

Abstract
RIP1 kinase is a key regulator of TNF-induced NFκB activation, apoptosis and necroptosis through its kinase and scaffolding activities. Dissecting the balance of RIP1 kinase activity and scaffolding function in vivo during development and TNF-dependent inflammation has been hampered by the perinatal lethality of RIP1-deficient mice. Here we generated RIP1 kinase-dead (Ripk1K45A) mice and showed they are viable and healthy, indicating that kinase activity of RIP1, but not its scaffolding function, is dispensable for viability and homeostasis. After validating that the Ripk1K45A mice were specifically protected against necroptotic stimuli in vitro and in vivo, we crossed these mice to SHARPIN-deficient cpdm mice, which develop severe skin and multi-organ inflammation that has been hypothesized to be mediated by TNF-dependent apoptosis and/or necroptosis. Remarkably, crossing Ripk1K45A mice to the cpdm strain protected against all cpdm-related pathology. Together, these data suggest that RIP1 kinase represents an attractive therapeutic target for TNF-driven inflammatory diseases.
Introduction
TNF is a pleiotropic cytokine that was first described as causing necrosis in tumors (1), but has since been recognized as a major driver of inflammation and human disease pathogenesis (2). TNF binding to its receptor, TNF receptor 1 (TNFR1), results in one of three distinct cellular fates, NFκB activation, apoptosis or necrosis. The serine/threonine kinase receptor-interacting protein 1 (RIP1) serves as a key decision checkpoint for these different fates through its function as a scaffolding protein and its kinase activity (3). Under conditions where RIP1 is highly ubiquitinated, TNFR1 engagement activates NFκB to promote gene transcription and cell survival. In situations where RIP1 is deubiquitinated, TNF initiates one of two programmed cell death pathways; apoptosis, or the recently discovered necroptosis pathway that requires conditions of caspase inhibition (4). In vitro experiments using the RIP1 kinase inhibitor Necrostatin-1 (Nec-1) have shown that RIP1 kinase activity is critical for TNF-induced necroptosis, but appears dispensable for NFκB activation and apoptosis (5). However, at present, the exact contribution of RIP1 kinase activity to viability, homeostasis and inflammation in vivo is unknown as RIP1-deficient mice die shortly after birth (6), and current pharmacological tools are unsuitable for chronic RIP1 inhibition (5).
TNFR1-mediated activation of NFκB is dependent upon the ubiquitination of RIP1 by the linear ubiquitin chain assembly complex (LUBAC), comprised of the enzymes HOIL, HOIP and SHARPIN (7). Cells lacking LUBAC function through deficiency of SHARPIN show no detectable NFκB activation following TNF stimulation, but are highly sensitive to TNF-induced apoptosis and RIP1-kinase dependent necroptosis (8). SHARPIN-deficient, chronic proliferative dermatitis (cpdm), mice develop a TNF-dependent multi-organ inflammatory pathology including a severe dermatitis (9). The in vitro data suggest that development of inflammation in cpdm mice is likely mediated by excessive TNF-dependent apoptosis and/or necroptosis. Both apoptosis and necroptosis have been implicated in driving inflammation in disease settings, and therefore the precise contributions of these cell death pathways in the pathogenesis of disease in the cpdm mouse remains unknown.
In this article, we generated RIP1 kinase-dead (Ripk1K45A) mice and showed that they are viable and healthy. Ripk1K45A mice showed a selective defect in TNF-induced necroptosis in vitro, and were resistant to a lethal TNF-dependent shock in vivo. Crossing Ripk1K45A mice with the cpdm line completely rescued the skin and tissue inflammatory pathology, highlighting the inflammatory potential of RIP1 kinase-dependent necroptosis and signaling in TNF-driven diseases.
Materials and Methods
Mice
RIP1 kinase-dead knock-in (Ripk1K45A) mice were generated as described in Figure 1A. Additional information is available upon request. cpdm mice were purchased from Jackson Laboratories. All animal procedures were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility at GlaxoSmithKline in accordance with the GlaxoSmithKline Policy on the Care, Welfare, and Treatment of Laboratory Animals, and were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at GlaxoSmithKline.
Figure 1.

Ripk1K45A mice are viable and lack pathology associated with the RIP1 KO mouse. (A) The Ripk1 gene-targeting vector was constructed from genomic C57Bl/6 mouse DNA. The K45A point mutation was inserted into Ripk1 exon 3 while a Neo cassette was inserted in intron 3 flanked by FRT sites for Flp-mediated excision. Exon 3 was flanked by loxP sites enabling access to its deletion through Cre-action. (B) Phospho-RIP1 ELISA analysis. Lysates were prepared from wt (black bars) or Ripk1K45A (white bars) macrophages (control), or cells stimulated with TNF or TNF and zVAD for 3 hours. Results are representative of two experiments. (C) Western blot analyses of RIP1 and RIP3 expression in the spleen, thymus, and lymph nodes (LN) from wt and Ripk1K45A mice. β-Actin is shown as a loading control. Data are representative of three experiments. (D) Histopathology analysis of spleen, thymus and white adipose tissue from 10 to 12 week old wt littermate and Ripk1K45A mice. Data are representative of three wt and 5 Ripk1K45A mice.
Preparation of bone marrow derived and peritoneal macrophages
Bone marrow-derived macrophages (BMDM) were prepared from bone marrow by differentiation with 10ng/ml M-CSF (R&D Systems). Thioglycolate-elicited peritoneal macrophages (TM) were obtained from mice that had been injected with 1.5ml 4% thioglycolate (Difco).
Cytokine and serum IgM analysis and Western blotting
Cytokines were measured using the Mouse Proinflammatory 7-Plex Ultra-Sensitive kit (Meso Scale Discovery). Serum IgM was measure by ELISA (eBioscience). To examine protein expression in tissues, lysates were run on SDS-PAGE and blotted onto nitrocellulose membranes (Invitrogen). Blots were probed for β-actin (Sigma), RIP1 (BD Biosciences) and RIP3 (Imgenex). For TNF activation experiments, BMDM were treated with 50ng/ml of TNF (R&D systems) and lysates were separated by SDS-PAGE and blotted onto a nitrocellulose membrane (Invitrogen). Blots were probed for IκB, phospho-IκB, tubulin, phospho-p38, total p38, phospho-JNK and total JNK (Cell Signaling), phospho-ERK and total ERK (Santa Cruz).
mRNA analysis
BMDM were treated for 2, 4, or 6 hours with TNF and zVAD. Total RNA was isolated using RNeasy mini kit (Qiagen). cDNA was generated using Taqman reverse transcription reagents (Invitrogen) and gene expression was evaluated with the Taqman PCR core reagent kit (Invitrogen) using the commercially available KC primer/probe set (Invitrogen, Mm 04207460_m1). Data were normalized to 18S rRNA.
Phospho-RIP1 ELISA
Phospho-RIP1 ELISAs were performed on lysates produced from TNF (20ng/ml) or TNF + zVAD (20μM, R&D Systems) stimulated TM using MULTI-ARRAY ® 96-well small spot plates (Meso-Scale Discovery) coated with anti-human RIP1 antibody (Abcam). Lysate samples were incubated, and phospho-RIP1 was detected with a novel rabbit anti-RIP1pS166 antibody (manuscript in preparation) and an anti-rabbit IgG, SULFO-tagged detection antibody (Meso-Scale Discovery). Plates were read on a Sector6000 and ECL counts were converted to ng using the standard curve.
Histology
Tissues for histologic examination were collected, immersion fixed, trimmed, decalcified (where applicable) until judged complete by palpation, and processed. Following fixation, tissue samples were embedded in paraffin wax, sectioned, stained with hematoxylin and eosin and examined by light microscopy.
In vitro analysis of cell death
Necroptotic and apoptotic cell death were induced with TNF (50ng/ml BMDM and 10ng/ml TM) and zVAD (50uM BMDM and 20uM TM) and TNF (50ng/ml) and cycloheximide (CHX; 12μg/ml, Sigma) treatment respectively. Cell death evaluation was performed 21 hours post stimulation by quantifying intracellular ATP levels using CellTiter-Glo Viability assay (Promega). Percent survival was calculated for each treatment (TNF treated cells were set to 100% survival). For Caspase 3/7 activity, cells were treated with TNF and CHX for 6 hours. Activity was measured using Caspase-Glo 3/7 assay (Promega). Data were normalized for protein levels. Supernatants were collected for cytokine analysis.
In vivo TNF shock
Mice were injected i.v. with 1.25mg/kg TNF (Cell Sciences) and 16.7mg/kg zVAD (Bachem). Temperature was monitored at regular intervals using a rectal probe. Mice were euthanized once a 7°C temperature loss from baseline was detected, in accordance with our IACUC protocol.
Cpdm mouse analysis
Cpdm mice and crosses were monitored by our veterinary staff and euthanized when the dermatitis was considered to be severe (covering ≥ 50% of the abdomen). Tissues were collected for histopathology.
Statistics
In vitro data are shown as mean +/− standard deviation. In vivo data are shown as mean +/− standard error of the mean and were analyzed using a one way ANOVA followed by Bonferroni’s post-hoc analysis. Cpdm dermatitis data were analyzed by Log-rank (Mantel-Cox) test. * represents a p-value of ≤ 0.05.
Results and Discussion
RIP1 kinase dead mice are viable and healthy
To study the role of RIP1 kinase activity in TNF signaling, we generated a kinase-dead knock-in (Ripk1K45A) mouse with a point mutation in the catalytic lysine (K45A) in exon 3 of the Ripk1 gene (Fig. 1A). Ripk1K45A mice were viable and born at the expected Mendelian ratios from interbreeding of heterozygous mice, and displayed normal litter sizes from homozygous breeding paradigms (data not shown). A similar RIP1 kinase-dead mouse with a D138N mutation was published while this manuscript was under review and was also shown to be viable and have a similar phenotype to our Ripk1K45A mice (10). This is in contrast to mice lacking RIP1 protein (RIP1 KO) that die one to three days after birth (6). We confirmed the previously reported RIP1 KO phenotype, as exon 3 of our Ripk1K45A construct was flanked by loxP sites (Fig. 1A), and no RIP1 KO homozygous mice were observed at weaning when knocking out the gene by crossing to a Cre/loxP-deleter transgenic line (data not shown).
To confirm that Ripk1K45A mice lacked kinase activity we used a newly generated antibody recognizing an autophosphorylation site on RIP1 (manuscript in preparation). Phospho-ELISA analysis detected RIP1 autophosphorylation in response to TNF and zVAD stimulation of cells from wt but not Ripk1K45A littermate controls (Fig. 1B). Despite lacking kinase activity, protein expression of RIP1, and its necroptosis inducing partner RIP3 (11), were normal in multiple tissues and cell types including spleen, thymus, lymph nodes and macrophages (Fig. 1C and data not shown). A thorough histopathological analysis was undertaken to explore if absence of kinase activity led to any pathologies despite the mice appearing grossly normal. Ripk1K45A mice were histologically indistinguishable from wild-type mice in any of the 20 tissues examined (Fig. 1D). This included organs where pathologies were observed in RIP1 KO mice when they were originally described (6). Additionally, no differences were observed in thymic double positive T cell populations between wild-type and Ripk1K45A mice (Supplemental Fig. 1). This contrasts to the defect in this population that was seen in RIP1 KO mice (6). By demonstrating that Ripk1K45A mice lacking RIP1 kinase activity are viable and healthy, we can attribute the perinatal lethality of the RIP1 deficient mice to an absence of RIP1 scaffolding function.
Ripk1K45A macrophages are selectively protected from TNF-induced necroptosis
We next set out to address which pathways downstream of TNFR1 were affected by the lack of RIP1 kinase activity in vitro. To this end, bone marrow derived (BMDM) and thioglycolate elicited macrophages (TM) were isolated from Ripk1K45A mice or wt littermate controls and stimulated with TNF and the caspase inhibitor zVAD to induce necroptosis. Ripk1K45A macrophages were completely protected from undergoing this form of cell death (Fig. 2A). In addition to being protected from cell death, Ripk1K45A macrophages showed diminished production of the chemokine KC as compared to wt controls (Fig. 2B), consistent with an emerging role for RIP1 kinase activity in the generation of inflammatory cytokines (12). Further investigation revealed that this RIP1-dependent cytokine production was likely to be regulated at the mRNA level, as wild-type mice showed roughly 2-fold higher levels of KC mRNA at 2 hours post stimulation, which preceded RIP1-dependence at the protein level which was not seen until 4 hours post stimulation (Fig. 2C).
Figure 2.

Macrophages from Ripk1K45A mice are protected from necroptosis, but not NFκB activation or apoptosis in vitro. (A) CTG viability analysis of BMDM or TM from wt (black bars) or Ripk1K45A (white bars) mice. Macrophages were left untreated (control) or stimulated with TNF and zVAD. (B) Measurement of KC release from BMDM treated as in (A). (C) KC mRNA and protein analysis from BMDM from wt or Ripk1K45A mice treated with TNF and zVAD for the indicated times (D) CTG viability analysis of BMDM from wt (black bars) or Ripk1K45A (white bars) mice. Macrophages were either left untreated (control) or stimulated with TNF or TNF and CHX for 21 hours. (E) Measurement of KC release from BMDM treated as in (D).
We next examined the effect of the absence of RIP1 kinase activity on TNF stimulation of the NFκB pathway and apoptosis. TNF stimulation resulted in an equal pro-survival signal (Fig. 2D) and cytokine production (Fig. 2E) in both the Ripk1K45A and wt control macrophages. Similarly, Ripk1K45A and wt control cells were equally susceptible to TNF and cycloheximide (CHX) induced apoptosis (Fig. 2D) and KC production (Fig. 2E). This lack of an effect on TNF-alone signaling was confirmed by examining TNF-driven IκB phosphorylation and degradation (Supplemental Fig. 2A) and p38, JNK or ERK activation out to 2 hours post TNF challenge (Supplemental Fig. 2B and data not shown). Similarly TNF and CHX induced caspase 3/7 activation was also unchanged (Supplemental Fig. 2C). Together, these results show that cells from the Ripk1K45A mice are selectively protected against RIP1 kinase-dependent cell death and signaling, in agreement with experiments using the RIP1 inhibitor Nec-1, as well as RIP3 deficient cells (5,13).
Ripk1K45A mice are protected from TNF and zVAD lethal shock
We next assessed the contribution of kinase activity in vivo. Previous work has shown that the administration of TNF and zVAD in vivo results in a shock like syndrome characterized by temperature loss, inflammatory cytokine production and death (14). This temperature loss has been shown to be partially rescued by blocking the necroptosis pathway through administration of the RIP1 kinase inhibitor Nec-1 or absence of the RIP3 protein (14). To follow up on these observations, TNF and zVAD were dosed to Ripk1K45A or wt control mice. wt mice lost 7°C on average three hours post injection, whereas Ripk1K45A mice were nearly completely resistant to the challenge, losing less than 1°C 3 hours post injection (Fig. 3), and surviving to termination of the study at 24 hours post challenge with no signs of morbidity (data not shown). The near complete protection from temperature loss in Ripk1K45A mice contrasts with only a partial protection in mice treated with Nec-1. This may be best explained by incomplete blockade of RIP1 kinase activity with Nec-1, as compounds with better potency and pharmacokinetics can completely block TNF and zVAD induced temperature loss (manuscript in preparation). Additionally, this discrepancy in the level of protection between the Ripk1K45A and RIP3 KO mice suggest that RIP1 kinase-dependent, RIP3-independent mechanisms may be involved in driving pathology in the model.
Figure 3.
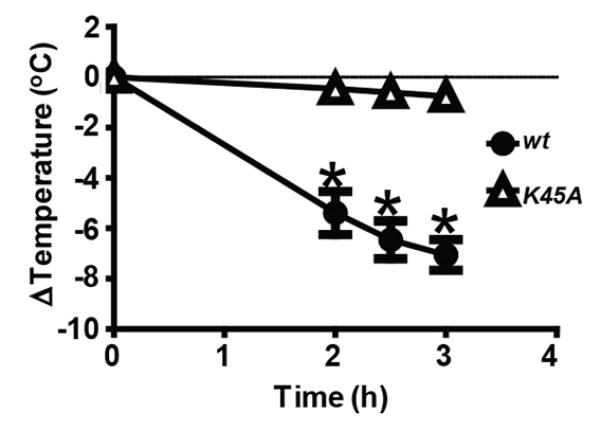
Ripk1K45A mice are protected from TNF and zVAD-induced shock. wt (closed circles) or Ripk1K45A (open triangles) mice were injected with TNF and zVAD. Temperature was monitored over three hours, and animals were euthanized after a 7°C loss. Data are representative of at least 3 experiments, each containing seven mice per group.
Ripk1K45A × cpdm cross is completely protected from inflammation
We next crossed the cpdm strain to the Ripk1K45A mice to understand the involvement of RIP1 kinase activity in driving the cpdm inflammatory phenotypes. cpdm mice develop a severe dermatitis beginning at 6-8 weeks of age (Fig. 4A). Ripk1K45A heterozygous SHARPIN-deficient mice showed a small, but significant delay in the onset of the severe dermatitis (Fig. 4B). Remarkably, cross of the cpdm to a Ripk1K45A homozygous background resulted in a complete protection from the dermatitis phenotype (Fig. 4A and B). In addition to the dermatitis, histopathological examination of the cpdm mice showed prominent inflammation in multiple tissues including skin, joint spaces, lung and liver (Fig. 4C). Histopathology changes consisted chiefly of multifocal to diffuse areas of inflammatory cell infiltration and edema, and localized epidermal ulceration and/or epidermal keratinocyte degeneration in the skin and inflammation of synovial membranes along with hyperplasia of the synovial epithelium in the joints (Fig. 4C). Additionally, lung inflammation was associated with interstitial peribronchiolar/peribronchial and perivascular areas, while multifocal inflammation with or without fibrosis was observed in the liver (Fig. 4C). Absence of RIP1 kinase activity protected against all cpdm-related inflammatory phenotypes at the histopathological level, with tissues being indistinguishable to those from non-cpdm mice (Fig. 4C). Furthermore, cpdm mice also display an increase in serum IgM levels compared to wild-type littermate controls that is completely reversed in the Ripk1K45A homozygous cpdm mice (Fig. 4D).
Figure 4.

RIP1 kinase activity drives inflammation in cpdm mice. (A) Representative photo of Ripk1K45A, cpdm and Ripk1K45A × cpdm mice at 7-8 weeks of age. (B) Cpdm and Ripk1K45A heterozygous mice were interbred to produce cpdm mice that were wt (WT/WT, black circles), heterozygous (WT/K45A, grey circles) or homozygous (K45A/K45A, white circles) for the Ripk1K45A gene. Dermatitis was considered to be severe (covering ≥ 50% of the abdomen). * indicates significantly different from RIP1 WT/WT (black circles) group. (C) Representative histology from cpdm or Ripk1K45A × cpdm mice at 7-8 weeks of age. Arrowheads in each panel point to, respectively, epidermal ulceration (black arrowhead) and dermal inflammation (red arrowhead) in skin, regions of inflammation in joint, regions of inflammation in lung and areas of inflammation in the liver. (D) Analysis of serum IgM levels.
Given the selective protection of cells from Ripk1K45A mice to TNF-induced necroptosis, this makes the complete rescue observed in the cpdm × Ripk1K45A mice that are sensitive to both TNF-mediated necroptosis and apoptosis surprising. This may highlight the potent inflammatory nature of necroptotic cell death, or alternatively, these results are consistent with emerging data suggesting that RIP1 kinase activity can also drive an apoptotic response under certain conditions (15). Together, these results show that the inflammatory phenotype of the cpdm mice is entirely RIP1 kinase-dependent. The absence of another LUBAC family member, HOIL, has been shown to cause autoinflammation and immunodeficiency in humans (16). Although these patients do not develop dermatitis, in some cases the inflammation was completely TNF-dependent (16). This makes it tempting to speculate that similarly to the SHARPIN-deficient mouse, many of the pathologies observed in HOIL-deficient patients are driven by aberrant RIP1 kinase activity.
The data in this paper highlight the utility of the Ripk1K45A mice as a tool to dissect the role of RIP1-dependent necroptosis and signaling in mediating inflammation driven by TNF and other factors that stimulate the RIP1 kinase pathway. Our data showing that RIP1 kinase activity completely rescues inflammation in the cpdm mouse add to the growing body of evidence that RIP1 kinase-dependent cell death and signaling is highly inflammatory (17). These data, coupled with the lack of pathology in the absence of RIP1 kinase signaling, makes RIP1 kinase an attractive target for the treatment of complex inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Drs. Amber Anderson and David Cooper for help with statistical analyses, Angela Dykon and Dr. Sean Maguire for help with animal care and Jon Renninger for his help in coordinating the histopathology.
Supported by N.I.H (PHS grants R01 AI20211 to E.S.M and DP1 OD012198 to W.J.K.).
Abbreviations used in the article
- BMDM
bone marrow derived macrophage
- CHX
cycloheximide
- Cpdm
chronic proliferative dermatitis
- CTG
CellTiter-Glo
- KO
knockout
- LUBAC
linear ubiquitin chain assembly complex
- Nec-1
Necrostatin-1
- RIP1
receptor interacting protein 1; RIP1
- TM
thioglycolate elicited macrophage
- TNFR
TNF receptor
- wt
wild-type
References
- 1.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U. S. A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death. Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 4.Vandenabeele P, Declercq W, Van HF, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 5.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 7.Emmerich CH, Schmukle AC, Walczak H. The Emerging Role of Linear Ubiquitination in Cell Signaling. Sci. Signal. 2011;4:re5. doi: 10.1126/scisignal.2002187. [DOI] [PubMed] [Google Scholar]
- 8.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 9.HogenEsch H, Gijbels M, Offerman E, van Hooft J, van Bekkum D, Zurcher C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am J Pathol. 1993;143:972–982. [PMC free article] [PubMed] [Google Scholar]
- 10.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 11.Chan FK, Baehrecke EH. RIP3 finds partners in crime. Cell. 2012;148:17–18. doi: 10.1016/j.cell.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNF[alpha] production. Cell Death Dis. 2012;3:e320. doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Duprez L, Takahashi N, Van HF, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Abhari BA, Cristofanon S, Kappler R, von SD, Humphreys R, Fulda S. RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene. 2012 doi: 10.1038/onc.2012.337. [DOI] [PubMed] [Google Scholar]
- 16.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israel L, Trevejo-Nunez G, Bogunovic D, Cepika AM, MacDuff D, Chrabieh M, Hubeau M, Bajolle F, Debre M, Mazzolari E, Vairo D, Agou F, Virgin HW, Bossuyt X, Rambaud C, Facchetti F, Bonnet D, Quartier P, Fournet JC, Pascual V, Chaussabel D, Notarangelo LD, Puel A, Israel A, Casanova JL, Picard C. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van LG, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.