

Abstract
A principal characteristic of redox signaling is that it involves an oxidation-reduction reaction or covalent adduct formation between the sensor signaling protein and second messenger. Non-redox signaling may involve alteration of the second messenger as in hydrolysis of GTP by G proteins, modification of the signaling protein as in farnesylation, or simple non-covalent binding of an agonist or second messenger. The chemistry of redox signaling is reviewed here. Specifically we have described how among the so-called reactive oxygen species, only hydroperoxides clearly fit the role of a second messenger. Consideration of reaction kinetics and cellular location strongly suggests that for hydroperoxides, particular protein cysteines are the targets and that the requirements for redox signaling is that these cysteines are in microenvironments in which the cysteine is ionized to the thiolate, and a proton can be donated to form a leaving group. The chemistry described here is the same as occurs in the cysteine and selenocysteine peroxidases that are generally considered the primary defense against oxidative stress. But, these same enzymes can also act as the sensors and transducer for signaling. Conditions that would allow specific signaling by peroxynitrite and superoxide are also defined. Signaling by other electrophiles, which includes lipid peroxidation products, quinones formed from polyphenols and other metabolites also involves reaction with specific protein thiolates. Again, kinetics and location are the primary determinants that provide specificity required for physiological signaling although enzymatic catalysis is not likely involved.
Keywords: reactive oxygen species, 4-hydroxynonenal, peroxidases, hydrogen peroxide, hydroperoxide, signal transduction
1. Introduction
This article concerns the chemistry of signaling by hydroperoxides (ROOH) and other electrophiles that modify activities of signaling proteins. Redox signaling is part of the normal physiology of all cells, including flow stimulated growth of endothelial cells, and plays a significant role in pathophysiological responses such as occur in ischemia/reperfusion injury, as well. The purpose of this review is to provide an overview of the chemistry of redox signaling while other reviews in this special issue will address how redox signaling is relevant to cardiovascular physiology. We will not attempt to provide a compendium of the increasing number of signaling pathways in which redox plays a role. Also, we cannot, in this limited space, provide a comprehensive coverage of all aspects of redox signaling. But, it is important to note that redox signaling is not strictly limited to the types of reactions that we will detail here.
Briefly, other areas of redox signaling that are of importance but cannot be addressed here are the production of superoxide (O2•−) and hydrogen peroxide (H2O2) by NADPH oxidases (NOX) and mitochondria; activation of the transcription factor hypoxia inducible factor 1α (HIF-1α) by hypoxia, which clearly involves redox reactions but not necessarily reactive species; signaling by products of enzymatically-oxidized lipids including prostaglandins and leukotrienes that signal through non-covalent binding to proteins, most commonly G-protein coupled receptors; the mechanism of the cyclooxygenases and lipoxygenases that produce prostaglandin and leukotriene hormones; and modulation of signaling by binding to metals such as occurs when nitric oxide (•NO) binds to the heme in guanylate cyclase. Rather, this chapter focuses on modification of signaling proteins through modification of one amino acid, cysteine. This does not mean that only cysteine can be involved in redox signaling; however, it is the chemical reactions of cysteine that predominantly defines redox signaling involving hydroperoxides and other electrophiles. Finally, we will not delve into the mechanism of formation of nitrosothiols (-SNO), which requires the oxidation of specific cysteines, as the mechanism(s) that occur under physiological conditions are not clear [1, 2]. Regardless the same question of which cysteine is modified to form a nitrosothiol is similar to that with hydroperoxides and other electrophiles. In contrast however, the specificity of nitrosothiol formation is different from hydroperoxides in that nitrosothiol formation does not require the thiol to be dissociated to the thiolate. Although a number of proteins can be modified by nitrosocysteine formation, only very recently has a study demonstrated specific roles in mitochondrial fatty acid metabolism [3].
Understanding the chemistry of redox signaling requires careful consideration of reaction kinetics. Redox signaling differs from other cellular signaling processes in using molecules with greater potential for non-specific reactions. Simply, an oxidant or other electrophile like H2O2 has many more potential targets than a molecule like cyclic adenosine monophosphate (cAMP). Yet, as will be described here, under physiological conditions such side reactions are not as common as may be thought. The biochemical and chemical principles described here can be found in textbooks that many biologists put on their shelves after taking their required chemistry courses and hoped would never have to be referred to again. But, the increasing interest in looking at redox signaling rather than oxidative stress as a focus, has given some of us chemistry-oriented biomedical scientists, the opportunity to consider this chemistry in terms of explaining how reactive molecules can be part of normal physiology or at least not lead to pathology. The reader is referred to a few reviews [4–10]
1.1 Redox reactions
In contrast with signaling by second messengers like cAMP and calcium, signaling by reactive species including ROOH or 4-hydroxy-2-nonenal (HNE), a major electrophilic product of lipid peroxidation, involves chemical modification of a target signaling protein cysteine residue rather than non-covalent binding to a protein. This has led to some mistakenly referring to cysteine-rich proteins as likely targets for signaling. As will be described below, it is the possession of particular cysteines rather than their abundance that makes a protein the target for redox signaling.
Redox signaling requires an electrophile (a molecule that attracts electrons) reacting with a nucleophile (a molecule that will give up electrons). There are thus two major types of reactions in redox signaling. One involves reactions in which the electrophile takes electrons away leaving the nucleophile in a more oxidized state. This is called an oxidation reaction and the electrophile is therefore referred to as an oxidant. The oxidant may take one electron (a free radical reaction) or two electrons. As free radical production more likely leads to further reaction, in signaling, it is two electron oxidations that predominate:
| <Reaction 1> |
where Nu− is a nucleophile and E is an oxidant-type electrophile. The second type of reaction that is common in redox signaling is an addition reaction in which a covalent bond is formed between the nucleophile and an electrophile:
| <Reaction 2> |
Nonetheless, redox reactions often involve hybrids between oxidation and addition in which an addition product is an intermediate.
2. General rules for signaling and application to redox signaling
2.1 Specificity
Signaling cannot depend upon random reactions. Protein kinase A has binding sites that specifically bind cAMP in preference to other small molecules. This binding takes place at specific sites on the regulatory subunit of this enzyme and at concentrations of cAMP within the physiological range that it is generated.
Similarly, the cysteines of a signaling protein that interact with ROOH or other electrophiles need to be specifically reactive compared with other cysteines and to be able to react at concentrations of electrophiles that are produced under physiologically relevant signaling conditions. The thiol group (-SH) of cysteine is not a good nucleophile. Thus, the reaction of thiols with electrophiles is relatively slow and will be in competition with the cysteine of glutathione (GSH), which in most cells is at least millimolar in concentration throughout the cell. Something else must be present in a protein to make some cysteines react with electrophiles in a specific way. The main determinants of that specificity are relative rate of reaction and location. This is because the overall governing principle is that the reaction of an electrophile with a target is determined by kinetics.
2.2 Kinetics
The rate at which an electrophile will react with its target is determined by second order reaction in which:
where [target] is the concentration of the specific protein cysteine and [electrophile] is the concentration of the electrophile. What makes specific cysteines have a higher rate constant is ionization to the thiolate form (-S−). Thiolates are excellent nucleophiles in contrast with the poorly nucleophilic thiol. But, for some reactions, including the breaking of an O-O bond in a hydroperoxide, the proximity of a specific environment or metal (often zinc) that can assist in the reaction is essential. This will be described in greater detail in the section 4.
2.3 Location
People in the business of selling real estate often say that the three most important things that determine how a property will attract attention are location, location and location. In this regard, it is important to realize that in signaling the location where the concentration of an electrophile will be greatest is at its source. In the cell, which has a high capacity for reduction of hydroperoxides and conjugation of electrophiles, steep gradients from the source into the rest of the cell exist. Thus, the target should be close to the source as it is in competition with the cellular mechanisms for elimination of the electrophile. As an example, if an oxidase generates H2O2, having the target at its highest concentration close to the source would give it a competitive advantage. In other words, the second order reactions that determine redox signaling are affected by location because gradients and competition for reaction with electrophiles requires that the target and source be close.
2.4 Reversibility and transient production of the second messenger
The opening of channels and the activation of calcium pumps tightly regulate intracellular calcium concentration so that it is only transiently elevated during physiological stimulation by agonists. Production of cAMP by adenylate cyclase and hydrolysis of cAMP by phosphodiesterases is similarly transiently altered up or down by agonists acting through G-protein coupled receptors. In both these cases, pathologies are associated with dysregulation; tetany with dysregulation of calcium [11] and cholera with a massive increase in cAMP [12]. In this regard, the transient production of H2O2 (largely indirectly from superoxide) by NOX and production of lipid hydroperoxides (LOOH) by lipoxygenases along with their removal by glutathione peroxidases and/or peroxiredoxins (see below) parallels the other second messengers.
Some electrophiles are also produced by enzymatic pathways but others including HNE are produced non-enzymatically. As described below, removal of an electrophile by a conjugation reaction catalyzed by a glutathione S-transferase (GST) is generally relatively slow and therefore differs from other second messenger signaling. The other major difference is that the reactions of electrophiles with targets may not be reversible in contrast with binding of second messengers like cAMP. We have yet to describe the reversible nature of hydroperoxide signaling. But, it is interesting that not all classic second messenger signaling is reversible. As an example, GTP binding to G proteins is ended by hydrolysis of the GTP to GDP and phosphate. The difference may be that the target signaling protein is not irreversibly modified. But, there are also many non-redox signaling pathways in which the protein is modified irreversibly as in the ubiquitinylation of cyclins during the cell cycle and farnesylation of Ras required for its activation.
2.5 Regulation by inhibition of the turn-off mechanism
The transient nature of many second messengers allows extension of the duration of signaling by inhibition of the enzyme that degrades the second messenger. Thus, sildenafil prolongs the effects of cyclic guanosine monophosphate by inhibiting the phosphodiesterase that catalyzes its hydrolysis [12]. There are also examples of inhibition of protein phosphatases prolonging the phosphorylation of signaling proteins such as in the inhibition of calcineurin by the immunosuppressant cyclosporine A [13]. This supplies a rationale for the proposal that inhibiting an enzyme that removes ROOH could regulate redox signaling [14]. But, as with the non-redox versions of this mechanism, the effect would be on the possible prolongation of a hydroperoxide-activated signal rather than necessary for activation of signaling. In other words, signaling does not require a flood of second messenger although breaking the dam can enhance or prolong the effect.
3. Hydroperoxides are the major second messengers among the reactive oxygen species
3.1 Hydroperoxides
As noted above, one essential component of signaling is specificity. In contrast with other oxygen centered molecules having the capacity to oxidize biological molecules, ROOH are unique in specificity. Contrary to common thought, ROOH are not unstable. Indeed, you can walk down to your local pharmacy and buy a bottle of H2O2 that has sat on the shelf for an indeterminate amount of time. As long as it does not come into contact with a transition metal or a thiol under basic conditions (as in the bleaching of hair using 30% H2O2), it is relatively unreactive outside of the active sites of specific enzymes and proteins having both a cysteine or a selenocysteine in the thiolate or selenate form, respectively, and additional features to break an O-O bond [2, 5]. Phospholipid hydroperoxides (PLOOH) have similar reactivity in terms of oxidation. One major difference between H2O2 and PLOOH is the tendency of the latter to rearrange and decompose in the hydroperoxyl or hydroxyl radical forms during lipid peroxidation [15, 16] [17] . Whether PLOOH themselves have a role in signaling is unknown. Certainly they contribute to the formation of electrophilic products, including HNE and phospholipid containing a greatly modified chain of fatty acid. A more detailed characterization of H2O2 chemistry and biochemistry is provided in a recent review [5].
3.2 Superoxide
What about the principal precursor of H2O2, superoxide? It has long been known that O2•− is also not a very reactive oxidant [18]. Winterbourn has measured the rate of thiol oxidation by O2•− or the hydroperoxyl radical (HO2•) formed by protonation of O2•−. These reactions produce a thiyl radical (-S•) that can initiate a chain reaction [19]. While the rate constant for the first reaction is perhaps as high as 103 M−1 sec−1, that is insignificant in contrast with the rate constant for the very abundant superoxide dismutases, which convert O2•− to O2 and H2O2 with rate constants greater than 109 M−1 sec−1 [20]. Furthermore, as the vast majority of cysteine in proteins is in the thiol form and in competition with 1mM or more GSH, the reaction would lack specificity. Thus, in terms of cysteine based signaling, O2•− functions just as a precursor of H2O2. Although there is the possibility that O2•− reacts with some target on the outside of cells, where much of it is generated by NADPH oxidases and where extracellular superoxide dismutase may be absent. It is also possible that O2•− can signal through interactions with iron-sulfur clusters in proteins such as occurs in its inactivation of mitochondrial aconitase [21] and the SoxR transcription factor in bacteria [22] but, beyond these, the potential for this mechanism of redox signaling has not gained as much attention as it may deserve.
3.2 Hydroxyl radical, singlet and peroxynitrite
If there is one molecule that is a complete mismatch for participation as a second messenger it is the hydroxyl radical (HO•). This remarkably reactive species, can remove an electron from almost any biological compound with a rate constant approaching the limit of diffusion. This lack of specificity eliminates a role for HO• in signaling.
Phagocytes can produce hypochlorous and hypobromous acids that react with H2O2 in very low yield to produce singlet oxygen. Ground state (triplet) oxygen is a diradical having two unpaired electrons and is far less reactive with organic molecules than are the two forms of singlet, one of which is not even a radical at all. Regardless, the chemistry of singlet oxygen, which is a dienophile that reacts far better with histidine than with cysteine, and its mechanism of production appear to rule out any significant role in signal transduction.
Peroxynitrite (ONOO−) is produced by a very rapid non-enzymatic reaction between nitric oxide (•NO) and O2•−. The rate constant for this reaction is greater than for the enzymatic dismutation of superoxide [23]. Peroxynitrite rapidly rearranges to nitrate (NO3−) [23], which is a weak base and mild oxidant. When peroxynitrite is protonated however, the resulting peroxynitrous acid (ONOOH) is an unusual kind of hydroperoxide that can be reduced by glutathione peroxidases (GPx) [24] or peroxiredoxins (Prdx) [25] to nitrite (NO2−) and water. ONOOH is also remarkably reactive and has the ability to act as though it were both free HO• and nitrogen dioxide (•NO2). In oxidizing compounds via its hydroxyl radical-like action, it has the same lack of respect for the rules of signaling that disqualifies HO• as a second messenger. The nitrogen dioxide aspect is somewhat more intriguing as its interaction with proteins can result in modifications such as tyrosine nitration, which may mimic phosphorylation in changing the neutral amino acid into an acidic residue. But, it remains to be seen whether tyrosine nitration is more than a marker of nitrosative stress and plays a significant role in signal transduction. A recent study of reversible tyrosine modification in regulation of cyclooxygenase activity suggests that the latter possibility needs further consideration [26].
4. The chemistry of cysteine
Cysteine is one of the more readily oxidized amino acids along with tyrosine, tryptophan, histidine and methionine. But, when cysteine is in the ionized form, thiolate, it is by far the most easily oxidized amino acid by ROOH and the best nucleophile in other reactions as well.
In terms of redox signaling, the thiolate form of cysteine fulfills several of the roles required to act as a specific target. As the pKa of cysteine in solution is generally around 8.3, the formation of the thiolate requires a peculiar environment. This can be found in microdomains of proteins where proton attraction lowers the pKa of cysteine, so that it dissociates to the thiolate. Alternatively, a metal, often zinc, is bound to cysteine so that the sulfur atom acquires a partial negative charge, making it a better nucleophile.
4.1 Reduction of hydroperoxides
While the formation of a thiolate is needed for cysteine to react at significant rates with electrophiles, for reaction with ROOH, the enhanced rate constant by dissociation is not sufficient to provide specificity. Simply, although the pKa of GSH is around 8.9, with 1 mM GSH in the cell, there will be about 10 μM glutathione in thiolate form (GS−) that will compete with the signaling protein’s thiolate. As the signaling protein concentration is likely in the picomolar range, the GS− will outcompete it. Thus, the need for the additional help in accelerating the oxidation of the signaling protein thiolate is clear.
The principal means for accelerating the oxidation of the thiolate evolved with the thiol peroxidases (TPx), namely the GPx and Prdx that have either a cysteine or selenocysteine (for most of the mammalian glutathione peroxidases) in their active site (Figure 1). In these enzymes, the first step, oxidation of the active site cysteine or selenocysteine occurs at 105 to 108 times faster than for a thiolate in free solution. How this is achieved is through helping break the O-O bond. It is a fundamental principle of chemistry that a reaction in which an RO− or HO− is a product (leaving group) will not occur or at least would be very slow. This means that the hypothetical reaction:
does not happen. Instead, for a reaction to occur, the leaving group must be protonated to become an alcohol (or water for H2O2) at the same time as the O-O bond is broken:
| <Reaction 3> |
Figure 1. Scheme of the catalysis in thiol peroxidases (TPx).
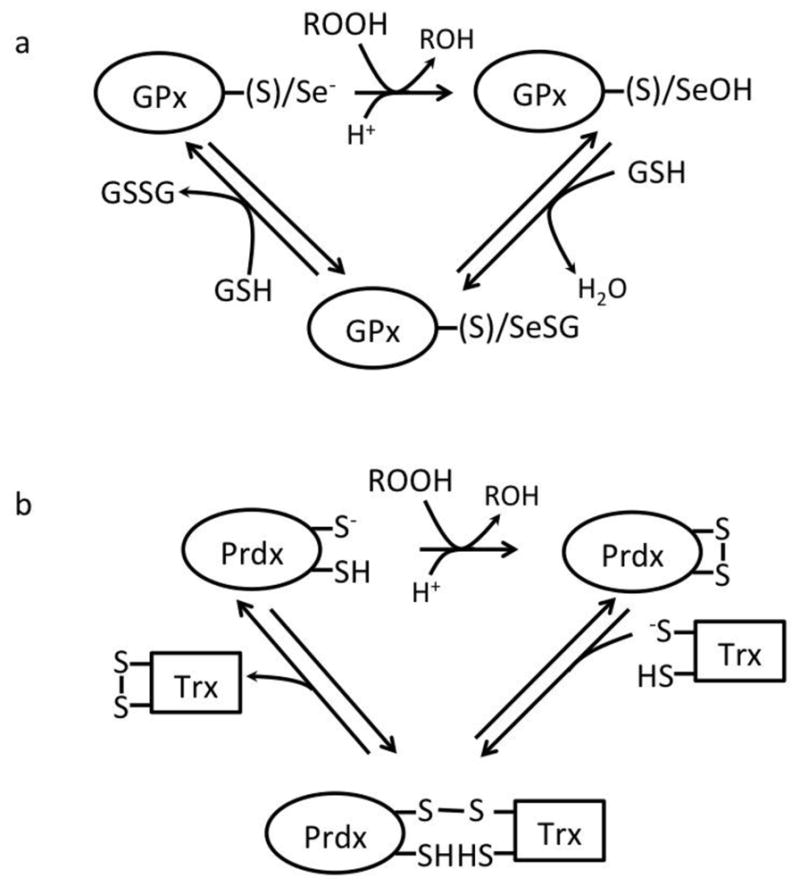
The catalytic cycle of glutathione peroxidases (GPx) and, (B) of atypical two cysteines peroxiredoxin (Prdx). (A) In the active site of GPx, the catalytic moiety is a selenium (-Se) of a selenocysteine or the sulphur (-S) of a cysteine, depending on the variant. The reduced, ground state enzyme is oxidised by a hydroperoxide (ROOH) yielding an oxidized intermediate, which is believed a selenenic (sulphenic) acid derivative of the seleno(cysteine) moiety. This is stepwise reduced by two GSH via a semi-reduced intermediate yielding a (S)Se-glutathionylated GPx. (B) The active site of the atypical two cysteines Prdx invariably contains the S moiety of a peroxidatic cysteine (-S− ), and a second cysteine thiol (-SH), acting as resolving. Upon oxidation by the ROOH, an intramolecular disulphide between the catalytic and the resolving cysteine is formed. One molecule of Trx regenerates the reduced enzyme via a thiol disulfide exchange, yielding a mixed disulfide with Prdx as a semi-reduced intermediate. In the typical 2 cysteine Prdx, the cycle and the substrates involved are identical, except that the resolving cysteine is in another Prdx molecule. Accordingly, an intermolecular disulfide is the oxidized species formed by the ROOH in these enzymes. Indeed, Prdx that contain one cysteine residue only, such as mammalian Prdx6, exist in mammals. These enzymes accept GSH as the reductant and, similarly to GPx, the semi-reduced intermediate is S-glutathionylated. As a peculiarity, the reduction of Prdx6 by GSH is preferentially assisted by GSTP (see section 4.2) [35]. Note that the two reductive steps are in principle reversible. The specificity for GSH of the TPxs may not always be strict. Indeed some TPx may accept protein thiols instead of GSH, yielding, theoretically, protein (mixed) disufides or S-thiolation of the protein substrate.
Obviously, if the proton were to be close to the thiolate/selenate, it would simply turn it into a far less reactive thiol/selenol. The trick these enzymes use, is to have a proton donating group delocalized among the residues of the active site close enough to the leaving –OH to protonate it. This does occur in free solution at pH 7 at about one millionth the frequency of what happens in the active site of the selenium-containing glutathione peroxidases and peroxiredoxins.
Once formed, the TPx-SOH (a sulfenic acid derivative) is highly reactive even with weak nucleophiles including GSH:
| <Reaction 4> |
although this is assisted in the same way in the active site of mammalian glutathione peroxidases and Prdx6, which catalyzes the same reaction and uses GSH as reductant as mammalian glutathione peroxidases. For these enzymes, a thiol-disulfide exchange with GSH results in the restoration of the TPx-S−/Se− form and release of glutathione disulfide, GSSG. Thus the overall reaction is:
| <Reaction 5> |
Alternatively, instead of using GSH, most of the mammalian peroxiredoxins use thioredoxin (Trx), a small protein with two critical cysteines that form an intramolecular disulfide bond. One of the cysteines is in the thiolate form. The initial reaction of these enzymes is basically the same as for the enzymes that use GSH to reduce hydroperoxides in <Reaction 3> except that the protein catalyst is a Prdx. This is followed by reactions that produce an overall reaction:
| <Reaction 6> |
Thiol peroxidases themselves can be regarded as hydroperoxide sensor and transducers; i.e. they may sense and transmit the signal carried by the signaling hydroperoxide to other targets. A few documented examples exist in this regard (section 5). In addition, parallel chemistry to that occurring in the reaction of TPx may as well occur in signaling proteins, in which the signaling protein directly reacts with ROOH rather than the competing TPx (section 5). It is obvious that, in signaling by a TPx-like reaction, the sensor protein must use the same strategy of the competing TPx for becoming reactive (i.e. having its cysteine accessible to the ROOH, dissociated and surrounded by residues competent for making available a mobile proton to form ROH as the leaving group).
4.2 Formation of protein S-adducts
Protein S-thiolation implies that the target protein cysteine forms a mixed disulfide with other protein thiols or GSH. Here we will focus on the formation of protein adducts with GSH (P-SSG), although the same chemistry is valid for protein thiolation in general.
Protein S-glutathionylation is a reversible modification emerging as a relevant aspect of redox signaling. It may modify enzyme activity, protein-protein and protein-DNA interactions [27]. Examples of proteins S-glutathionylated include actin [28], NO synthase [29], ryanodine receptors [30] [31], and the protein phosphatase 1 B (PTP1B) [32]. Whether all of the S-glutathionylated adducts found in cells results from enzymatic catalysis is unknown. Although glutaredoxins (GRx) [33] or GSTs catalyze S-glutathionylating reactions, the capability of these enzymes to promote this reaction appears limited. Yet, GRx mostly control the glutathionylation status of proteins that are already glutathionylated [34] and, among GSTs, so far only GSTP (also known as GSTπ) has been shown to directly participate in the S-glutathionylation of Prdx6 [35]. This reaction indeed mimics a typical nucleophilic glutathione conjugation reaction, which requires the sulfenic form of the target protein [36] (reaction 14). In addition, other candidates for catalysis of protein S-glutathionylation have been proposed, namely mononooxygenase-like enzymes, flavoprotein sulfhydryl oxidase and TPx (reviewed in [37]), but their role in glutathionylation remains hypothetical. Clearly, further investigation is required to elucidate whether protein glutathionylation is a catalyzed process, and how specificity and speed are achieved. The mechanisms yielding protein S-glutathionylation are briefly described below.
In the P-SSG adducts, sulfur is oxidized to an oxidation state of −1. Thus protein glutathionylation requires electrophiles reacting with the protein thiolate (PS−). In the simplest case, GSSG produced by GPx or Prdx6 activity (reaction 5) can exchange its disulfide with the protein thiolate yielding a glutathionylated adduct (P-SSG):
| <Reaction 9> |
Whether this reaction occurs in vivo is matter of debate: the uncatalyzed reaction is slow at physiologic pH, GSSG is supposed to be low in the cytosol, as it is kept reduced by glutathione reductase, or extruded from cells. Alternatively, the reaction might be relevant only in the endoplasmic reticulum (ER) or the Golgi apparatus, which contain relatively higher concentration of GSSG [38]. Similarly, the protein thiolate can form a disulfide in the presence of a nitrosothiol, such as S-nitroso glutathione (G-SNO):
| <Reaction 10> |
In the case of P-SSG adduct formation by GSH, the target protein thiolate (PS−) must be oxidized first to the sulfenic acid (P-SOH). Thus, the reaction is similar to that carried out by TPx (reaction 3). ROOH, ONOOH, are the oxidants in the reaction with the target protein thiolate yielding the sulfenic acid derivative [39, 40] (reactions 11 and 12). Alternatively, a cyclic sulfenyl amide (P-SNP) (reaction 13) can be formed [39]. This product has been described so far upon the reaction of PTP1B with H2O2, where the sulfenic acid derivative of catalytic cysteine attacks a backbone amide. The formation of this cyclic amide protects P-SOH by further oxidation and inhibits the catalytic activity in a way that could be physiologically relevant [41].
| <Reaction 11> |
| <Reaction 12> |
| <Reaction 13> |
where parenthesis indicates that the reaction occurs within the same protein.
These oxidized intermediates are effectively reduced by GSH, yielding the glutathione adduct:
| <Reaction 14> |
| <Reaction 15> |
Moreover, •NO or higher oxides of nitrogen (NOx) produced by metal catalysis [42] yield P-SNO as the oxidized intermediate eventually leading to P-SSG.
| <Reaction 16> |
| <Reaction 17> |
P-SSG adducts can be also formed by free radical reactions. This implies the formation of the thiyl radical either of GSH or the protein target (GS• or PS•) by superoxide or OH•. Interestingly, the mechanism of glutathionyl transfer by Grx requires GS•, which forms a disulfide anion radical intermediate (Grx-SSG•−) on Grx catalytic cysteine thiolate. This forms the PS•, which becomes glutathionylated by interacting with a second Grx-SSG•− in a reaction that regenerates the reduced Grx [34].
4.3 Conjugation with electrophiles
Lipid peroxidation products including HNE are often associated with toxicity. Toxicity of electrophiles results from their conjugation with guanine in DNA or lysine, histidine or cysteine in proteins. Yet, under physiological conditions, our plasma contains 300–700 nM HNE, which appears to be non-toxic [43]. Many other electrophiles are produced in metabolism without any deleterious consequences. Furthermore, the antioxidant role of polyphenols is largely due to their oxidation to quinones, which are strong electrophiles [44]. Thus, electrophiles, while potentially toxic, are well tolerated. This is largely due to their conjugation to GSH and export from the cell.
As in reaction with hydroperoxides, when cysteine of GSH is in the thiolate form it has much greater nucleophilicity (capacity to donate electrons). This markedly accelerates conjugation to cysteine relative to other amino acids or guanine:
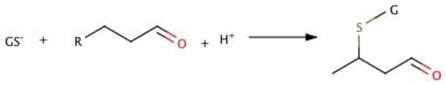 |
<Reaction 18> |
where RC=CH=O is an α,β-unsaturated aldehyde such as HNE. GSTs catalyze Reaction 18; however, for most substrates, the non-enzymatic rate of conjugation of GSH to GST substrates can be more rapid at the high GSH concentration than for the GST catalyzed rate [45]. Thus, in signaling by Michael addition to a thiolate in a protein, there must be something in its environment that will accelerate the rate constant enough to outcompete 10 μM GS−.
Another reaction of thiolate that results in conjugation is the SN2 type substitution reaction that results in alkylation of the cysteine:
| <Reaction 19> |
where R-X is an alkylating agent and X− is the leaving group in the nucleophilic substitution reaction.
GSTs can also catalyze alkylation reactions using GSH. The same issues of competition between non-enzymatic and enzymatic reactions apply. Similarly, competition between GS− and PS− also requires that the protein’s target Cys needs assistance to compete with GS−. One factor that can markedly increase the nucleophilicity of a cysteine is binding to zinc, which gives the sulfur a partial negative charge while also providing a positively charged attractant for the departing leaving group.
5. Signaling by hydroperoxides and peroxynitrite
As outlined above, for their high reactivity toward different ROOH and ONOOH, thiol peroxidases appear tailored to fulfill the criteria required for sensing and appropriately transducing a redox signal. Signal transduction by TPx may occur because the peroxidase directly promote a thiol disulfide exchange, or indirectly, by its products. In the first case the signaling protein thiol is used alternatively to GSH or Trx in reactions 5 or 6 respectively, to reduce the oxidized peroxidatic cysteine or selenocysteine of the sensor TPx. This yields disulfides or glutathionylated targets: in this regard few examples exist for disulfide formation, but positive evidence that TPx are involved in protein S-glutathionylation, is still lacking (see also Figure 1 and section 4.2). On the other hand, if TPx indirectly mediates transduction, the product of the peroxidatic reaction i.e. ROH, or GSSG or TrxS2 (reaction 5 and 6) may act as the transducer.
The best example of a direct involvement of a peroxidase as a sensor and transducer in a signaling reaction has been documented for the yeast GPx3 (alias Orp1). GPx3 is a member of the invertebrate GPx and, as such, normally uses Trx as reducing agent [46]. The peroxidatic Cys 36 of yeast GPx3, upon oxidation by H2O2, instead of reacting with the N terminal Cys of the CXXC motif of Trx, forms a disulfide with Cys 598 of the yeast transcription factor Yap1, producing a heterodimer. The Yap Cys residue 303 later resolves this, yielding Yap containing the Cys598-Cys 303 disulfide and releasing reduced GPx3. The oxidation of Yap1 leads to transcriptional gene activation [47]. Furthermore, in maturating mammalian spermatozoa, when GSH becomes limiting, a selenocysteine-containing GPx, GPx4, transfers oxidative potential of hydroperoxides to the cysteine-rich proteins of the mitochondrial capsule, including the ‘sperm mitochondrial capsule cys rich protein’ (SMCP). In this process GPx4 is self-incorporated into the immature sperm mitochondrial capsules together with the cysteine-rich proteins via mixed Se-S bonds, thus moonlighting into a structural component of the structure [48, 49]. More recently GPx7, a mammalian ER peroxidase, has been shown to oxidize protein disulfide isomerase (PDI) [50] providing oxidized PDI for protein folding, and chaperone glucose regulated protein 78 (GRP78), enhancing its activity [51].
Another example of signaling in which an antioxidant enzyme is the sensor is the activation of apoptosis signal-regulating kinase-1 (ASK1). The reduced form of Trx binds to ASK1, which prevents its activation [52]. Activation of ASK1 by H2O2 generated in macrophages by stimulation of NOX 2, demonstrated transient dissociation of the ASK1/Trx complex that allowed ASK1 activation [53]. As Prdx accelerate the oxidation of Trx by H2O2 approximately 107 fold, and the enzymes are abundant, it seemed very likely that the endogenous activation of ASK1 was an example of Prdx acting as the transducer of redox signaling rather than an inhibitor by removal of H2O2. More recently, it was demonstrated that Prdx1 appears to be the isoform that activates ASK1 while Prdx2 appears to inhibit ASK1 activation [54] suggesting a more direct interaction between the Prdx and ASK1 than simply the oxidation of the bound Trx. It has also been suggested that a Trx binding protein (TXNIP) may facilitate the oxidation of Trx and its removal from ASK1 [55]. Indeed, TXNIP may be involved in multiple aspects of signaling by H2O2 [56].
6. Signaling by electrophiles
Activation of nuclear erythroid-2 like factor-2 (Nrf2) and increased transcription through the electrophile response element (EpRE, also known as the antioxidant response element) may be the most studied response to electrophiles in which cysteine modification is involved. The modification is not however, of Nrf2 itself but of a particular protein, Kelch-like ECH-associated protein 1 (Keap1), which usually facilitates the rapid turnover of Nrf2 by ubiquitin-dependent degradation. Although Nrf2 activation can result from signaling by high concentration of exogenous H2O2 through formation of a disulfide between Cys489 on two Keap1 molecules [57], it appears that the more physiological Keap1 modification by electrophiles is Michael addition or other alkylation of cysteines on a single Keap1 molecule. The cysteine modified by Michael addition or alkylation depends upon the electrophile and its concentration [7, 58], [59–64] with Cys151, Cys273, Cys288 being identified [58, 59].
Although Keap1 appears to have particularly reactive cysteines that can compete with the much higher concentration of GS− in cells to form conjugates with electrophiles, the location of Keap1 near the plasma membrane [65] would allow it another competitive advantage in sensing exogenous electrophiles or those formed by lipid peroxidation of the plasma membrane. Many other targets of electrophilic signaling, particularly by HNE have been identified [66–69] but the sites of modification and how they outcompete GS− remains to be clarified [66].
7. Conclusion: The false dichotomy between antioxidant defense and redox signaling
Despite their previous recognition as damaging oxidants, recent evidence suggests that ROOH and ONOOH participate or influence signaling cascades under physiological conditions. In agreement with this new perception, the reaction of thiol peroxidases should also be re-considered, beyond the ‘antioxidant defense’, classically proposed. Indeed, location of and expression of multiple forms of glutathione peroxidases and peroxiredoxins suggests the specific evolution of some of these enzymes as signaling rather than defensive enzymes. Participation of TPx in redox signaling seems rationale because it provides the specificity and the speed that a signaling mechanism requires. However, to date a direct participation to signaling has been demonstrated only in few cases (see section 5). On the other hand, evidence is accumulating that these peroxidases may also signal by the reaction they catalyze and/or its product. When a ROOH or ONOOH is reduced in a specific location, an antioxidant function is achieved because the oxidant is transformed, but, also, redox signaling is attenuated or prevented. This is clearly demonstrated by the phenotype of GPx1 over-expressing mice. Interestingly, these animals develop insulin resistance and obesity, which definitely point for a role of H2O2 in insulin activity under normal conditions [70]. As well, a thiol product (or ROH/HNO2) is generated that may signal by itself, as in the case of Prdx activation of ASK1 through Trx oxidation (see section 5). Thus, the exact same reaction and even the exact same enzyme can function as either a signaling and/or antioxidant enzyme: redox signaling emerges therefore as not strictly committed to the so-called ‘antioxidant defense’. By no means an antioxidant reaction can be considered straightforwardly beneficial because it protects from damaging oxidants, at least under physiological conditions. Certainly, we can expect that future work in this area will reveal more examples and greater insight into the mechanisms of hydroperoxide signaling.
With regard to signaling by electrophiles, the evidence for participation of enzymes is far less clear. The GSTs, which would seem likely candidates, as they conjugate electrophiles to GSH, are not exceptionally good as catalysts. More likely, the combination of the target being close to where the electrophile is generated and a relatively faster rate of reaction of the protein thiolate compared with GS−, the principal competitor, accounts for the chemistry of electrophilic signaling. Keap1, with its particularly reactive cysteines and location near the plasma membrane would allow it to act as a sensor of electrophilic lipid peroxidation products. What is obvious is that the chemistry of signaling by electrophiles; i.e., conjugation to protein thiolates is the same as that in the protection provided by conjugation of electrophiles to GSH, whether non-enzymatic or catalyzed by GSTs. As with ROOH signaling, the separation between antioxidant defense and redox signaling is a matter of degree rather than a difference in chemistry. Thus, as in all physiological processes, going too far in one direction or the other can result in aberrant consequences we refer to as pathology.
Highlights.
Redox signaling involves cysteine oxidation or addition reactions by electrophiles
Relative reaction rate and location are the main determinants of specificity
For signaling by cysteine oxidation, rate is achieved by dissociation and protonation of the leaving RO−
For signaling by addition reactions, rate is achieved by dissociation
Thiol peroxidases can function as the signaling proteins in hydroperoxide signaling
Acknowledgments
Supported by ES020942 to HJF and Progetto Strategico di Ateneo (Università di Padova), grant n D170.0520PRAT09 to FU
Abbreviations
- cAMP
Adenosine monophosphate
- ASK1
Apoptosis signal-regulating kinase-1
- P-SNP
Cyclic sulfenyl amide
- GSH
Glutathione (reduced)
- GSSG
Glutathione (oxidized)
- GPx
Glutathione peroxidase(s)
- GST
Glutathione S-transferase(s)
- GS•
Glutathione thiyl radical
- GSTP
Glutathione S-transferase P
- GRx
Glutaredoxin(s)
- H2O2
Hydrogen peroxide
- ROOH
Hydroperoxide(s)
- HNE
4-Hydroxy-2-nonenal
- HO•
Hydroxyl radical
- HIF-1α
Hypoxia inducible factor 1α
- Keap1
Kelch-like ECH-associated protein 1
- Nrf2
nuclear erythroid-2 like factor-2
- NOX
NADPH oxidase(s)
- •NO
Nitric oxide
- -SNO
Nitrosothiol(s)
- Prdx
Peroxiredoxin(s)
- ONOO−
Peroxynitrite
- ONOOH
Peroxynitrous acid
- PLOOH
Phospholipid hydroperoxides
- P-SSG
Protein adduct with GSH
- PDI
Protein disulfide isomerase
- PTP1B
Protein phosphatase 1 B
- PS−
Protein thiolate
- PS•
Protein thiyl radical
- -SOH
Sulfenic acid derivative
- O2•−
Superoxide
- -SH
Thiol
- TPx
Thiol peroxidase(s)
- -S−
Thiolate
- Trx
Thioredoxin
- TXNIP
Trx binding protein
Footnotes
Disclosures: none declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li Q, Lancaster JR., Jr A Conspectus of Cellular Mechanisms of Nitrosothiol Formation from Nitric Oxide. Forum on immunopathological diseases and therapeutics. 2012;3:183–91. doi: 10.1615/ForumImmunDisTher.2012006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toppo S, Flohé L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochimica et biophysica acta. 2009;1790:1486–500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Science signaling. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13:361–6. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–42. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forman HJ. Reactive oxygen species and alpha, beta-unsaturated aldehydes as second messengers in signal transduction. Annals of the New York Academy of Sciences. 2010;1203:35–44. doi: 10.1111/j.1749-6632.2010.05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–82. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nature chemical biology. 2008;4:278–86. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 9.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flohe L. Changing paradigms in thiology from antioxidant defense toward redox regulation. Methods in enzymology. 2010;473:1–39. doi: 10.1016/S0076-6879(10)73001-9. [DOI] [PubMed] [Google Scholar]
- 11.Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- 12.Moss J, Vaughan M. ADP-ribosylation of guanyl nucleotide-binding regulatory proteins by bacterial toxins. Adv Enzymol Relat Areas Mol Biol. 1988;61:303–79. doi: 10.1002/9780470123072.ch6. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 14.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–3. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 15.Porter NA. A perspective on free radical autoxidation: the physical organic chemistry of polyunsaturated fatty acid and sterol peroxidation. The Journal of organic chemistry. 2013;78:3511–24. doi: 10.1021/jo4001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esterbauer H, Zollner H, Schaur RJ. Aldehydes formed by lipid peroxidation: mechanisms of formation, occurrence and determination. In: Vigo-Pelfrey C, editor. Membrane Lipid Oxidation. Boca Raton, FL: CRC Press; 1990. pp. 239–68. [Google Scholar]
- 17.Maiorino M, Coassin M, Roveri A, Ursini F. Microsomal lipid peroxidation: effect of vitamin E and its functional interaction with phospholipid hydroperoxide glutathione peroxidase. Lipids. 1989;24:721–6. doi: 10.1007/BF02535211. [DOI] [PubMed] [Google Scholar]
- 18.Sawyer DT, Valentine JS. How super is superoxide? Accounts of Chemical Research. 1981;14:393–400. [Google Scholar]
- 19.Winterbourn CC, Metodiewa D. Reaction of superoxide with glutathione and other thiols. Methods in enzymology. 1995;251:81–6. doi: 10.1016/0076-6879(95)51112-1. [DOI] [PubMed] [Google Scholar]
- 20.Forman HJ, Fridovich I. Superoxide dismutase: a comparison of rate constants. Archives of biochemistry and biophysics. 1973;158:396–400. doi: 10.1016/0003-9861(73)90636-x. [DOI] [PubMed] [Google Scholar]
- 21.Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. The Journal of biological chemistry. 1995;270:13399–405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 22.Fujikawa M, Kobayashi K, Kozawa T. Direct oxidation of the [2Fe-2S] cluster in SoxR protein by superoxide: distinct differential sensitivity to superoxide-mediated signal transduction. The Journal of biological chemistry. 2012;287:35702–8. doi: 10.1074/jbc.M112.395079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. American Journal of Physiology. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 24.Sies H, Sharov VS, Klotz LO, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. The Journal of biological chemistry. 1997;272:27812–7. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 25.Trujillo M, Ferrer-Sueta G, Radi R. Kinetic studies on peroxynitrite reduction by peroxiredoxins. Methods in enzymology. 2008;441:173–96. doi: 10.1016/S0076-6879(08)01210-X. [DOI] [PubMed] [Google Scholar]
- 26.Deeb RS, Nuriel T, Cheung C, Summers B, Lamon BD, Gross SS, et al. Characterization of a cellular denitrase activity that reverses nitration of cyclooxygenase. American journal of physiology Heart and circulatory physiology. 2013;305:H687–98. doi: 10.1152/ajpheart.00876.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl M, Mata-Cabana A, Kieselbach T. The disulfide proteome and other reactive cysteine proteomes: analysis and functional significance. Antioxidants & redox signaling. 2011;14:2581–642. doi: 10.1089/ars.2010.3551. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, et al. Reversible glutathionylation regulates actin polymerization in A431 cells. The Journal of biological chemistry. 2001;276:47763–6. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 29.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–8. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aracena P, Sanchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. The Journal of biological chemistry. 2003;278:42927–35. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. Journal of molecular and cellular cardiology. 2005;39:982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Salsman SJ, Hensley K, Floyd RA. Sensitivity of protein tyrosine phosphatase activity to the redox environment, cytochrome C, and microperoxidase. Antioxidants & redox signaling. 2005;7:1078–88. doi: 10.1089/ars.2005.7.1078. [DOI] [PubMed] [Google Scholar]
- 33.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxidants & redox signaling. 2005;7:348–66. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 34.Gallogly MM, Starke DW, Mieyal JJ. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxidants & redox signaling. 2009;11:1059–81. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radical Biology and Medicine. 2005;38:1422–32. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 36.Board PG, Menon D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochimica et biophysica acta. 2013;1830:3267–88. doi: 10.1016/j.bbagen.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 37.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Current opinion in pharmacology. 2007;7:381–91. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO reports. 2006;7:271–5. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochimica et biophysica acta. 2013 doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. Journal of Biological Chemistry. 1991;266:4244–50. [PubMed] [Google Scholar]
- 41.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxidants & redox signaling. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 42.Gow AJ, Buerk DG, Ischiropoulos H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. The Journal of biological chemistry. 1997;272:2841–5. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 43.Kirichenko A, Li L, Morandi MT, Holian A. 4-hydroxy-2-nonenal-protein adducts and apoptosis in murine lung cells after acute ozone exposure. Toxicol Appl Pharmacol. 1996;141:416–24. doi: 10.1006/taap.1996.0307. [DOI] [PubMed] [Google Scholar]
- 44.Forman HJ, Davies KJ, Ursini F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radical Biology and Medicine. 2013 doi: 10.1016/j.freeradbiomed.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coles B, Wilson I, Wardman P, Hinson JA, Nelson SD, Ketterer B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped-flow kinetic study. Archives of biochemistry and biophysics. 1988;264:253–60. doi: 10.1016/0003-9861(88)90592-9. [DOI] [PubMed] [Google Scholar]
- 46.Maiorino M, Ursini F, Bosello V, Toppo S, Tosatto SC, Mauri P, et al. The thioredoxin specificity of Drosophila GPx: a paradigm for a peroxiredoxin-like mechanism of many glutathione peroxidases. Journal of molecular biology. 2007;365:1033–46. doi: 10.1016/j.jmb.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 47.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–81. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 48.Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, et al. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 1999;285:1393–6. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- 49.Maiorino M, Roveri A, Benazzi L, Bosello V, Mauri P, Toppo S, et al. Functional interaction of phospholipid hydroperoxide glutathione peroxidase with sperm mitochondrion-associated cysteine-rich protein discloses the adjacent cysteine motif as a new substrate of the selenoperoxidase. The Journal of biological chemistry. 2005;280:38395–402. doi: 10.1074/jbc.M505983200. [DOI] [PubMed] [Google Scholar]
- 50.Bosello-Travain V, Conrad M, Cozza G, Negro A, Quartesan S, Rossetto M, et al. Protein disulfide isomerase and glutathione are alternative substrates in the one Cys catalytic cycle of glutathione peroxidase 7. Biochimica et biophysica acta. 2013;1830:3846–57. doi: 10.1016/j.bbagen.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 51.Wei PC, Hsieh YH, Su MI, Jiang X, Hsu PH, Lo WT, et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Molecular cell. 2012;48:747–59. doi: 10.1016/j.molcel.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal- regulating kinase (ASK) 1. Embo J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu H, Zhang H, Iles KE, Rinna A, Merrill G, Yodoi J, et al. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free radical research. 2006;40:865–74. doi: 10.1080/10715760600758514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radical Biology and Medicine. 2012;53:1522–30. doi: 10.1016/j.freeradbiomed.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 55.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxidants & redox signaling. 2012;17:1738–47. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Chng WJ. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion. 2013;13:163–9. doi: 10.1016/j.mito.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 57.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of biological chemistry. 2010;285:8463–71. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and Cellular Biology. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. The Journal of biological chemistry. 2005;280:31768–75. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 60.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chemical Research in Toxicology. 2005;18:1917–26. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 61.Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–73. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 62.Luo Y, Eggler AL, Liu D, Liu G, Mesecar AD, van Breemen RB. Sites of alkylation of human Keap1 by natural chemoprevention agents. Journal of the American Society for Mass Spectrometry. 2007;18:2226–32. doi: 10.1016/j.jasms.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohnuma T, Nakayama S, Anan E, Nishiyama T, Ogura K, Hiratsuka A. Activation of the Nrf2/ARE pathway via S-alkylation of cysteine 151 in the chemopreventive agent-sensor Keap1 protein by falcarindiol, a conjugated diacetylene compound. Toxicology and Applied Pharmacology. 2010;244:27–36. doi: 10.1016/j.taap.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 64.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chemical Research in Toxicology. 2008;21:705–10. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 65.Velichkova M, Hasson T. Keap1 in adhesion complexes. Cell motility and the cytoskeleton. 2003;56:109–19. doi: 10.1002/cm.10138. [DOI] [PubMed] [Google Scholar]
- 66.Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, et al. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radical Biology and Medicine. 2004;37:607–19. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 67.Forman HJ, Dickinson DA, Iles KE. HNE--signaling pathways leading to its elimination. Molecular aspects of medicine. 2003;24:189–94. doi: 10.1016/s0098-2997(03)00013-x. [DOI] [PubMed] [Google Scholar]
- 68.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, et al. HNE interacts directly with JNK isoforms in human hepatic stellate cells. Journal of Clinical Investigation. 1998;102:1942–50. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. The Journal of biological chemistry. 1999;274:2234–42. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 70.McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, Liu F, et al. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8852–7. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]