mis-assignments exist in the database, it is possible to have more than one way of assigning a pair of chemical shifts within a residue. For example, when 58.8, 32.0, 33.4 and 18.0 ppm are inputted into PLUQ, the top two results are helical Met Cα–Cγ–Cβ–Cε and helical Met Cα–Cβ–Cγ–Cε. In other words, the PLUQ program makes sub-lists for each of the categories when the individual atom assignment is switched within a group.
PLUQ queries are fast. A typical evaluation of 3 or 4 chemical shifts takes less than a second on a computer with a 3.33 GHz core Intel processor and 12 GB of memory. PLUQ and PACSYlite are available online at http://www.public.iastate.edu/~hongweb/pluq.html. PLUQ can also be imported into other Python programs as a module.
Results
Secondary structure definition and construction of 2D correlation maps
The purpose of this work is to facilitate assignment of both amino acid type and the secondary structure. The secondary structure of the protein residues in the PACSY database (Lee et al. 2012) is defined using STRIDE. In addition to the canonical α-helix (H), β-sheet (E), and random coil (C) conformations, STRIDE also includes the less common structural motifs of π-helix (I), 310-helix (G), isolated β-bridge (B or b), and turn (T) (Heinig and Frishman 2004). These structural elements are statistically rare in the database, and when their chemical shifts are plotted separately, they do not cluster into defined regions of the spectra but are dispersed throughout. The lack of clear trends may reflect the complexity of some of these secondary structure elements such as turn (Creighton 1993). Therefore, we incorporated these minor structural categories into the broader categories as follows: helix includes H, G, I; sheet includes E, B, b and coil includes C and T. For all 2D 13C correlation maps, α-helical chemical shifts are shown in red, random-coil shifts in blue, and β-strand shifts in green.
Table 1 lists the statistics of the PACSYlite database. In total, 4,406 proteins and 262,209 residues are included in PACSYlite. These are selected from the full PACSY database of 7,395 proteins because they have 13C and 15N chemical shifts, while the remaining proteins only have 1H chemical shifts. In constructing the 2D correlation maps from PACSYlite, we initially found chemical shift correlations that were well outside the characteristic chemical shift regions. These were tracked to parsing errors in the original PACSY database, which were subsequently corrected by the authors of PACSY (Lee et al. 2012), With these modifications, few reported chemical shifts lie outside the characteristic chemical shift regions of the amino acids.
Since random coils contain a wide range of torsion angles, including those of β-sheet and α-helix, the coil chemical shifts have significant overlap with the chemical shifts of the other two structural motifs. To prevent masking the information in the correlation maps, we plotted the chemical shift maps so that the contour lines show through while the fill color represents the regions of the most populated secondary structure category. The lowest contour level is plotted with a thicker linewidth to make the range easily visible.
Representative 2D 13C–13C correlation maps of amino acid residues
Figure 1 shows expanded views of selected regions of the 2D13C–13C correlation maps of Ala, Gly, Phe and Arg. A complete set of 2D correlation maps for all 20 amino acids is shown in supplementary Figures S1-S20. In general, conformation dependence of the 13C chemical shifts is manifested as a tilt of the whole intensity pattern from the two axes of the spectra. For Ala, the Cα–Cβ correlation signals for helix and sheet can be distinguished unambiguously, but the coil chemical shift region overlaps with the helix and sheet regions. A similar situation is observed for the Gly CO–Cα correlation, except that the coil region covers a larger area, as expected. Interestingly, the helical and coil Ala chemical shifts show bimodal distributions. For the helical Ala, the main Cα–Cβ chemical shift peak is centered at 55 and 18 ppm, while a second Cα–Cβ peak is found at 53 and 15 ppm. The reason for this second correlation peak is unclear at present, since the average (ϕ, ψ) angles and their standard deviations, which are (−64.9 ± 11.5°, −39.6 ± 12.2°) for the major conformer and (−63.6 ± 13.3°, −40.6 ± 12.9°) for the minor conformer, do not differ significantly (Fig. S21). For coil Ala, a similar situation is observed, with the minor Cα–Cβ peak showing smaller chemical shifts than the major peak. Phe Cα–Cβ chemical shifts exhibit a narrow distribution with well-resolved helix and sheet regions and a relatively small coil chemical shift distribution. This profile differs from those of Trp and Tyr (Fig. S19 and S20), suggesting that Phe (ϕ, ψ) torsion angles are more tightly clustered than those of the other two aromatic amino acids. Figure 1d shows the CO–Cα correlation of Arg, which is representative of non-Gly amino acids: the helical and sheet signals are well separated and both overlap partially with the coil chemical shifts.
Fig. 1.

Selected regions of several 2D 13C-13C chemical shift correlation maps. a Ala Cα-Cβ. b Gly CO–Cα. c Phe Cα-Cβ. d Arg Cα-CO. The chemical shifts are color-coded in red for helix, green for sheet and blue for coil in this paper. The ranges of the linear contour levels are indicated for each panel. Although there are regions where secondary structures can be discerned unambiguously, the coil chemical shift range overlaps significantly with the helix and sheet chemical shift regions. Dashed boxes in a indicate second maxima in the helix and coil chemical shift distributions of Ala
Figures 2, 3, 4, 5 show 2D 13C–13C correlation maps of selected residues. To save space and allow comparison of chemical shifts of similar residues, we display two amino acids per figure, split along the diagonal. Figure 2 shows the 2D maps of the methyl-rich Ile and Val. Both residues have α-helical Cα chemical shifts of >60 ppm and methyl peaks around 20 ppm. However, specific differences exist. For example, the Cβ chemical shift distribution is narrower in Val than in Ile. This may be related to the narrower rotameric distribution of Val: 90 % of α-helical Val’s and 72 % of β-sheet Val adopt the trans χ1 rotamer, while the dominant mt rotamer (χ1 = −60°, χ2 = 180°) of Ile occurs in 81 % of helical Ile and 58 % of β-sheet Ile (Hong et al. 2009; Lovell et al. 2000). The Cα–Cβ cross peak of helical Val is usually resolved from the Cα–Cβ peaks of sheet and coil Val, but the sheet/coil Val Cα–Cβ peaks can overlap with helical Glu, Gln, Lys, Met (Figs. 4, 5) and coil Met (Fig. S11). To detect Val signals selectively, C–H spectral editing is an attractive possibility (Schmidt-Rohr et al. 2012). Figure 2 shows that sidechain Cγ1 and/or Cγ2 chemical shifts also have moderate conformation dependence. When the Cγ1 chemical shift is larger than 30 ppm for Ile or the Cγ1/2 shift is larger than 23.5 ppm for Val (shaded gray area), the secondary structure can be definitively assigned to helix. Again, this trend likely reflects backbone dependence of the sidechain rotamer populations. α-helical Val is dominated (90 %) by trans-rotamers (χ1 = 180°) while β-sheet Val contains a non-negligible percentage of the χ1 = −60° rotamer (20 %) (Lovell et al., 2000). The Cγ1/2 chemical shifts of Val depend on the χ1 angle (Hong et al. 2009) and thus indirectly depend on the backbone conformation. Comparison of all 20 correlation maps indicates that Ile Cδ and Cγ2 correlation signals are well resolved from all other amino acids and thus serve as unique identifiers of Ile.
Fig. 2.

2D 13C-13C correlation maps of Ile and Val. Gray bars denote the methyl Cγ1/2 chemical shifts that can be uniquely assigned to α-helical conformation. The values are greater than 30 ppm for Ile and greater than 23.5 ppm for Val
Fig. 3.

2D 13C-13C correlation maps of Leu and Pro. Note that the Pro chemical shifts are dominated by the coil conformation
Fig. 4.

2D 13C-13C correlation maps of Lys and Arg. Lys exhibits a characteristic pattern of three nearly equidistant Cα-sidechain correlation strips that are orthogonal to the ridge formed by the Cε correlation peaks. Note the bimodal distribution of β-sheet Arg Cα-Cβ chemical shifts
Fig. 5.

2D 13C-13C correlation maps of sidechain-carbonyl-containing Glu and Gln. Despite the similarity of their chemical shifts, the 36–40 ppm region is uniquely Glu Cβ (gray bars). Chemical shifts above 180 ppm generally result from Cδ
Pattern recognition of these chemical shift correlation maps gives information about the uniqueness of residue type assignment. By showing all cross peaks within three bonds, which can be readily measured using appropriate 13C spin diffusion mixing times, these 2D maps facilitate resonance assignment by making related correlation signals apparent. For example, combining the Leu map (Fig. 3) and the Asp map (Fig. S3), we can see that while a (54, 46) ppm cross peak can be either β-sheet Leu or β-sheet Asp Cα–Cβ, if the same 46-ppm frequency is correlated with signals between 20 and 30 ppm, then the peak must be assigned to Leu. Another example is Pro. Comparison of all correlation maps show that a peak within 2.5 ppm of (64.0, 50.0) ppm can be assigned to Pro Cα–Cδ more than 99.7 % of the time, thus can be used as a unique reporter of Pro. Figure 3 shows that the Pro chemical shift distribution is dominated by coil, whose chemical shift range fully encompasses that of helix and sheet; thus little conformational information can be obtained from Pro chemical shifts.
Figure 4 shows the 2D correlation maps of the cationic Lys and Arg. Arg has a characteristic Cα–Cδ correlation pattern with a narrow Cδ shift range centered at 43 ppm, and β-sheet Arg Cα–Cβ has a distinct bimodal distribution. For Lys, Cα correlates with the sidechain Cβ, Cδ and Cγ chemical shifts in an equi-distant pattern, and all these sidechain peaks further correlate with a narrow Cε ridge around 42 ppm.
Due to their structural similarities, the amino acid pairs Glu/Gln and Asp/Asn are difficult to distinguish by chemical shifts. Figure 5 shows that the 2D spectral patterns of Glu/Gln are almost identical and their chemical shifts are highly overlapped. Nevertheless, the 2D maps indicate that if a Glx Cβ chemical shift is larger than 36.2 ppm, then the peak can be definitively assigned to Glu. If this is not the case, one can distinguish Glu and Gln using the CN-free spectral editing experiment (Schmidt-Rohr et al. 2012), where the sidechain 15N of Gln can be used to suppress the Cδ signal through the 13C–15N dipolar interaction. As a result, the only peaks remaining in the carbonyl region will belong to Asp and Glu, which can be distinguished based on the Cβ–Cγ cross peak.
Many interesting conformational dependences of 13C chemical shifts can be discerned from these 2D correlation maps (Fig. S1-S20). These maps are useful for looking for expected cross peaks of a tentative assignment, for extracting unambiguous residue-type assignments, and for optimizing isotopic labeling patterns. If a peptide or protein is known to have a single conformation, then one should consider only the chemical shift distributions of that conformation.
Combining the correlation maps of all 20 amino acids, we ask which chemical shifts can be uniquely assigned to a single amino acid type, without using more than one correlation peak. Dividing the 5–75 ppm region of the 2D spectrum into bins of 1 × 1 ppm, and superimposing all 20 correlation maps, we find that Thr, Ser, Ala, Ile, Val, Leu and Arg have some well resolved and isolated chemical shifts (Fig. 6), while amino acids such as Met, Glu, Asn, and Lys have small areas of chemical shifts with only minor overlap. Figure 6 can thus be used for rapid type assignment in these regions of the 2D 13C–13C correlation spectra.
Fig. 6.

13C chemical shift regions that can be uniquely assigned to a single residue type. PLUQ was run for 1 × 1 ppm bins within 5–75 ppm using the “all” options, which give amino acid types irrespective of the secondary structure. A bin was filled if a single residue type was found or if the most probable residue type is more than 10 times more likely than the second most likely amino acid. Only bins with more than 10 data points were considered, to eliminate outliers in the database
PLUQ assignment of residue type and secondary structure
The 2D correlation maps exhibit the rich information content of the PACSYlite database. Similar statistical information about 15N chemical shift distribution is also available, although we do not consider them here. Since manual amino-acid type assignment requires a probabilistic judgment of the likelihood of a certain set of correlated chemical shifts belonging to a residue type, we need to combine the statistics of all 20 correlation maps. To achieve this, we created the PLUQ program, which gives tentative assignment of the amino acid type and the associated secondary structure based on input 13C and 15N chemical shifts. Figure 7 shows screenshots of three representative PLUQ queries, where two or three chemical shifts are inputted. When two chemical shifts are inputted, which is the minimum number, the output assignment is ranked according to the number of times that the pair of chemical shifts is found in the database. Figure 7a shows the query result for (46.5, 53.5) ppm: the assignment with the highest number of hits is β-sheet Leu Cβ–Cα, followed by coil Leu, whose number of hits is only ~10 % of the first ranked assignment. The next most probable amino acid type is Asp, but the probability is only ~5 % that of Leu.
Fig. 7.
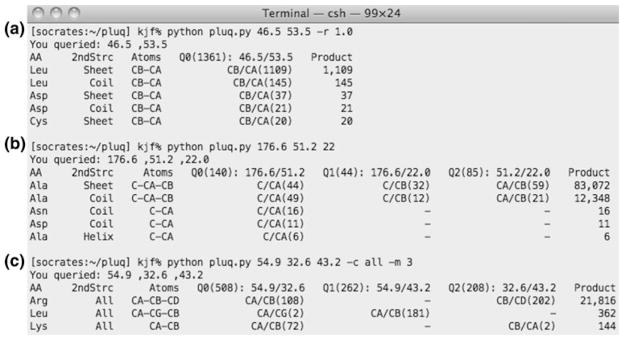
Representative PLUQ queries. (a) Cβ and Cα chemical shifts of a β-sheet Leu with a chemical shift search range (the “-r 1.0” option) of ± 1.0 ppm. (b) CO, Cα and Cβ chemical shifts of a beta-sheet Ala. (c) Cα, Cβ and Cδ chemical shifts of Arg, where all secondary structures were grouped together using the “-c all” option and only the top three most probable amino acid types were outputted using the “–m 3” option. Note that Arg Cα - Cδ assignment was not outputted because the default bond number was 2. It can be changed to 3 using the option “-b 3”
When three or more correlated chemical shifts are inputted, PLUQ outputs the assignments of pairwise combinations of the chemical shifts. If the sub-queries give the same residue type and secondary structure in the highest rank, then the product of the numbers of times each pair of chemical shift is found in the database is used to represent the score of the first rank. The next most probable assignments are scored in the same way. For example, for input chemical shifts of 175.6, 51.2, and 22.0 ppm (Fig. 7b), the sub-query of (175.6, 51.2) ppm was assigned to β-sheet Ala (CO, Cα) 53 times, the (175.6, 22.0) ppm sub-query was assigned to β-sheet Ala (CO, Cβ) 41 times, and the (51.2, 22.0) ppm peak was assigned to β-sheet Ala (Cα, Cβ) peak 59 times. The product of these three sub-queries, 128,207, is the score for the β-sheet Ala assignment. The second ranked assignment is coil Ala, which has a much lower score of 12,012. Thus, we can confidently conclude that these three chemical shifts most likely result from β-sheet Ala. Since most manual resonance assignment protocols do not directly involve secondary structure, the PLUQ information content is much higher. In the Ala case, if only amino acid type is needed, then the top two choices can be grouped into one, as Ala.
The PLUQ scoring system is analogous but not identical to the joint probability. When no result is found for a specific sub-query, the score is implicitly assumed to be 1 instead of 0. This choice reduces the chance of mis-assignment due to erroneous grouping of multiple chemical shifts by the user, which is easy to happen in spectra with broad linewidths. The scores are not normalized but simply reflect the number of times each pair of chemical shift is found in the database. Since the database sizes differ for different amino acids (Table 1), it is best to compare the relative scores within a query rather than between different queries.
The PLUQ program by default restricts pairwise correlations of chemical shifts to 2 bonds and uses a chemical shift search range of ±0.25 ppm. Unless otherwise noted, for all queries shown in this work, we used the default “strc” option of PLUQ to combine structural subcategories into the three broad categories of helix, sheet and coil. These options can be modified if desired.
Clearly, the more chemical shifts that are inputted per spin system, the higher the PLUQ scores. However, if many chemical shifts are known for a spin system, then it is also easy to assign the spin system manually. Therefore, in this work we consider PLUQ queries of only two and three chemical shifts, which represent the realistic situations of type-assigning a single peak in a 2D spectrum, two correlated peaks in a 2D spectrum, or a single peak in a 3D spectrum.
We tested the accuracy of PLUQ assignment on four proteins with high-quality structures and manually assigned chemical shifts deposited in the BMRB. These proteins include the mixed-conformation GB1 (Franks et al. 2005), the predominantly β-sheet VDAC (Hiller et al. 2008) and Het-S proteins (Wasmer et al. 2008), and the predominantly α-helical type-III needle protein (Loquet et al. 2012). For each protein we queried the Cα–Cβ chemical shifts and CO–Cα–Cβ chemical shifts, and compared the PLUQ output with both the manual assignment result and the STRIDE-determined secondary structure. For GB1, VDAC and Het-S, the STRIDE definition of secondary structure matched the experimental results. However, for the type-III needle protein, STRIDE did not indicate the α-helical nature of the protein for unknown reasons, thus we used the DSSP-based secondary structure definition instead (Kabsch and Sander 1983). Table 2 shows the PLUQ output for GB1 using Cα and Cβ chemical shifts. In total, 48 % of the peaks are correctly assigned to both amino acid type and secondary structure in the first ranked result. This is a remarkable success rate, since no other chemical shifts were used and a single 13C–13C 2D spectrum was sufficient to give this result. For the other peaks, 25 % had the correct amino acid type and secondary structure in the second ranked result. Table 2 shows that incorrect prediction in the first rank usually occurs for structurally similar amino acid pairs such as Tyr/Phe, Glu/ Gln, and Asp/Asn. In a few places when the PLUQ-predicted secondary structure differed from the STRIDE-defined secondary structure, there is reasonable doubt about the STRIDE definition. For example for residue 7, PLUQ predicted a coil Leu as the first choice, while STRIDE assigned Leu7 as β-sheet. Since residues 8–13 exhibit coil/ turn structure in the protein, the inconsistent assignment may be related to the question of which structural parameters best reflect the conformation. If we consider only the correct residue-type assignment, then the percentage of the correct prediction in the first rank increases to 56 %. The queried database does contain the four proteins tested here. However, these proteins represent only a very small fraction of all residues in the database, thus the effect of this inclusion on the test result is very small.
Table 2.
PLUQ assignment based on the Cα–Cβ chemical shifts of GB1 (Franks et al. 2005)
| Residue No. | Amino Acid | Secondary structure | Result #1 | Result #2 | Result #3 |
|---|---|---|---|---|---|
| 1 | Met | Coil | Lys-C(131) | Glu-S(85) | Gln-S(58) |
| 2 | Gln | Sheet | Arg-C(159) | Glu-C(147) | His-C(73) |
| 3 | Tyr | Sheet | Phe-S(183) | Leu_C(107) | Tyr-S(26) |
| 4 | Lys | Sheet | Glu-S(183) | Lys-S(107) | Glu-C(98) |
| 5 | Leu | Sheet | Asp-C(57) | Leu-S(55) | Leu-C(40) |
| 6 | Ile | Sheet | Glu-H(46) | Tyr-H(25) | Phe-H(23) |
| 7 | Leu | Sheet | Leu-C(124) | Leu-S(44) | Leu-H(30) |
| 8 | Asn | Coil | Asn-C(124) | Asn-S(9) | Asp-C(7) |
| 10 | Lys | Turn | Lys-H(206) | Lys-C(73) | Met-H(63) |
| 11 | Thr | Turn | Thr-C(259) | Thr-S(259) | Thr-H(12) |
| 12 | Leu | Turn | Leu-C(112) | Leu-S(48) | Leu-H(21) |
| 13 | Lys | Coil | Asn-C(441) | Asn-H(45) | Asp-C(28) |
| 15 | Glu | Sheet | Glu-S(66) | Gln-S(56) | Gln-C(28) |
| 16 | Thr | Sheet | Thr-C(45) | Thr-S(35) | Ile-C(1) |
| 17 | Thr | Sheet | Thr-S(26) | Thr-C(18) | - |
| 18 | Thr | Sheet | Thr-S(94) | Thr-C(76) | Thr-H(4) |
| 19 | Glu | Sheet | Lys-C(32) | Gln-S(26) | Arg-C(26) |
| 20 | Ala | Coil | Ala-S(49) | Ala-C(10) | Arg-C(8) |
| 21 | Val | Coil | Pro-C(710) | Val-C(51) | Val-S(47) |
| 22 | Asp | Coil | Asp-C(59) | Leu-S(36) | Asp-S(36) |
| 23 | Ala | Helix | Ala-H(400) | Ala-C(138) | Asp-H(2) |
| 24 | Ala | Helix | Ala-H(370) | Ala-C(135) | Asp-H(2) |
| 25 | Thr | Helix | Thr-H(45) | Thr-H(9) | Thr-C(2) |
| 26 | Ala | Helix | Ala-H(408) | Ala-C(16) | Ala-S(1) |
| 27 | Glu | Helix | Glu-H(468) | Gln-H(116) | Glu-C(75) |
| 28 | Lys | Helix | Lys-H(162) | Val-C(92) | Met-H(29) |
| 29 | Val | Helix | Val-H(267) | Pro-H(60) | Met-H(29) |
| 30 | Phe | Helix | Glu-C(25) | Tyr-C(15) | Phe-H(12) |
| 31 | Lys | Helix | Lys-H(99) | Val-C(18) | Arg-H(16) |
| 32 | Gln | Helix | Glu-H(342) | Gln-H(153) | Glu-C(66) |
| 33 | Tyr | Helix | Tyr-H(24) | Ile-S(17) | Val-S(11) |
| 34 | Ala | Helix | Ala-H(173) | Ala-C(13) | Val-C(2) |
| 35 | Asn | Helix | Asp-H(66) | Asp-C(56) | Phe-C(39) |
| 36 | Asp | Helix | Asn-H(144) | Leu-H(21) | Asp-H(19) |
| 37 | Asn | Helix | Asp-C(143) | Asn-C(63) | Leu-C(25) |
| 39 | Val | Coi | Pro-C(73) | Val-C(58) | Val-S(44) |
| 40 | Asp | Coi | Asp-C(80) | Leu-C(60) | Asp-S(25) |
| 42 | Glu | Sheet | Glu-S(59) | Arg-C(51) | Met-C(36) |
| 43 | Trp | Sheet | Lys-C(83) | Gln-H(47) | Gln-C(51) |
| 44 | Thr | Sheet | Thr-S(12) | Thr-C(8) | Val-C(1) |
| 45 | Tyr | Sheet | Leu-H(257) | Leu-C(33) | Asp-H(16) |
| 46 | Asp | Sheet | Asp-C(8) | Asp-S(6) | Leu-C(5) |
| 47 | Asp | Turn | Leu-C(105) | Leu-S(45) | Leu-H(28) |
| 48 | Ala | Turn | Ala-H(111) | Ala-C(78) | Ala-S(2) |
| 49 | Thr | Turn | Thr-C(38) | Thr-S(32) | Thr-H(5) |
| 50 | Lys | Turn | Arg-S(76) | Leu-S(38) | Arg-C(38) |
| 51 | Thr | Sheet | Thr-S(24) | Thr-C(16) | Thr-H(3) |
| 52 | Phe | Sheet | Phe-S(59) | Leu-C(41) | Tyr-S(37) |
| 53 | Thr | Sheet | Thr-S(147) | Thr-C(61) | Thr-S(1) |
| 54 | Val | Sheet | Lys-H(147) | Lys-C(92) | Cys-C(49) |
| 55 | Thr | Sheet | Thr-S(47) | Thr-C(11) | - |
| 56 | Glu | Coil | Gln-H(79) | Lys-C(68) | Lys-H(32) |
Bold green indicates correct assignment of both amino acid type and secondary structure in the first ranked result. Italic blue indicates the correct amino acid and secondary structure assignment as the second ranked result. Orange indicates an additional correct assignment using switched Cβ and Cα frequencies for Thr. Purple indicates the correct amino acid type assignment but incorrect secondary structure assignment in the top three results, which is rare
Table 3 summarizes PLUQ statistics for all four proteins. When only Cα and Cβ chemical shifts were queried, 41–60 % of the first ranked results gave the correct amino acid type as well as secondary structure. When the top three results were combined, PLUQ was correct 73–82 % of the time. When three chemical shifts CO–Cα–Cβ were used in the query, then the first choice was correct 57–61 % of the time, and within the top three results 77–85 % contained the correct assignment. The more α-helical type-III needle protein and GB1 have significantly better assignment statistics than the more β-sheet VDAC and Het-S proteins. This can be attributed to the fact that α-helical residues have narrower chemical shift distributions than sheet and coil residues. Thus, the well-known difficulty of resolving the chemical shifts of α-helical proteins is an advantage for database-driven amino acid type assignment.
Table 3.
Percentages of residues with the correct PLUQ assignment of both residue type and secondary structure for several proteins, using Cα–Cβ chemical shifts and CO–Cα–Cβ chemical shifts in the BMRB
| Protein | Protein: BMRB ID, PDB ID | Cα–Cβ | CO/N–Cα–Cβ |
|---|---|---|---|
| GB1 | BMRB-15156, PDB-2GI9 | 48 %, 75 % | 60 %, 82 % |
| VDAC | BMRB-16381, PDB-2K4T | 41 %, 73 % | 57 %, 77 % |
| Het-S | BMRB-11064, PDB-2LBU | 43 %, 79 % | 61 %, 84 % |
| Type III needle | BMRB-18276, PDB-2LPZ | 60 %, 82 % | 58 %, 85 % |
The VDAC BMRB entry does not contain CO chemical shifts; thus the amide 15N chemical shifts were used instead. In each column, the first number is the percentage of the correct assignment in the first ranked results, and the second number is the percentage of the correct assignment among the top three results
Discussion
The rapid and unbiased assignment of amino acid types by PLUQ dovetails semi-automated sequential assignment strategies such as the recently introduced Monte Carlo simulated annealing assignment method (Hu et al. 2011a, b; Tycko and Hu 2010). This algorithm requires input peak lists with tentative amino-acid type assignments. If the tentative type assignments are incomplete, then sequential assignment will fail for the spin system of interest. At the same time, the input peak list should not include highly unlikely type assignments. If a large number of type assignments are included for each spin system, while the program can find a high-scoring sequential assignment, the number of independent sets of assignments that agree with the input will increase artificially, and many assignments will have similar scores, making it difficult to distinguish among the possibilities (Hu et al. 2011a, b).
To decide how many PLUQ-prediction results to retain as input for subsequent sequential assignment, the user should evaluate the PLUQ scores carefully. In general, if the first ranked result has a much higher score than the second and other lower ranked results, then it is reasonable to retain only the first ranked result. For example, for GB1, the predictions of Asn8 and Ala26 based on Cα–Cβ chemical shifts gave first-ranked scores that are an order of magnitude larger than the second ranked scores (Table 2), indicating high confidence of the first prediction. For the (54.9, 32.6, 43.2) ppm spin system shown in Fig. 7c, the first ranked result, Arg, is scored 60 times more likely than the second ranked result of Leu, thus it is reasonable to keep only the Arg result. On the other hand, if the scores of the top few predictions are similar, as seen, for example, for Leu5, Glu15, Thr18, and Asn35 in GB1 (Table 2), then it is important to include all these predictions until the point where a significant drop in the score is reached. The exact ratio between the lowest score of the retained predictions and the highest score of the discarded predictions is best kept empirical at this point, and may be adjusted based on how sequential assignment turns out. If the sequential assignment gives questionable outcomes or low-quality structures, then the type assignments may have been too exclusive, i.e. the score ratio between the retained and discarded predictions may have been set too low, and the user should include more type-assignment outputs from PLUQ.
Although our analysis and discussion here focus on 2D 13C–13C correlation spectra, PLUQ queries and the correlation maps can also be used to help assign 3D NCC spectra, where the 15N chemical shift dimension serves to reduce the resonance overlap in the 2D 13C–13C planes. 15N chemical shifts can also be directly inputted into PLUQ to assign a spin system.
The secondary structure information obtained from PLUQ is complementary to existing chemical-shift based structure prediction programs. One of the secondary structure prediction programs commonly used by solid-state NMR spectroscopists is the TALOS+ program, which requires chemical shifts of three consecutive residues to predict the structure of the central residue (Cornilescu et al. 1999; Shen et al. 2009). However, often the neighboring residues’ chemical shifts are unknown, due to site-specific labeling of a chemically synthesized peptide, amino-acid-type labeling of an expressed protein, or missing chemical shifts in the multidimensional NMR spectra. PLUQ allows secondary structure to be predicted for scattered uniformly labeled residues, thus giving preliminary conformation information that can guide further experiments. Moreover, TALOS and other programs such as chemical shift index (Wishart and Sykes 1994) give secondary structure information only after resonance assignment, while PLUQ predicts amino acid assignment as well as secondary structure. Very recently, a program called RESPLS (Ikeda et al. 2013) was introduced to obtain secondary structure of proteins by using nine-residue peptide fragments with known Cα, Cβ and CO chemical shifts to fit broad-line experimental 2D spectra. PLUQ differs from this method by making explicit residue-type assignments without homology modeling.
Conclusions
We have introduced and demonstrated a robust program to assign the amino acid type and secondary structure of proteins based on correlated 13C (and 15N) chemical shifts measured in multidimensional MAS solid-state NMR spectra. PLUQ takes advantage of the large number of experimentally determined 13C and 15N chemical shifts in the PACSY database and the correlations between these chemical shifts. 2D 13C–13C correlation maps generated from this database show interesting conformation dependence of the 13C chemical shifts. They are more comprehensive and direct to use than chemical shift tables for amino-acid type assignment, since they allow visual inspection and pattern recognition. The PLUQ program queries the database to predict the amino acid types and secondary structures. For an input of only Cα and Cβ chemical shifts, PLUQ gives the correct assignment in 40–60 % of the first ranked results and in 73–82 % of the top three results. Inputting more chemical shifts increases the assignment accuracy. Spectral regions with nearly unique assignments are identified. This combined aminoacid type and secondary-structure assignment approach is expected to be particularly useful for disordered proteins, and can be used in combination with sequential assignment programs to facilitate protein structure determination by solid-state NMR.
Supplementary Material
Acknowledgments
This research was supported by NIH grant GM088204. We are grateful to Mr. Wonghee Lee for correcting parsing errors in the original PACSY database.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s10858-013-9732-z) contains supplementary material, which is available to authorized users.
References
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Creighton TE, editor. Proteins: Structures and molecular properties. W.H. Freeman and Co; New York: 1993. [Google Scholar]
- Eghbalnia HR, Bahrami A, Wang LY, Assadi A, Markley JL. Probabilistic identification of spin systems and their assignments including coil-helix inference as output (PISTACHIO) J Biomol NMR. 2005;32:219–233. doi: 10.1007/s10858-005-7944-6. [DOI] [PubMed] [Google Scholar]
- Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM. Magic-angle spinning solid-state NMR spectroscopy of the beta1 immunoglobulin binding domain of protein G (GB1): 15 N and 13C chemical shift assignments and conformational analysis. J Am Chem Soc. 2005;127:12291–12305. doi: 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;39:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M. Solid-state NMR studies of the structure, dynamics, and assembly of beta-sheet membrane peptides and alpha-helical membrane proteins with antibiotic activities. Acc Chem Res. 2006;39:176–183. doi: 10.1021/ar040037e. [DOI] [PubMed] [Google Scholar]
- Hong M, Mishanina TV, Cady SD. Accurate measurement of methyl 13C chemical shifts by solid-state NMR for the determination of protein sidechain conformation: the influenza M2 transmembrane peptide as an example. J Am Chem Soc. 2009;131:7806–7816. doi: 10.1021/ja901550q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhang Y, Hu F. Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem. 2012;63:1–24. doi: 10.1146/annurev-physchem-032511-143731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu KN, McGlinchey RP, Wickner RB, Tycko R. Segmental polymorphism in a functional amyloid. Biophys J. 2011a;101:2242–2250. doi: 10.1016/j.bpj.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu KN, Qiang W, Tycko R. A general Monte Carlo/simulated annealing algorithm for resonance assignment in NMR of uniformly labeled biopolymers. J Biomol NMR. 2011b;50:267–276. doi: 10.1007/s10858-011-9517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Egawa A, Fujiwara T. Secondary structural analysis of proteins based on (13)C chemical shift assignments in unresolved solid-state NMR spectra enhanced by fragmented structure database. J Biomol NMR. 2013;55:189–200. doi: 10.1007/s10858-012-9701-y. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Lee W, Yu W, Kim S, Chang I, Lee W, Markley JL. PACSY, a relational database management system for protein structure and chemical shift analysis. J Biomol NMR. 2012;54:169–179. doi: 10.1007/s10858-012-9660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics. 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. Atomic model of the type III secretion system needle. Nature. 2012;486:276–279. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins: Structure Function, and Genetics. 2000;40:389–408. [PubMed] [Google Scholar]
- Markley JL, Ulrich EL, Berman HM, Henrick K, Nakamura H, Akutsu H. BioMagResBank (BMRB) as a partner in the Worldwide Protein Data Bank (wwPDB): new policies affecting biomolecular NMR depositions. J Biomol NMR. 2008;40:153–155. doi: 10.1007/s10858-008-9221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott AE. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Strauss CEM, Misura KMS, Baker D. Protein structure prediction using ROSETTA. Methods Enzymology. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Schmidt-Rohr K, Fritzsching KJ, Liao SY, Hong M. Spectral editing of two-dimensional magic-angle-spinning solid-state NMR spectra for protein resonance assignment and structure determination. J Biomol NMR. 2012;54:343–353. doi: 10.1007/s10858-012-9676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu GH, Eletsky A, Wu YB, Singarapu KK, Lemak A, Ignatchenko A, Arrow-smith CH, Szyperski T, Montelione GT, Baker D, Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R, Hu KN. A Monte Carlo/simulated annealing algorithm for sequential resonance assignment in solid state NMR of uniformly labeled proteins with magic-angle spinning. J Magn Reson. 2010;205:304–314. doi: 10.1016/j.jmr.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Wenger RK, Yao HY, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins-Structure Function and Bioinformatics. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. The C-13 Chemical-Shift Index - a Simple Method for the Identification of Protein Secondary Structure Using C-13 Chemical-Shift Data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- Ye CH, Fu RQ, Hu JZ, Hou L, Ding SW. C-13 Chemical-Shift Anisotropies of Solid Amino-Acids. Magn Reson Chem. 1993;31:699–704. [Google Scholar]
- Zawadzka-Kazimierczuk A, Kozminski W, Billeter M. TSAR: a program for automatic resonance assignment using 2D cross-sections of high dimensionality, high-resolution spectra. J Biomol NMR. 2012;54:81–95. doi: 10.1007/s10858-012-9652-3. [DOI] [PubMed] [Google Scholar]
- Zimmerman DE, Kulikowski CA, Huang YP, Feng WQ, Tashiro M, Shimotakahara S, Chien CY, Powers R, Montelione GT. Automated analysis of protein NMR assignments using methods from artificial intelligence. J Mol Biol. 1997;269:592–610. doi: 10.1006/jmbi.1997.1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.