

Abstract
Organophosphate (OP) compounds are used as insecticides, acaracides, and chemical agents and share a common neurotoxic mechanism of action. The biochemical alterations leading to many of the deleterious effects have been studied in neuronal cell lines, however, non-neuronal toxic effects of OPs are far less well characterized in vitro, and specifically in cell lines representing oral routes of exposure. To address this void, the human salivary gland (HSG) cell line, representing likely interactions in the oral cavity, was exposed to the representative OP paraoxon (PX; O,O-diethyl-p-nitrophenoxy phosphate) over a range of concentrations (0.01 μM to 100 μM) and analyzed for cytotoxicity. PX induced cytotoxicity in HSG cells at most of the exposure concentrations as revealed by MTT assay, however, the release of LDH only occurred at the highest concentration of PX tested (100 μM) at 48 h. Slight increases in cellular ATP levels were measured in PX-exposed (10 μM) HSG cells at 24 h. Exposing HSG cells to 10 μM PX also led to an increase in DNA fragmentation prior to loss of cellular membrane integrity implicating reactive oxygen species (ROS) as a trigger of toxicity. The ROS genes gss, gstm2, gstt2 and sod2 were upregulated, and the presence of superoxide following 10 μM PX exposure was determined via dihydroethidium fluorescence studies further implicating PX-induced oxidative stress in HSG cells.
Keywords: paraoxon, HSG cells, oxidative stress, organophosphate, MTT, DNA fragmentation
1. Introduction
Organophosphate compounds (OPs) are widely used to control insect pests but are also known as a chemical class used as warfare agents. Exposure to OPs can result in neurotoxicity (Ballantyne and Marrs, 1992; Casida and Quistad, 2004; Fukuto, 1990) that is typically attributed to the inhibition of acetylcholinesterase (AChE) (Scheme 1). In contrast to the warfare agents, OP insecticides must be converted from the less reactive thionate (P=S) into the oxon (P=O) form to inhibit AChE. When AChE is inhibited, an accumulation of acetylcholine (ACh) occurs resulting in an overstimulation of acetylcholine receptors (AChR’s) (Ballantyne and Marrs, 1992; Broomfield et al., 1995). OPs have also been shown to interact with a number of cholinergic and non-cholinergic protein targets (Bomser and Casida, 2000; Bomser and Casida, 2001; Ehrich et al., 1994; Proskocil et al., 2010; Quistad et al., 2001; Richards et al., 1999; Schuh et al., 2002).
Scheme 1.
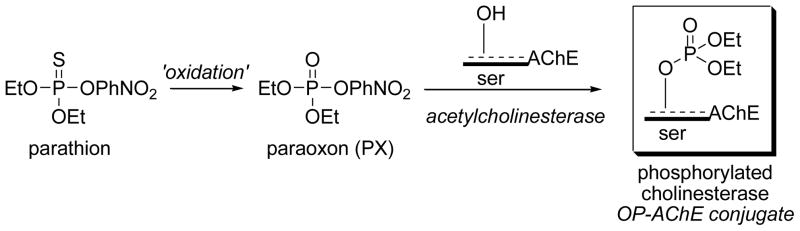
Oxidation of parathion to paraoxon and the inhibition of acetylcholinesterase by paraoxon to form an OP-AChE adduct.
The formation of oxons occurs during in vivo metabolic oxidation but also during the manufacture, storage, or environmental lifetime, for example in chlorinated water (Kamel et al., 2009). Therefore, direct exposure to OP oxons may occur prior to in vivo metabolism. Skin absorption is the most common pathway to OP exposure, yet more OP poisonings occur when pesticides enter the mouth. Food and water contaminated with OPs, airborne OPs (spraying or foggers), and hand to mouth transfer in infants/children (Chambers et al., 2007) each contribute to oral and inhalation exposures of parent and oxon forms of the insecticide.
A number of studies have examined the in vitro interactions between OPs and cell lines. Altered signaling pathways in neuronal and glial cells (Garcia et al., 2001; Hong et al., 2003; Qian et al., 2007; Schuh et al., 2002) adverse effects on mitochondrial integrity and ATP production (Hong et al., 2003; Knoth-Anderson et al., 1992; Massicotte et al., 2005), and an increase in stress response (Garcia et al., 2001; Sachana et al., 2001) have been reported. In PC-12 cells, chlorpyrifos exposure induced apoptosis via mitochondrial damage (Lee et al., 2012).
Paraoxon (PX) is the oxidative metabolite of parathion (Scheme 1), an inhibitor of AChE, and has been used as a model OP compound to investigate and identify toxicologically relevant targets in cell lines of neuronal origin (Bharate et al., 2010; Casida and Quistad, 2005; Pope, 1999; Richards et al., 1999) including the SH-SY5Y cell line (Bharate et al., 2010; Carlson and Ehrich, 1999; Ehrich et al., 1997; Prins et al., 2010; Saleh et al., 2003), and non-neuronal HepG2 cells (Hreljac et al., 2008). PX inhibits AChE to form a diethoxyphosphorylated serine residue (Scheme 1) that is identical to the OP-AChE conjugates formed from the oxidative metabolites of diazinon, phorate, chlorpyriphos, etc. PX inhibits the AChE present in SH-SY5Y cells within minutes at sub-micromolar levels but the loss of cell viability occurs at millimolar levels (24–48 h) suggesting deleterious biochemical mechanisms that may be downstream effects of AChE inhibition or may result from modification of proteins other than AChE (Ehrich et al., 1997). Likewise, protein expression changes occur when SH-SY5Y cells were treated with micromolar PX (Prins et al., 2010). However, none of these studies address possible interactions of OP compounds in the oral cavity.
In order to elucidate possible non-neuronal effects resulting from OP oral exposure, the human salivary gland (HSG) cell line was selected. The HSG cell line was derived from isolated epithelial cells from the irradiated submandibular salivary gland of a squamous cell carcinoma. This cell line is characterized as an epithelial duct cell type and expresses fibrinolytic activity (Shirasuna et al., 1981). HSG cells have been used in cellular response studies of toll-like receptor-mediated immune responses (Kawakami et al., 2007), growth factor stimulation of signal transduction pathways (Crema et al., 2006), and ATP-dependent activation of potassium channels (Liu et al., 1999). In the event of a human exposure to foodstuffs or aerosols of OP insecticides, the salivary glands would be a likely target in the oral cavity. Therefore, the HSG cell line was selected an appropriate model to study non-neuronal OP toxicity responses. In this study, HSG cells were exposed to paraoxon (Scheme 1), and select cytotoxic assays were conducted and validated by gene expression changes to assess toxicity resulting from low-level exposure.
2. Material and methods
2.1. Reagents and chemicals
Ethyl paraoxon was purchased (catalog number PS-610; Chem Service, Inc. West Chester, PA).
2.2. Culture of human salivary gland cells
The human salivary gland (HSG) cell line was established from an irradiated human salivary gland (Shirasuna et al., 1981), and was generously provided by Dr. Bruce Baum (National Institute of Health/National Institute of Dental and Cranial Facial Research (NIH/NIDCR). HSG cells were cultured using Dulbecco’s Modified Eagle’s medium and Ham’s F-12 nutrient mixture (DMEM/F12) (GIBCO BRL, Grand Island, NY) supplemented with 5% fetal bovine serum (FBS) (Hyclone; Thermo Scientific, Rockford, IL), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine in a CO2 incubator maintained at 5% CO2 and 37 °C. The medium was changed every two days, and cells were allowed to reach 80% confluence before exposure to PX.
2.3 Acetylcholinesterase activity in HSG cells
Total cellular proteins were harvested from HSG cells by lysis (150 mM NaCl, 1% Triton X-100, 50 mM Tris pH 8.0) and used immediately for analysis. Ellman assays (Ellman et al., 1961) were based on published reports, and were performed by incubation of crude protein lysates with 5,5-dithiobis(2-nitrobenzoic acid) (final concentration, 0.32 mM in 0.1 M potassium phosphate buffer, pH 7.6) and initiated with acetylthiocholine iodide (ATCh-I) (final concentration, 0.75 mM) in a final volume of 200 μl. (George et al., 2003). All kinetic assays performed under these conditions were linear with respect to time and corrected for protein concentration (Redinbaugh and Turley, 1986; Zhang and Halling, 1990). Negative controls were lysates plus DTNB (no ATCh-I). Changes in absorbance were measured at OD 412 nm on a VersaMax microplate reader with Softmax Pro V. 3.0 software. The optimum lengths of time for incubations were determined empirically to allow maximum changes in absorbance for samples versus blanks. Recombinant mouse AChE (rMoAChE) was used as a positive control for AChE activity. Ellman assays were performed in triplicate.
2.3. HSG cell exposure to PX
A 10 mM stock solution of PX was prepared in 100% ethanol and concentrations from 0.01 μM to 100 μM were prepared by dilution of the stock into DMEM/F12 medium (50:50), 1% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. Prior to PX treatment (24 h), the FBS in the HSG cell culture media was reduced from 5% to 1%. Culture media was replaced with PX culture media and allowed to incubate at 37°C for the various time points described below.
2.4. MTT and LDH cell viability assays
Effects of PX exposure on HSG cell viability was estimated using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and LDH (lactate dehydrogenase) release assays (Cell Proliferation Kit (MTT) and Cytotoxicity Detection Kit (LDH), Roche Applied Science, Indianapolis, IN). Cells were grown in 96-well culture plates to 80% confluence before exposure to PX. For both MTT and LDH assays, cells were exposed to PX concentrations (or ethanol alone as the vehicle control) ranging from 0.01 μM to 100 μM for 24, or 48 h with a 2% Triton X-100 solution in assay medium used as a positive control for cell death (all additions were at final v/v of 0.1%.
For the MTT assay, cells were rinsed several times with culture medium prior to PX exposure, the culture medium was removed and 100 μl of fresh medium containing the various PX concentrations or Triton X-100 was added to each well (n = 6–8 for each PX concentration and Triton X-100). At various incubation time points, 10 μl of the supplied MTT labeling reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to each well, incubated for 4 h, after which 100 μl of solubilizing solution was added to each well. Plates were incubated overnight and then measured at 575 nm (formazan) using a VersaMax microplate reader with Softmax Pro V. 3.0 software. Cytotoxicity was determined by comparing the absorbance readings of the wells containing the PX-treated cells with those of the vehicle- (0.1% ethanol) treated cells.
For the LDH assay, cells were rinsed several times with culture medium prior to PX exposure. Culture medium was removed and 100 μl of fresh medium containing the various PX concentrations was added to each well (n = 6–8 for each PX concentration and Triton X-100). At the appropriate time points, 100 μl of supernatant was removed from each well and transferred into the corresponding wells of a new 96-well plate. LDH activity was determined by adding 100 μl of reaction mixture (included with kit) to each well and incubating for 20 min in the dark at room temperature. Absorbance measurements were taken at 490 nm on a microplate reader with a reference wavelength at 650 nm. Cytotoxicity was determined by comparing the absorbance readings of the wells containing the PX-treated cells with those of the vehicle- (0.1% ethanol) treated cells.
2.5. Assay for cellular ATP levels
Cellular levels of ATP were measured using an ATP Bioluminescence Assay Kit (HS II, Roche Applied Science, Indianapolis, IN). Briefly, HSG cells were cultured in a 96-well plate to 80% confluence, exposed to 10 μM PX or ethanol control for 8–24 h, and harvested and lysed. Total protein was quantitated by BCA assay. The level of cellular ATP was calculated relative to a standard wherein the bioluminescence was quantified on a Luminex 100. Samples were analyzed as n = 4–6.
2.6. DNA fragmentation assay
Cellular DNA fragmentation was measured using a Cellular DNA Fragmentation ELISA kit (Roche Applied Science, Indianapolis, IN). In this assay, 5′-bromo-2′-deoxy-uridine (BrdU)-labeled DNA fragments in cell lysates or cell supernatants indicate the cell death type (e.g., apoptosis or necrosis). HSG cells were cultured in 96-well culture plates to a density of 80% confluence in culture media containing 10 μM BrdU-labeling solution. Cells were incubated overnight (~16 h), and BrdU media was replaced with culture media containing 10 μM PX (or 0.1% ethanol) for 2–24 h (n = 6–8). BrdU-labeled DNA fragments released from the HSG cells were measured from 100 μL samples of cell culture media supernatant at various time points. Samples for each time point were stored at −20 °C prior to analysis. After removal of all the remaining supernatant, 200 μL of cell lysis buffer was added to each 100 μL sample well followed by a 30 min incubation to lyse the remaining cells. Lysate (100 μL) was removed and placed in a new 96-well plate to measure the amount of BrdU-labeled DNA fragment in the cytosol. The amounts of BrdU-labeled DNA released were measured by sandwich ELISA using antibodies against DNA and BrdU. Absorbance was measured at 450 nm using a microplate reader with a reference wavelength at 690 nm.
2.7. Reactive oxygen species analysis
The presence of reactive oxygen species, superoxide and peroxide, was determined using dihydroethidium (DHE) (Sigma-Aldrich, St. Louis, MO) and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (C400, Molecular Probes, Grand Island, NY). For superoxide analysis, HSG cells were cultured in plates containing glass cover slips until 75% confluence was reached. At this point, culture medium was removed; cells were rinsed with phosphate-buffered saline (PBS), and media containing 10 μM PX was added to cells for 4 h. After 4 h, the PX-treated media was removed, cells rinsed with PBS, and media plus 5 μM DHE was added for 30 min. Cells were rinsed with PBS and imaged for fluorescence at 525/610 nm (excitation/emission) using a NIKON epifluorescence microscope. Image Pro Analyzer 6.3 was used for DHE quantitation using the Integrated Optical Density (IOD) tool. For peroxide analysis, HSG cells were seeded in black 96 well plates to a density of 80% confluence and allowed to acclimate for 24 h. Cells were incubated with 10 μM PX for 4 h. With 30 min remaining in the incubation, H2DCF-DA was added at a final concentration of 10 μM. Cells were washed with PBS and fluorescence intensity was measured at 488/525 nm excitation/emission using a Molecular Devices fluorescent microplate reader.
2.8. Quantitative real-time PCR
HSG cells were exposed to 10 μM PX or ethanol (control) as described for 4 and 24 h. Total RNA was extracted from control and 10 μM PX-exposed cells with TRIzol/chloroform, DNaseI treated, and column purified (Omega Bio-Tek, Inc., Norcross, GA). PCR primers were designed using the Roche Universal Probe Library (UPL) Assay Design Center (Supplemental Table 1). Primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA). All primer sets spanned an exon–exon junction to reduce errors due to contaminating genomic DNA. Probes were selected from the Roche UPL. Amplification and detection of the fluorescence were measured using a Stratagene Mx3005p (Stratagene, La Jolla, CA). All signals were normalized to glyceraldehyde 3-phosphate dehydrogenase (gapdh). The fold change and p-values were determined by comparing mRNA expression in PX-treated to control cells. Once data had been normalized (ΔCt), the average of 4 replicates in each group was used to calculate the ΔΔCt and fold change (fold change = 2(ΔΔCt)). p-values were calculated using ΔΔCt values, n = 4.
2.9. Statistical analyses
For the MTT and LDH cytotoxicity assays, statistical significances were determined by one-way ANOVA followed by a Tukey-Kramer post hoc test. For the cellular ATP, DNA fragmentation, and the q-rt-PCR experiments, statistical significances were determined using Student’s t-test. For Mean IOD quantification of DHE fluorescence, statistical significance was determined by a two-tailed, unpaired t-test.
3. Results
3.1. AChE activity in HSG cells
The activity of AChE present in HSG cells was measured using a colorimetric assay (Ellman et al., 1961) at 5 min and 1 h, and afforded an acetylthiocholine hydrolysis rate of about 0.15 mU/μg (nanomole/min/μg). The low hydrolysis rate is just above background, non-enzymatic hydrolysis and slightly above the limit of detection (Supplemental Data Fig. 1). Enzyme activity was measured at 5 min and 1 h to determine any time-dependent variability in the observed AChE activity due to the lysis conditions, and to correlate the activity with prior studies (Strömbland, 1959) that measured the AChE activity in salivary gland cells over a 30 min time course. Because AChE activity was not significantly above baseline in HSG cells, the inhibition by PX could not be accurately measured and no further experiments with AChE were conducted.
3.2. HSG cell viability – MTT and LDH assays
Results from the MTT assay showed that there was no cytotoxicity at 24 h exposure to 0.01 to 0.1 μM PX concentrations (Fig. 1; Panel A). However, significant levels of cytotoxicity were observed at 24 h in HSG cells when treated with 1 μM, 10 μM and 100 μM PX (17%, 25%, and 35%, respectively) (Fig. 1; Panel A). Likewise, HSG cells at 48 h post-exposure to PX also showed significant cytotoxicity at doses of 1–100 μM (17–32%) as compared to control. In contrast, when LDH release was measured as an indicator of cytotoxicity, only 100 μM PX at 24 h showed deleterious effects (Fig. 1; Panel B). After 48 h of exposure, the cytotoxicity measured at 100 μM PX treatment increased to 13%. From this data, 10 μM PX was selected for exposure experiments at 48 h since this concentration caused low, yet significant cytotoxicity in the MTT assay.
Fig. 1.

Cytotoxicity of paraoxon (PX) on HSG cells. Cells were treated with PX and analyzed at 24 h (squares) and 48 h (triangles). Cytotoxicity was estimated by a MTT colorimetric assay (A), and an LDH release assay (B). Results are presented as average with standard deviation compared to cells treated with vehicle (0.1% ethanol). Triton X-100 was used as a positive control for cell death (100% cytotoxicity). *A significant difference between the treatment and negative control (no PX) was determined (p < 0.05) using one-way ANOVA followed by a Tukey-Kramer post host test (n = 6–8).
3.3. Cellular ATP levels
The results from the MTT assay indicate that mitochondria function may be impaired by PX exposure. If mitochondria function is affected, a corresponding decrease in cellular ATP could result. In order to further explore this possibility, cellular ATP levels were measured following 10 μM PX exposure (Fig. 2). However, no change in cellular ATP levels was found up to 12 h of exposure. By 24 h, a slight, yet significant increase in cellular ATP levels was observed.
Fig. 2.
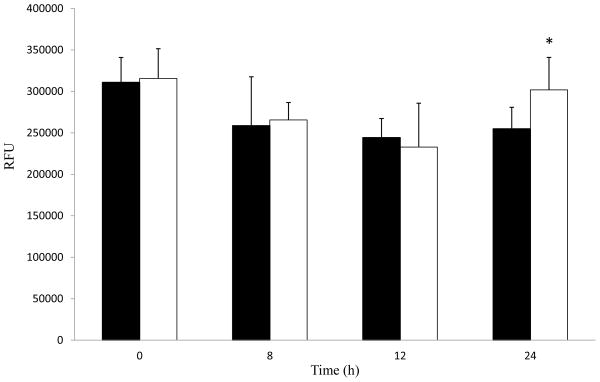
Paraoxon (PX) exposure slightly alters cellular ATP levels in HSG cells. Cells were treated with 0.1% ethanol as control (black bars) or 10 μM PX (white bars). Cellular lysates were analyzed for cellular ATP levels (RFU, relative fluorescence units) from 8 to 24 h. Samples were analyzed and results shown are averages with standard deviations. Samples were analyzed for significance with Student’s t-test, * represents p values < 0.05 (n = 4–6).
3.4. DNA fragmentation assay
In order to further characterize the cytotoxicity as apoptotic or necrotic, cellular DNA fragmentation resulting from PX exposure was determined using an ELISA to detect BrdU- labeled DNA fragments in HSG cell lysates or in cell culture supernatants. DNA fragments detected in cell lysates indicate apoptosis, whereas DNA fragments detected in cell culture supernatants is indicative of loss of membrane integrity and necrosis. HSG cells were exposed to 10 μM PX for 2–24 h and DNA fragmentation was measured as BrdU incorporation. In PX-treated cell lysates, a significant amount of BrdU was observed within 12–24 h (Fig. 3A). There was a slight, yet significant increase in DNA fragments early (6–8 h post-exposure) in the supernatant fractions of the control cells (Fig. 3B), which decreased after 8 h.
Fig. 3.

Paraoxon (PX) exposure induces DNA fragmentation in HSG cell lysates. Cell lysates (Panel A) or cell culture supernatants (Panel B) were analyzed for BrdU incorporation at 2 to 24 h following 10 μM PX treatment (or ethanol control). Black squares, control lysates; black triangles, control supernatants; white squares, PX lysates; white triangles, PX supernatants. Results were analyzed using Student’s t-test and data are represented as average with standard deviation. * represents p values < 0.05, ** represents p values < 0.01, and *** represents p values < 0.001 (n = 6–8).
3.5. Reactive oxygen species analysis
To determine if reactive oxygen species (ROS) contributed to the cytotoxicity of PX-treated HSG cells, analyses for the presence of superoxide and peroxide were conducted. Superoxide oxidizes dihydroethidium (DHE) into ethidium that subsequently intercalates into DNA and stains the nucleus a fluorescent red (Bindokas et al., 1996). At 4 h following 10 μM PX exposure, nuclear fluorescence increased 2.6-fold in the PX-treated HSG cells compared to control cells indicating the formation of superoxide anion (Fig. 4). Conversely, an assay for peroxide using 2′,7′-dichlorofluorescein diacetate (H2DCF-DA), which reacts with intracellular H2O2 to form the fluorescent compound dichlorofluorescein (Saulsbury et al., 2009) indicated no difference between control and PX-exposed cells (Supplemental Table 2).
Fig. 4.

Paraoxon (PX) exposure induces superoxide formation in HSG cells. Cells were treated with 0.1% ethanol as control (Panel A) or 10 μM PX (Panel B) and treated with DHE at 4 h. Quantification of fluorescent staining (Panel C) shows that the mean IOD was significantly higher in PX-treated cells compared to controls (Student’s t-test, *** p<0.001).
3.6. q-RT-PCR analysis
To further substantiate the possibility that PX exposure induces oxidative stress, the change in transcription of several genes associated with oxidative stress was assessed by q-RT-PCR. The genes selected for the q-RT-PCR experiments were based on a previous study that identified transcriptional changes in oxidative stress genes in PC12 cells after chlorpyrifos exposure (Slotkin and Seidler, 2009). The results from the q-RT-PCR experiments indicated that the genes gstt2 (glutathione S-transferase theta 2) and sod2 (superoxide dismutase 2) were significantly upregulated following exposure to 10 μM PX for 4 h. Exposure to 10 μM PX for 24 h resulted in significant upregulation of the genes gss (glutathione synthetase), gstm2 (glutathione S-transferase mu 2), and gstt2 (Fig. 5).
Fig 5.

Expression of oxidative stress genes in paraoxon-treated HSG cells. Cells were treated with either ethanol (0.1%) as a control or 10 μM PX for 4h (white bars) or 24 h (black bars). Fold-change measured by q-RT-PCR of gpx7, gss, gstm2, gstt2, sod2, and txnrd2 was measured. Expression was normalized to gapdh expression. Once data had been normalized (ΔCT), the average of 4 replicates in each group was used to calculate the ΔΔCt and fold change (fold-change = 2(ΔΔCT)). p-Values were calculated using ΔΔCt values, n=4, compared to the control using Student’s t-test. * represents p values < 0.05, ** represents p values < 0.01.
4. DISCUSSION
This study tested the cellular response of the HSG cell line to a sub-lethal dose of the model organophosphate anticholinesterase agent, paraoxon (PX) – a dose that reduces mitochondrial function in approximately 20% of cells but does not cause cell death. Acetylcholinesterase (AChE) activity was determined to be negligible in the HSG cell line although substrate hydrolysis occured slightly above background, possibly indicative of non-specific esterases. Previous reports have identified AChE activity in salivary glands or saliva using different enzyme activity assays (Ng et al., 2009; Strömbland, 1959). When AChE activity was measured manometrically (activity was expressed in ~ml CO2 evolved/30 min/g of tissue), contamination by blood may have played a significant role in the enzyme activity since the samples were minced whole salivary glands (Strömbland, 1959). A more recent study measured extremely low AChE activity levels in salivary glands using a sensitive fluorescence-based assay (Amplex Red ACh/AChE Assay from Molecular Probes), which supports our conclusion (Ng et al., 2009). Based on the AChE activity assays and data from previous reports, we hypothesize that the cellular toxicity measured in this report most likely occurs through mechanisms other than AChE inhibition.
In order to determine if PX affected HSG cell viability, MTT assays and LDH release assays were employed to assess cytotoxicity. Significant increases in PX-induced cytotoxicity occurred in the HSG cells at most of the exposure concentrations tested beginning at 1 μM PX concentration at 24 and 48 h time points, as revealed by MTT assay. In contrast, significant changes in LDH concentrations only occurred at the highest concentration of PX tested (100 μM) following 48 h of exposure. The discrepancy in the results obtained from the MTT and LDH assays may be explained, in part, by differences in how cytotoxicity is determined. The MTT assay assesses mitochondria function whereas the LDH assay measures loss of cell membrane integrity. Therefore, the disparate assay results possibly suggest that exposure to PX may lead to impaired mitochondrial function (MTT reduction) in HSG cells prior to reduction in membrane integrity (LDH release).
If exposure to PX results in mitochondrial dysfunction, a resultant decrease in cellular ATP levels would be expected. However, there was no significant decrease in cellular ATP levels at 24 h, clearly contrary to expectation. These results may suggest that mitochondria are sensitive to PX exposure through mechanisms that are unrelated to ATP levels. For comparison, exposure to 100 μM of PX reduced HSG viability to 65% of control at 24 h, as analyzed by MTT assay, which is much less than that observed for SH-SY5Y neuroblastoma cells (Bharate et al., 2010; Prins et al., 2010). After 24 h exposure to 100 μM PX, SH-SY5Y viability was reduced to 20% of control (Bharate et al., 2010). The difference in cytotoxicity susceptibility between HSG and SH-SY5Y cells may be due to the inhibition of AChE and related OP-susceptible proteins in SH-SY5Y cells that are not present in HSG cells.
The amount of DNA fragmentation was measured to differentiate apoptotic or necrotic mechanisms. The results of the BrdU-labeling study indicate that DNA fragmentation is occurring prior to loss of cellular membrane integrity, and therefore, cell cytotoxicity is most likely occurring via an apoptotic pathway (Elmore, 2007). However, BrdU-labeled DNA fragments in the cell lysates appear after 12 h of exposure, suggesting a time-dependent cytotoxicity possibly through the generation of ROS.
The levels of ROS like peroxide and superoxide can be increased by environmental stressors resulting in damage to cellular components, including damage to DNA, RNA, and proteins (Roberts et al., 2010). Cellular damage induced by ROS is cumulatively referred to as oxidative stress. In order to determine if PX induced ROS formation and subsequent oxidative stress, the HSG cells were analyzed for the presence of superoxide and peroxide (Roberts et al., 2010). The results of the DHE and H2DCF-DA assays indicated that superoxide but not peroxide formed in PX-treated HSG cells, suggesting that the cells underwent oxidative stress within 4 h of exposure to 10 μM PX. This outcome raises the possibility that oxidative stress is a trigger of cytotoxicity (Lukaszewicz-Hussain, 2010).
Cells defend themselves against ROS damage by upregulating proteins such as superoxide dismutase (sod2) and glutathione metabolism enzymes (gss, gstm2, and gstt2) that play an important role as cellular antioxidants (Laskin et al., 2010). In order to further establish that oxidative stress may play a role in PX induced cytotoxicity, q-RT-PCR was used to monitor the expression levels for several genes associated with oxidative stress. The genes gstt2 (glutathione S-transferase theta 2) and sod2 (superoxide dismutase 2) were upregulated following exposure to 10 μM PX for 4 h, and at 24 h PX exposure induced an upregulation of the genes gss (glutathione synthetase), gstm2 (glutathione S-transferase mu 2), and gstt2. This further confirms that oxidative stress plays an important role in PX induced cytotoxicity in HSG cells.
Although these studies suggest that oxidative stress plays a role in the cytotoxic mechanism in HSG cells exposed to PX, additional studies must be undertaken to exclude other possible biochemical routes. PX is a potent phosphorylating agent and likely to covalently modify a number of proteins and biomolecules. Therefore, it may be difficult to elucidate whether PX-induced oxidative stress in HSG cells results from direct phosphorylation of biomolecules involved in regulating oxidative species or indirect modification of biomolecules resulting from upregulation of oxidative stress proteins. This report demonstrates the potential utility of the HSG cell line as an effective in vitro model system to examine the non-cholinergic effects of exposure to organophosphate chemicals, which will ultimately lend to the discovery of novel mechanisms of toxicity and lead to the discovery of new biomarkers of OP exposure.
Supplementary Material
Supplemental Fig. 1. HSG cells express very low amounts of AChE activity. Ellman assays were performed on crude protein lysates of HSG cells. Various amounts of protein were analyzed at three time points. Black bars, 8.45 mg/ml protein; white bars, 84.5 μg/ml protein; gray bars, 420 μg/ml protein. ACh hydrolysis activity is expressed as nanomoles substrate hydrolyzed/min/mg. Samples were analyzed in triplicate and the results shown are the average activity with standard deviation; ND: not detectable.
Supplemental Table 1. Primers used for q-RT-PCR analysis. Nucleotide sequences of right and left primers are shown. The sizes of the Amplicon and Roche UPL probes are noted.
Supplemental Table 2. Paraoxon does not induce peroxide production in HSG cells. Samples were analyzed in triplicate (n = 3) and results are shown as averages with standard deviation.
Highlights.
Human salivary gland (HSG) cells show a cytotoxic response to paraoxon exposure when analyzed by MTT assays but not LDH assays;
Paraoxon induces the formation of superoxide in HSG cells as demonstrated by dihydroethidium analysis;
Genes associated with reactive oxygen species were upregulated in paraoxon-treated HSG cells.
Acknowledgments
This work was supported by NIH UO1ES016102 and P30NS055022. The project described was supported by Award Numbers P20RR017670 and P20RR015583 from the National Center for Research Resources.
Footnotes
Conflict of interest statement
No conflict of interest exists with any authors or this institution.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ballantyne B, Marrs TC. Clinical and experimental toxicology of organophosphates and carbamates. Butterworth Heinemann; Oxford; Boston: 1992. [Google Scholar]
- Bharate SB, Prins JM, George KM, Thompson CM. Thionate versus Oxon: comparison of stability, uptake, and cell toxicity of ((14)CH(3)O)(2)-labeled methyl parathion and methyl paraoxon with SH-SY5Y cells. J Agric Food Chem. 2010;58:8460–8466. doi: 10.1021/jf100976v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomser J, Casida JE. Activation of extracellular signal-regulated kinases (ERK 44/42) by chlorpyrifos oxon in Chinese hamster ovary cells. J Biochem Mol Toxicol. 2000;14:346–353. doi: 10.1002/1099-0461(2000)14:6<346::AID-JBT7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Bomser JA, Casida JE. Diethylphosphorylation of rat cardiac M2 muscarinic receptor by chlorpyrifos oxon in vitro. Toxicol Lett. 2001;119:21–26. doi: 10.1016/s0378-4274(00)00294-0. [DOI] [PubMed] [Google Scholar]
- Broomfield CA, Millard CB, Lockridge O, Caviston TL. Enzymes of the Cholinesterase Family. Plenum Press; New York: 1995. [Google Scholar]
- Carlson K, Ehrich M. Organophosphorus compound-induced modification of SH-SY5Y human neuroblastoma mitochondrial transmembrane potential. Toxicol Appl Pharmacol. 1999;160:33–42. doi: 10.1006/taap.1999.8741. [DOI] [PubMed] [Google Scholar]
- Casida JE, Quistad GB. Organophosphate toxicology: safety aspects of nonacetylcholinesterase secondary targets. Chem Res Toxicol. 2004;17:983–998. doi: 10.1021/tx0499259. [DOI] [PubMed] [Google Scholar]
- Casida JE, Quistad GB. Serine hydrolase targets of organophosphorus toxicants. Chem Biol Interact. 2005;157–158:277–283. doi: 10.1016/j.cbi.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Chambers JE, Boone JS, Davis MK, Moran JE, Tyler JW. Assessing transferable residues from intermittent exposure to flea control collars containing the organophosphate insecticide chlorpyrifos. Journal of exposure science & environmental epidemiology. 2007;17:656–666. doi: 10.1038/sj.jes.7500570. [DOI] [PubMed] [Google Scholar]
- Crema VO, Hamassaki DE, Santos MF. Small Rho GTPases are important for acinus formation in a human salivary gland cell line. Cell Tissue Res. 2006;325:493–500. doi: 10.1007/s00441-006-0192-6. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Correll L, Veronesi B. Acetylcholinesterase and neuropathy targe esterase inhibitions in neuroblastoma cells to distinguish organophosphorus compounds causing acute and delayed neurotoxicity. Fundam Appl Toxicol. 1997;38:55–63. doi: 10.1006/faat.1997.2330. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Intropido L, Costa LG. Interaction of organophosphorus compounds with muscarinic receptors in SH-SY5Y human neuroblastoma cells. J Toxicol Environ Health. 1994;43:51–63. doi: 10.1080/15287399409531903. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicologic Pathology. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuto TR. Mechanism of action of organophosphorus and carbamate insecticides. Environ Health Perspect. 1990;87:245–254. doi: 10.1289/ehp.9087245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia SJ, Seidler FJ, Crumpton TL, Slotkin TA. Does the developmental neurotoxicity of chlorpyrifos involve glial targets? Macromolecule synthesis, adenylyl cyclase signaling, nuclear transcription factors, and formation of reactive oxygen in C6 glioma cells. Brain Res. 2001;891:54–68. doi: 10.1016/s0006-8993(00)03189-9. [DOI] [PubMed] [Google Scholar]
- George KM, Schule T, Sandoval LE, Jennings LL, Taylor P, Thompson CM. Differentiation between acetylcholinesterase and the organophosphate-inhibited form using antibodies and the correlation of antibody recognition with reactivation mechanism and rate. J Biol Chem. 2003;278:45512–45518. doi: 10.1074/jbc.M304781200. [DOI] [PubMed] [Google Scholar]
- Hong MS, Hong SJ, Barhoumi R, Burghardt RC, Donnelly KC, Wild JR, Venkatraj V, Tiffany-Castiglioni E. Neurotoxicity induced in differentiated SK-N-SH-SY5Y human neuroblastoma cells by organophosphorus compounds. Toxicol Appl Pharmacol. 2003;186:110–118. doi: 10.1016/s0041-008x(02)00016-9. [DOI] [PubMed] [Google Scholar]
- Hreljac I, Zajc I, Lah T, Filipic M. Effects of model organophosphorous pesticides on DNA damage and proliferation of HepG2 cells. Environ Mol Mutagen. 2008;49:360–367. doi: 10.1002/em.20392. [DOI] [PubMed] [Google Scholar]
- Kamel A, Byrne C, Vigo C, Ferrario J, Stafford C, Verdin G, Siegelman F, Knizner S, Hetrick J. Oxidation of selected organophosphate pesticides during chlorination of simulated drinking water. Water Res. 2009;43:522–534. doi: 10.1016/j.watres.2008.10.038. [DOI] [PubMed] [Google Scholar]
- Kawakami A, Nakashima K, Tamai M, Nakamura H, Iwanaga N, Fujikawa K, Aramaki T, Arima K, Iwamoto N, Ichinose K, Kamachi M, Ida H, Origuchi T, Eguchi K. Toll-like receptor in salivary glands from patients with Sjogren’s syndrome: functional analysis by human salivary gland cell line. J Rheumatol. 2007;34:1019–1026. [PubMed] [Google Scholar]
- Knoth-Anderson J, Veronesi B, Jones K, Lapadula DM, Abou-Donia MB. Triphenyl phosphite-induced ultrastructural changes in bovine adrenomedullary chromaffin cells. Toxicol Appl Pharmacol. 1992;112:110–119. doi: 10.1016/0041-008x(92)90286-2. [DOI] [PubMed] [Google Scholar]
- Laskin JD, Black AT, Jan YH, Sinko PJ, Heindel ND, Sunil V, Heck DE, Laskin DL. Oxidants and antioxidants in sulfur mustard-induced injury. Ann N Y Acad Sci. 2010;1203:92–100. doi: 10.1111/j.1749-6632.2010.05605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Park JH, Shin IC, Koh HC. Reactive oxygen species regulated mitochondria-mediated apoptosis in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol. 2012;263:148–162. doi: 10.1016/j.taap.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Liu X, Singh BB, Ambudkar IS. ATP-dependent activation of K(Ca) and ROMK-type K(ATP) channels in human submandibular gland ductal cells. J Biol Chem. 1999;274:25121–25129. doi: 10.1074/jbc.274.35.25121. [DOI] [PubMed] [Google Scholar]
- Lukaszewicz-Hussain A. Role of oxidative stress in organophosphate insecticide toxicity – Short review. Pesticide Biochemistry and Physiology. 2010;98:145–150. [Google Scholar]
- Massicotte C, Knight K, Van der Schyf CJ, Jortner BS, Ehrich M. Effects of organophosphorus compounds on ATP production and mitochondrial integrity in cultured cells. Neurotox Res. 2005;7:203–217. doi: 10.1007/BF03036450. [DOI] [PubMed] [Google Scholar]
- Ng V, Koh D, Wee A, Chia SE. Salivary acetylcholinesterase as a biomarker for organophosphate exposure. Occup Med (Lond) 2009;59:120–122. doi: 10.1093/occmed/kqn164. [DOI] [PubMed] [Google Scholar]
- Pope CN. Organophosphorus pesticides: do they all have the same mechanism of toxicity? J Toxicol Environ Health B Crit Rev. 1999;2:161–181. doi: 10.1080/109374099281205. [DOI] [PubMed] [Google Scholar]
- Prins JM, George KM, Thompson CM. Paraoxon-induced protein expression changes to SH-SY5Y cells. Chem Res Toxicol. 2010;23:1656–1662. doi: 10.1021/tx100192f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proskocil BJ, Bruun DA, Thompson CM, Fryer AD, Lein PJ. Organophosphorus pesticides decrease M2 muscarinic receptor function in guinea pig airway nerves via indirect mechanisms. PLoS One. 2010;5:e10562. doi: 10.1371/journal.pone.0010562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Venkatraj J, Barhoumi R, Pal R, Datta A, Wild JR, Tiffany-Castiglioni E. Comparative non-cholinergic neurotoxic effects of paraoxon and diisopropyl fluorophosphate (DFP) on human neuroblastoma and astrocytoma cell lines. Toxicol Appl Pharmacol. 2007;219:162–171. doi: 10.1016/j.taap.2006.11.030. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Sparks SE, Casida JE. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol Appl Pharmacol. 2001;173:48–55. doi: 10.1006/taap.2001.9175. [DOI] [PubMed] [Google Scholar]
- Redinbaugh MG, Turley RB. Adaptation of the bicinchoninic acid protein assay for use with microtiter plates and sucrose gradient fractions. Anal Biochem. 1986;153:267–271. doi: 10.1016/0003-2697(86)90091-6. [DOI] [PubMed] [Google Scholar]
- Richards P, Johnson M, Ray D, Walker C. Novel protein targets for organophosphorus compounds. Chem Biol Interact. 1999;119–120:503–511. doi: 10.1016/s0009-2797(99)00064-2. [DOI] [PubMed] [Google Scholar]
- Roberts RA, Smith RA, Safe S, Szabo C, Tjalkens RB, Robertson FM. Toxicological and pathophysiological roles of reactive oxygen and nitrogen species. Toxicology. 2010;276:85–94. doi: 10.1016/j.tox.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachana M, Flaskos J, Alexaki E, Glynn P, Hargreaves AJ. The toxicity of chlorpyrifos towards differentiating mouse N2a neuroblastoma cells. Toxicol In Vitro. 2001;15:369–372. doi: 10.1016/s0887-2333(01)00038-8. [DOI] [PubMed] [Google Scholar]
- Saleh AM, Vijayasarathy C, Fernandez-Cabezudo M, Taleb M, Petroianu G. Influence of paraoxon (POX) and parathion (PAT) on apoptosis: a possible mechanism for toxicity in low-dose exposure. J Appl Toxicol. 2003;23:23–29. doi: 10.1002/jat.880. [DOI] [PubMed] [Google Scholar]
- Saulsbury MD, Heyliger SO, Wang K, Johnson DJ. Chlorpyrifos induces oxidative stress in oligodendrocyte progenitor cells. Toxicology. 2009;259:1–9. doi: 10.1016/j.tox.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Schuh RA, Lein PJ, Beckles RA, Jett DA. Noncholinesterase mechanisms of chlorpyrifos neurotoxicity: altered phosphorylation of Ca2+/cAMP response element binding protein in cultured neurons. Toxicol Appl Pharmacol. 2002;182:176–185. doi: 10.1006/taap.2002.9445. [DOI] [PubMed] [Google Scholar]
- Shirasuna K, Sato M, Miyazaki T. A neoplastic epithelial duct cell line established from an irradiated human salivary gland. Cancer. 1981;48:745–752. doi: 10.1002/1097-0142(19810801)48:3<745::aid-cncr2820480314>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Oxidative and excitatory mechanisms of developmental neurotoxicity: transcriptional profiles for chlorpyrifos, diazinon, dieldrin, and divalent nickel in PC12 cells. Environ Health Perspect. 2009;117:587–596. doi: 10.1289/ehp.0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strömbland BCR. Cholinesterase in human salivary glands. Experientia. 1959;15:426–427. doi: 10.1007/BF02157694. [DOI] [PubMed] [Google Scholar]
- Zhang JX, Halling PJ. pH and buffering in the bicinchoninic acid (4,4′-dicarboxy-2,2′-biquinoline) protein assay. Anal Biochem. 1990;188:9–10. doi: 10.1016/0003-2697(90)90520-j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. HSG cells express very low amounts of AChE activity. Ellman assays were performed on crude protein lysates of HSG cells. Various amounts of protein were analyzed at three time points. Black bars, 8.45 mg/ml protein; white bars, 84.5 μg/ml protein; gray bars, 420 μg/ml protein. ACh hydrolysis activity is expressed as nanomoles substrate hydrolyzed/min/mg. Samples were analyzed in triplicate and the results shown are the average activity with standard deviation; ND: not detectable.
Supplemental Table 1. Primers used for q-RT-PCR analysis. Nucleotide sequences of right and left primers are shown. The sizes of the Amplicon and Roche UPL probes are noted.
Supplemental Table 2. Paraoxon does not induce peroxide production in HSG cells. Samples were analyzed in triplicate (n = 3) and results are shown as averages with standard deviation.