

Abstract
This article reviews the current understanding of the mechanisms of calcineurin inhibitor–induced hypertension. Already early after the introduction of cyclosporine in the 1980s, vasoconstriction, sympathetic excitation and sodium retention by the kidney had been shown to play a role in this form of hypertension. The vasoconstrictive effects of calcineurin inhibitors are related to interference with the balance of vasoactive substances, including endothelin and nitric oxide. Until recently, the renal site of the sodium-retaining effect of calcineurin inhibitors was unknown. We and others have shown that calcineurin inhibitors increase the activity of the thiazide-sensitive sodium chloride cotransporter through an effect on the kinases WNK and SPAK. Here, we review the pertinent literature on the hypertensinogenic effects of calcineurin inhibitors, including neural, vascular and renal effects, and we propose an integrated model of calcineurin inhibitor–induced hypertension.
Keywords: Cyclosporine, Sodium chloride cotransporter, Tacrolimus, Vasoconstriction, WNK kinase
Introduction
Calcineurin is a calcium- and calmodulin-dependent serine-threonine phosphatase that is also referred to as protein phosphatase 3. Calcineurin consists of 2 subunits called calcineurin A and calcineurin B. Calcineurin A is the calmodulin-binding catalytic subunit, while calcineurin B is the calcium-binding regulatory subunit. In turn, calcineurin A and B consist of 3 (α, β and γ) and 2 isoforms (α and β), respectively, all of which are encoded by separate genes. Although calcineurin is present in most cell types, and it is also important during development (1), it is best known for its role in the immune response. That is, when an antigen-presenting cell interacts with the T-cell receptor on T cells, the cytoplasmic level of calcium rises, and this activates calcineurin. Subsequently, calcineurin dephosphorylates a transcription factor called the cytoplasmic nuclear factor of activated T cells (NFATc). Dephosphorylation of NFATc causes it to be translocated into the cell nucleus, where it increases transcriptional activation of early cytokine genes for interleukin-2, interleukin-3, interleukin-4 and tumor necrosis factor alpha. Together, this effect stimulates the proliferation and differentiation of leukocytes.
Calcineurin is the target of a drug class known as calcineurin inhibitors (CNIs), which includes cyclosporine and tacrolimus (2). CNIs mainly inhibit the pathway described above in T-helper cells, and they are, therefore, potent immunosuppressive drugs. CNIs are routinely used to prevent rejection after transplantation and occasionally to treat autoimmune disease. Cyclosporine and tacrolimus were both isolated from fungi found in soil, and tacrolimus is in fact a macrolide antibiotic. Although cyclosporine and tacrolimus share the same target protein, they have different cytoplasmic-binding proteins, namely cyclophilins for cyclosporine, and FK-binding proteins (FKBPs) for tacrolimus (3). To date, cyclosporine and tacrolimus are still considered the cornerstone of the immunosuppressive regimen after transplantation, including kidney transplantation. Their proven efficacy in preventing rejection, however, comes at the cost of adverse effects. Of these effects, hypertension is among the most common and prominent side effect. Hypertension after kidney transplantation is important because it increases not only graft failure but also recipient mortality (4). This topic, therefore, has important clinical implications and was extensively reviewed recently (5, 6). The current review focuses on the pathogenesis of CNI-induced hypertension.
Calcineurin inhibitor–induced hypertension
It is difficult to assess the contribution of CNIs to the development of hypertension after kidney transplantation, because of the major physiological alterations after transplantation and concurrent factors contributing to hypertension. These factors include delayed graft function, volume overload, steroid treatment and the presence of hypertension prior to kidney transplantation (5). In certain other patient populations treated with CNIs, these factors play a less important role – for example, in heart, liver or bone marrow transplant recipients, and in patients with psoriasis. A long-term follow-up study of 1,000 liver transplant patients treated with tacrolimus showed that 29% had hypertension after 3 months, and 46% had hypertension after 60 months. In patients with a bone marrow or heart transplant, hypertension was relatively uncommon prior to the introduction of cyclosporine (~10%), but increased to 30%–60% and 70%–100%, respectively, after cyclosporine became the mainstay of treatment (7). In patients with psoriasis treated long-term with cyclosporine, 21% developed hypertension (8). A meta-analysis of all published trials using cyclosporine also showed an unequivocal hypertensive effect of cyclosporine (9). Depending on whom you ask, the primary cause of hypertension is believed to reside in the nervous system, the vasculature or the kidney. These different perspectives are also present in discussions of the pathogenesis of CNI-induced hypertension, which is said to be caused by effects on sympathetic nerve activity, vascular tone or kidney sodium transport. Here, we review this topic in all 3 aspects, highlighting recent insights (Fig. 1).
Fig. 1.

Model for the effects of calcineurin inhibitors on vascular tone, sympathetic nervous system and kidney sodium retention. Question marks refer to interactions that have been shown in other studies, but not in the context of CNIs (10, 11). Ang II = angiotensin II; NCC = sodium chloride cotransporter; ROS = reactive oxygen species; SNS = sympathetic nervous system; SPAK = Ste20-related kinase; WNK = with no K (K = lysine).
When analyzing the available evidence, a few observations stand out. First, cyclosporine has been studied more extensively than tacrolimus, which was introduced later. Second, there are important differences between cyclosporine and tacrolimus. This is most likely due to differences in their binding proteins; indeed, the complex between the drug and the binding protein has also been shown to be biologically active (12). Third, different mechanisms appear to play a role in the acute hypertensive effects of CNIs compared with their more chronic effects (13). Fourth, CNI-induced hypertension with preserved kidney function should be distinguished from CNI-induced nephrotoxicity, which is also associated with hypertension. Finally, many studies showed effects of CNIs on elements of the blood pressure control system, but did so without analyzing the functional contribution of this effect on blood pressure.
Vascular effects of calcineurin inhibitors
Vasoconstriction
It has been demonstrated that cyclosporine causes both systemic and renal vasoconstriction (14, 15). Because captopril does not prevent cyclosporine-induced renal vasoconstriction, it does not appear to be mediated by angiotensin II (15). Instead, renal vasoconstriction is thought to be caused by the production of endothelin in larger preglomerular arteries where it acts via the endothelin A receptor (16–18). In liver transplant patients treated with cyclosporine or tacrolimus, urinary endothelin remains elevated for at least 2 years after transplantation and is independent of plasma endothelin levels (19). The role of endothelin in CNI-induced hypertension was illustrated by rat studies in which cotreatment with an endothelin receptor antagonist partially prevented cyclosporine- and tacrolimus-induced hypertension (20, 21). Still, the question remains whether endothelin contributes to CNI-induced hypertension through renal or systemic vasoconstriction (22). Although chronic renal vasoconstriction with renal hypoperfusion clearly contributes to cyclosporine-induced nephrotoxicity (23), its role in hypertension is less clear. For example, Kaye and colleagues showed that although cyclosporine caused acute renal vasoconstriction in heart transplant patients, arterial blood pressure remained unchanged (14). The opposite is also true: cyclosporine can induce hypertension before renal vasoconstriction occurs (24).
Impaired vasodilation
Some studies have suggested that CNIs impair vasodilation rather than cause vasoconstriction. In subcutaneous resistance vessels of normal subjects, cyclosporine decreased spontaneous relaxation and inhibited endothelium-dependent relaxation, but did not change the resting tone of the vessels (25). Studies in isolated rat mesenteric artery resistance vessels showed that cyclosporine impaired vasodilation by reducing nitric oxide (26). Forearm blood flow studies in kidney transplant patients maintained on cyclosporine confirmed impaired vasodilation, which was also attributed to a reduction in basal and stimulated nitric oxide production (27). The effects of CNIs on nitric oxide could be due to an inhibition of inducible nitric oxide synthase in vascular smooth muscle, although this effect could only be demonstrated for cyclosporine (28). Tacrolimus did, however, decrease both the level and activity of endothelial nitric oxide synthase in the aorta of rats (20). Polymorphisms in genes encoding endothelial nitric oxide synthase also protected against hypertension after kidney transplantation (29).
CNIs and the renin-angiotensin system
Besides these direct effects of CNIs on the vasculature, it remains unclear if and, if so, how CNIs activate the renin-angiotensin system (30). Although some studies found CNIs to increase plasma renin activity (31, 32), others observed suppressed levels (13, 33, 34). Some studies showed that CNIs inhibit the mineralocorticoid receptor, and this effect was held to be responsible for signs of hypoaldosteronism such as hyperkalemia and acidosis (35, 36). Indeed, treatment with fludrocortisone alleviated these symptoms in kidney transplant patients (36). On the other hand, in rats, treatment with the mineralocorticoid receptor antagonist eplerenone also prevented some of the adverse effects of CNIs, including hypertension and reductions in glomerular filtration rate and renal blood flow (37). In rats, cyclosporine led to higher angiotensin II concentrations in plasma and kidney, and this may add to hypertension through either vasoconstriction or the production of reactive oxygen species (38). In smooth muscle cells, cyclosporine caused a twofold increase in the number of angiotensin II type 1 receptors, suggesting that the effects of angiotensin II could be augmented, even when its plasma levels are unchanged (39). More recently, one study suggested that inhibition of NFATc by CNIs represses the hepatic nuclear factor 4 alpha and that this would activate angiotensin II type 1 receptors to cause vasoconstriction (40).
Differences in vascular effects between cyclosporine and tacrolimus
In comparison with cyclosporine, the effects of tacrolimus on vascular tone are less consistent. For example, the intravenous infusion of tacrolimus in healthy volunteers caused hypertension and reduced fractional lithium excretion (an index of proximal tubular sodium reabsorption), but had no effect on glomerular filtration rate or renal plasma flow (41). Similarly, Nankivell and colleagues showed that cyclosporine, but not tacrolimus, caused hypoperfusion of the transplanted kidney when assessed by quantitative color Doppler imaging (42). In a study in liver transplant patients, cyclosporine and tacrolimus did reduce glomerular filtration rate and renal blood flow similarly, but tacrolimus caused less hypertension, possibly because it had less of an effect on systemic vascular resistance (43). Indeed, studies in which patients were converted from cyclosporine to tacrolimus also showed a reduction in blood pressure (44, 45).
Sodium-retaining effects of calcineurin inhibitors
A classic study by Curtis and coworkers showed cyclosporine-induced hypertension to be sodium-dependent (33) (Fig. 2). In this study, 15 kidney transplant patients who used cyclosporine and had hypertension were compared with 15 matched controls who also had hypertension, but who were treated with azathioprine. In the patients treated with cyclosporine, a low sodium diet lowered blood pressure, whereas a high sodium diet raised blood pressure to hypertensive levels. By contrast, these changes in dietary sodium had no effect on blood pressure in the patients treated with azathioprine.
Fig. 2.
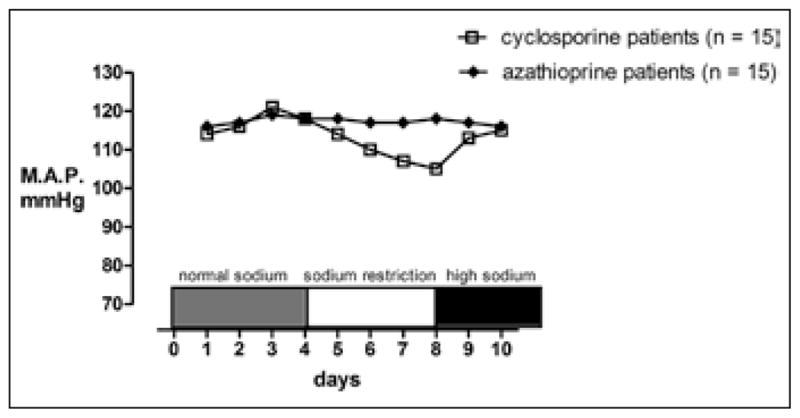
Mean arterial pressures (MAP) (average of 5 measurements each day for each patient) of the cyclosporine- and azathioprine-treated groups. Patients had similar MAP values on the control diet (150 mEq). With sodium restriction, the cyclosporine group had a significant (p<0.01) fall in blood pressure compared with either their baseline values or with the values of the azathioprine group, which did not decrease with sodium restriction. Adapted from (33) with permission from Elsevier.
Sodium retention by the sympathetic nervous system
Despite the clear association between CNIs, sodium and blood pressure, the mechanism by which CNIs cause renal sodium retention has long remained elusive. In part, this may be due to the fact that renal sodium handling is closely linked to both renal blood flow and the sympathetic nervous system. For example, an early study in rats showed that cyclosporine-induced sodium retention was caused by activation of the sympathetic nervous system, because denervation abrogated the sodium-retaining effect (46). The molecular basis for this effect was partly unraveled when the phosphoprotein synapsin was linked to CNI-induced hypertension (47). Synapsin is found on microvesicles in renal sensory nerve endings and has been implicated in neurotransmitter release. CNI-induced hypertension was attenuated in synapsin knockout mice (47), although renal sodium excretion was not studied in these animals. In healthy males, acute cyclosporine, but not acute tacrolimus, caused an increase in muscle sympathetic nerve activity, whereas sustained cyclosporine and tacrolimus both suppressed muscle sympathetic nerve activity (13). Furthermore, during sustained CNI treatment, body weight slightly increased and plasma renin activity decreased, indicating that the subjects had become volume expanded. This suggests that, at least for cyclosporine, hypertension switched from being caused by sympathetic excitation to being caused by salt and water retention (48), which might account for the suppression of sympathetic nerve activity. In a study design similar to the one used by Curtis and colleagues, Magina et al showed that blood pressure in patients with psoriasis on cyclosporine was salt-sensitive (49); however, they were unable to detect changes in plasma or urinary norepinephrine concentrations.
Effect of CNIs on renal tubular sodium transport
Several studies have sought to identify the renal site of the sodium-retaining effect of CNIs (recently reviewed in (50)). In principle, one would expect this effect to be on one of the major apical sodium transporters along the nephron. These include the sodium-hydrogen exchanger type 3 (NHE3) in the proximal tubule, the sodium-potassium-chloride co-transporter type 2 (NKCC2) in the loop of Henle, the sodium chloride cotransporter (NCC) in the distal convoluted tubule or the epithelial sodium channel (ENaC) in the collecting duct. Using fractional lithium clearances, Canzanello et al suggested cyclosporine-induced sodium retention to be due to increased sodium reabsorption in the proximal tubule (51), which might reflect an increase in sympathetic nerve activity and its facilitatory interaction with angiotensin II. In rats treated with CNIs, however, NHE3, the main sodium transporter in the proximal tubule, was either unaffected (52) or even down-regulated (53). Instead, Esteva-Font et al showed that cyclosporine increased the abundance of NKCC2 in the loop of Henle in rats (52). The functional significance of this finding on the hypertensive effect of cyclosporine, however, was not studied. Previously, Aker et al had shown that in MDCK cells, cyclosporine inhibited NKCC2, whereas tacrolimus stimulated NKCC2 (54). In A6 distal nephron cells, Wang et al showed that cyclosporine increased the open probability of ENaC (55). According to their studies, the stimulatory effect on ENaC was caused indirectly, because cyclosporine inhibited the ATP-binding cassette A1 transporter to promote cholesterol uptake.
Effect of CNIs on the sodium chloride cotransporter
Recently, we also studied the relationship between CNIs and sodium retention by the kidney (56). Our studies were prompted by the notion that the renal tubular disorders caused by CNIs bear a striking resemblance to a rare hereditary form of hypertension called familial hyperkalemic hypertension (FHHt, also called Gordon syndrome or pseudohypoaldosteronism type 2) (57). Both CNIs and FHHt are characterized by hypertension, hyperkalemia, renal tubular acidosis and hypercalciuria (57). Because FHHt is caused by mutations in WNK kinases that activate NCC (58), our hypothesis was that CNIs could also activate this pathway. We first demonstrated that wild-type mice treated with the CNI tacrolimus developed salt-sensitive hypertension, hyperkalemia, renal tubular acidosis and hypercalciuria, recapitulating the FHHt phenotype. We then showed that tacrolimus increased WNK3, WNK4, the Ste 20-related kinase SPAK and the phosphorylated form of NCC in kidney homogenates. We did not find an effect of tacrolimus on NKCC2 or its phosphorylated form. To exclude a secondary effect of tacrolimus on NCC we confirmed that calcineurin A-α, the major isoform of calcineurin, colocalized with NCC in the distal convoluted tubule. As more functional evidence for the involvement of NCC, we showed that tacrolimus failed to cause hypertension in NCC knockout mice, but caused an exaggerated hypertensive response in transgenic mice overexpressing NCC.
Patients with CNI-induced hypertension showed a greater increase in fractional chloride excretion after receiving a thiazide diuretic, suggesting increased NCC activity (59). Bio-impedance measurements suggested that these patients were volume expanded, which was reflected in their plasma aldosterone levels, which were reduced. These patients also had a higher expression of NCC and phosphorylated NCC in their transplant kidney biopsies. Together, these findings suggested CNI-induced hypertension to be, at least in part, a salt-sensitive form of hypertension mediated by the WNK-SPAK-NCC pathway. Another group found similar results using cyclosporine in rats (60). Therapeutically, this implies that CNI-induced hypertension could be especially sensitive to thiazide diuretics. From a more mechanistic point of view, the question remains how calcineurin interacts with the kinases SPAK and WNK. Although this should be resolved by future studies, some of the possibilities are depicted in Figure 3.
Fig. 3.

Hypothetical model for the effect of tacrolimus on the sodiumchloride cotransporter (NCC). An epithelial cell in the distal convoluted tubule (DCT) is shown. Tacrolimus (also FK506) binds to its intracellular binding protein (FKBP). This complex inhibits calcineurin (CaN). Because calcineurin is a phosphatase, it may inhibit some kinases under basal conditions. In the DCT, the kinases WNK3, WNK4 and SPAK interact to phosphorylate and activate NCC. Therefore, tacrolimus may prevent inhibition of these kinases by calcineurin, allowing phosphorylation and activation of NCC. This model is largely based on experiments reported in (56).
CNI-induced hypertension: toward an integrated model
How to reconcile the various effects of CNIs on vascular tone, the sympathetic nervous system and kidney sodium handling? In Figure 1 we propose an integrated model for CNI-induced hypertension based on the available evidence. This model should be considered as a hypothetical model, and it will require experimental validation for some of the interactions shown. Interestingly, however, several recent studies point toward remarkable interactions among the 3 systems controlling blood pressure. For example, studies in SPAK knockout mice not only showed that SPAK regulated the phosphorylation of NCC in the kidney (10, 61), but also that SPAK was expressed in vascular smooth muscle, where it colocalized with NKCC1 (10). SPAK-deficient mice exhibited a blunted response in vascular contractility to the α1-adrenergic agonist phenylephrine and the NKCC1 inhibitor bumetanide (10). A primary effect of CNIs on SPAK could explain how CNIs might affect vascular tone and kidney sodium reabsorption in parallel, suggesting a common pathway for mechanisms of hypertension that were previously considered as distinct. Alternatively, the changes in vasoreactivity during treatment with CNIs could be secondary to increased sodium retention by the kidney. For example, patients in whom NCC is genetically activated or inactivated also show changes in vasoreactivity, even though NCC is not expressed in vascular tissue (62, 63). Similar to the potential interactions between renal sodium handling and vascular tone, an interaction between the sympathetic nervous system and renal sodium handling was reported recently (11). In this study, stimulation of the β2-adrenergic receptor led to decreased transcription of the gene encoding WNK4 by increasing histone acetylation. In turn, suppression of WNK4 activated NCC, thereby causing salt-sensitive hypertension. Extrapolating these results to CNI-induced hypertension may explain the link between the sympathetic nervous system and sodium retention by the kidney, as originally reported in 1985 (46, 64). As such, these new studies help explain earlier observations and should ultimately lead not only to a better understanding of CNI-induced hypertension, but also to better treatments (6, 65).
Acknowledgments
Financial support: E.J.H. is supported by a grant from The Netherlands Organisation for Scientific Research (NWO, Veni grant).
Footnotes
Conflict of interest statement: None.
References
- 1.Nishiyama T, Yoshizaki N, Kishimoto T, Ohsumi K. Transient activation of calcineurin is essential to initiate embryonic development in Xenopus laevis. Nature. 2007;449(7160):341–345. doi: 10.1038/nature06136. [DOI] [PubMed] [Google Scholar]
- 2.Stepkowski SM. Molecular targets for existing and novel immunosuppressive drugs. Expert Rev Mol Med. 2000;2(4):1–23. doi: 10.1017/S1462399400001769. [DOI] [PubMed] [Google Scholar]
- 3.Marks AR. Cellular functions of immunophilins. Physiol Rev. 1996;76(3):631–649. doi: 10.1152/physrev.1996.76.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Kasiske BL, Anjum S, Shah R, et al. Hypertension after kidney transplantation. Am J Kidney Dis. 2004;43(6):1071–1081. doi: 10.1053/j.ajkd.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Chatzikyrkou C, Menne J, Gwinner W, et al. Pathogenesis and management of hypertension after kidney transplantation. J Hypertens. 2011;29(12):2283–2294. doi: 10.1097/HJH.0b013e32834bd1e7. [DOI] [PubMed] [Google Scholar]
- 6.Marasà M, Remuzzi G, Cravedi P. Hypertension after kidney transplantation: an important, but still neglected issue. J Hypertens. 2011;29(12):2310–2311. doi: 10.1097/HJH.0b013e32834d7815. [DOI] [PubMed] [Google Scholar]
- 7.Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc. 1994;69(12):1182–1193. doi: 10.1016/s0025-6196(12)65772-3. [DOI] [PubMed] [Google Scholar]
- 8.Jain A, Reyes J, Kashyap R, et al. What have we learned about primary liver transplantation under tacrolimus immunosuppression? Long-term follow-up of the first 1000 patients. Ann Surg. 1999;230(3):441–449. doi: 10.1097/00000658-199909000-00016. [discussion: 448–449] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robert N, Wong GW, Wright JM. Effect of cyclosporine on blood pressure. Cochrane Database Syst Rev. 2010;(1):CD007893. doi: 10.1002/14651858.CD007893.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang SS, Lo YF, Wu CC, et al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21(11):1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mu S, Shimosawa T, Ogura S, et al. Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17(5):573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 12.Schoeber JP, van de Graaf SF, Lee KP, Wittgen HG, Hoenderop JG, Bindels RJ. Conditional fast expression and function of multimeric TRPV5 channels using Shield-1. Am J Physiol Renal Physiol. 2009;296(1):F204–F211. doi: 10.1152/ajprenal.90473.2008. [DOI] [PubMed] [Google Scholar]
- 13.Klein IH, Abrahams AC, van Ede T, Oey PL, Ligtenberg G, Blankestijn PJ. Differential effects of acute and sustained cyclosporine and tacrolimus on sympathetic nerve activity. J Hypertens. 2010;28(9):1928–1934. doi: 10.1097/HJH.0b013e32833c20eb. [DOI] [PubMed] [Google Scholar]
- 14.Kaye D, Thompson J, Jennings G, Esler M. Cyclosporine therapy after cardiac transplantation causes hypertension and renal vasoconstriction without sympathetic activation. Circulation. 1993;88(3):1101–1109. doi: 10.1161/01.cir.88.3.1101. [DOI] [PubMed] [Google Scholar]
- 15.Murray BM, Paller MS, Ferris TF. Effect of cyclosporine administration on renal hemodynamics in conscious rats. Kidney Int. 1985;28(5):767–774. doi: 10.1038/ki.1985.196. [DOI] [PubMed] [Google Scholar]
- 16.Cavarape A, Endlich K, Feletto F, Parekh N, Bartoli E, Steinhausen M. Contribution of endothelin receptors in renal microvessels in acute cyclosporine-mediated vasoconstriction in rats. Kidney Int. 1998;53(4):963–969. doi: 10.1111/j.1523-1755.1998.00852.x. [DOI] [PubMed] [Google Scholar]
- 17.Lanese DM, Conger JD. Effects of endothelin receptor antagonist on cyclosporine-induced vasoconstriction in isolated rat renal arterioles. J Clin Invest. 1993;91(5):2144–2149. doi: 10.1172/JCI116440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perico N, Dadan J, Remuzzi G. Endothelin mediates the renal vasoconstriction induced by cyclosporine in the rat. J Am Soc Nephrol. 1990;1(1):76–83. [PubMed] [Google Scholar]
- 19.Textor SC, Burnett JC, Jr, Romero JC, et al. Urinary endothelin and renal vasoconstriction with cyclosporine or FK506 after liver transplantation. Kidney Int. 1995;47(5):1426–1433. doi: 10.1038/ki.1995.200. [DOI] [PubMed] [Google Scholar]
- 20.Takeda Y, Miyamori I, Furukawa K, Inaba S, Mabuchi H. Mechanisms of FK 506-induced hypertension in the rat. Hypertension. 1999;33(1):130–136. doi: 10.1161/01.hyp.33.1.130. [DOI] [PubMed] [Google Scholar]
- 21.Takeda Y, Miyamori I, Wu P, Yoneda T, Furukawa K, Takeda R. Effects of an endothelin receptor antagonist in rats with cyclosporine-induced hypertension. Hypertension. 1995;26(6 Pt 1):932–936. doi: 10.1161/01.hyp.26.6.932. [DOI] [PubMed] [Google Scholar]
- 22.Forslund T, Hannonen P, Reitamo S, Fyhrquist F. Hypertension in cyclosporin A-treated patients is independent of circulating endothelin levels. J Intern Med. 1995;238(1):71–75. doi: 10.1111/j.1365-2796.1995.tb00901.x. [DOI] [PubMed] [Google Scholar]
- 23.Gaston RS. Chronic calcineurin inhibitor nephrotoxicity: reflections on an evolving paradigm. Clin J Am Soc Nephrol. 2009;4(12):2029–2034. doi: 10.2215/CJN.03820609. [DOI] [PubMed] [Google Scholar]
- 24.Sturrock ND, Lang CC, Struthers AD. Cyclosporin-induced hypertension precedes renal dysfunction and sodium retention in man. J Hypertens. 1993;11(11):1209–1216. [PubMed] [Google Scholar]
- 25.Richards NT, Poston L, Hilton PJ. Cyclosporine A inhibits relaxation but does not induce vasoconstriction in human subcutaneous resistance vessels. J Hypertens. 1989;7(1):1–3. doi: 10.1097/00004872-198901000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Roullet JB, Xue H, McCarron DA, Holcomb S, Bennett WM. Vascular mechanisms of cyclosporin-induced hypertension in the rat. J Clin Invest. 1994;93(5):2244–2250. doi: 10.1172/JCI117222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris ST, McMurray JJ, Rodger RS, Farmer R, Jardine AG. Endothelial dysfunction in renal transplant recipients maintained on cyclosporine. Kidney Int. 2000;57(3):1100–1106. doi: 10.1046/j.1523-1755.2000.00937.x. [DOI] [PubMed] [Google Scholar]
- 28.Marumo T, Nakaki T, Hishikawa K, Suzuki H, Kato R, Saruta T. Cyclosporin A inhibits nitric oxide synthase induction in vascular smooth muscle cells. Hypertension. 1995;25(4 Pt 2):764–768. doi: 10.1161/01.hyp.25.4.764. [DOI] [PubMed] [Google Scholar]
- 29.Basset EA, Berthoux P, Cécillon S, et al. Hypertension after renal transplantation and polymorphism of genes involved in essential hypertension: ACE, AGT, AT1 R and ecNOS. Clin Nephrol. 2002;57(3):192–200. doi: 10.5414/cnp57192. [DOI] [PubMed] [Google Scholar]
- 30.Lassila M. Interaction of cyclosporine A and the renin-angiotensin system; new perspectives. Curr Drug Metab. 2002;3(1):61–71. doi: 10.2174/1389200023337964. [DOI] [PubMed] [Google Scholar]
- 31.Ciresi DL, Lloyd MA, Sandberg SM, Heublein DM, Edwards BS. The sodium retaining effects of cyclosporine. Kidney Int. 1992;41(6):1599–1605. doi: 10.1038/ki.1992.231. [DOI] [PubMed] [Google Scholar]
- 32.Kurtz A, Della Bruna R, Kühn K. Cyclosporine A enhances renin secretion and production in isolated juxtaglomerular cells. Kidney Int. 1988;33(5):947–953. doi: 10.1038/ki.1988.92. [DOI] [PubMed] [Google Scholar]
- 33.Curtis JJ, Luke RG, Jones P, Diethelm AG. Hypertension in cyclosporine-treated renal transplant recipients is sodium dependent. Am J Med. 1988;85(2):134–138. doi: 10.1016/s0002-9343(88)80331-0. [DOI] [PubMed] [Google Scholar]
- 34.Edwards BD, Chalmers RJ, O’Driscoll J, Lawson RS, Testa HJ, Ballardie FW. Modulation of abnormalities in renal haemodynamics and vasoactive mediators by nifedipine in patients with psoriasis on low-dose cyclosporin. Nephrol Dial Transplant. 1993;8(10):1071–1078. [PubMed] [Google Scholar]
- 35.Deppe CE, Heering PJ, Viengchareun S, Grabensee B, Farman N, Lombès M. Cyclosporine a and FK506 inhibit transcriptional activity of the human mineralocorticoid receptor: a cell-based model to investigate partial aldosterone resistance in kidney transplantation. Endocrinology. 2002;143(5):1932–1941. doi: 10.1210/endo.143.5.8821. [DOI] [PubMed] [Google Scholar]
- 36.Heering PJ, Kurschat C, Vo DT, Klein-Vehne N, Fehsel K, Ivens K. Aldosterone resistance in kidney transplantation is in part induced by a down-regulation of mineralocorticoid receptor expression. Clin Transplant. 2004;18(2):186–192. doi: 10.1046/j.1399-0012.2003.00154.x. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen FT, Jensen BL, Marcussen N, Skøtt O, Bie P. Inhibition of mineralocorticoid receptors with eplerenone alleviates short-term cyclosporin A nephrotoxicity in conscious rats. Nephrol Dial Transplant. 2008;23(9):2777–2783. doi: 10.1093/ndt/gfn204. [DOI] [PubMed] [Google Scholar]
- 38.Nishiyama A, Kobori H, Fukui T, et al. Role of angiotensin II and reactive oxygen species in cyclosporine A-dependent hypertension. Hypertension. 2003;42(4):754–760. doi: 10.1161/01.HYP.0000085195.38870.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avdonin PV, Cottet-Maire F, Afanasjeva GV, Loktionova SA, Lhote P, Ruegg UT. Cyclosporine A up-regulates angiotensin II receptors and calcium responses in human vascular smooth muscle cells. Kidney Int. 1999;55(6):2407–2414. doi: 10.1046/j.1523-1755.1999.00481.x. [DOI] [PubMed] [Google Scholar]
- 40.Niehof M, Borlak J. HNF4alpha dysfunction as a molecular rational for cyclosporine induced hypertension. PLoS ONE. 2011;6(1):e16319. doi: 10.1371/journal.pone.0016319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golbaekdal K, Nielsen CB, Pedersen EB. The acute effects of FK-506 on renal haemodynamics, water and sodium excretion and plasma levels of angiotensin II, aldosterone, atrial natriuretic peptide and vasopressin in pigs. J Pharm Pharmacol. 1996;48(11):1174–1179. doi: 10.1111/j.2042-7158.1996.tb03916.x. [DOI] [PubMed] [Google Scholar]
- 42.Nankivell BJ, Chapman JR, Bonovas G, Gruenewald SM. Oral cyclosporine but not tacrolimus reduces renal transplant blood flow. Transplantation. 2004;77(9):1457–1459. doi: 10.1097/01.tp.0000121196.71904.e0. [DOI] [PubMed] [Google Scholar]
- 43.Textor SC, Wiesner R, Wilson DJ, et al. Systemic and renal hemodynamic differences between FK506 and cyclosporine in liver transplant recipients. Transplantation. 1993;55(6):1332–1339. doi: 10.1097/00007890-199306000-00023. [DOI] [PubMed] [Google Scholar]
- 44.Artz MA, Boots JM, Ligtenberg G, et al. Improved cardiovascular risk profile and renal function in renal transplant patients after randomized conversion from cyclosporine to tacrolimus. J Am Soc Nephrol. 2003;14(7):1880–1888. doi: 10.1097/01.asn.0000071515.27754.67. [DOI] [PubMed] [Google Scholar]
- 45.Artz MA, Boots JM, Ligtenberg G, et al. Conversion from cyclosporine to tacrolimus improves quality-of-life indices, renal graft function and cardiovascular risk profile. Am J Transplant. 2004;4(6):937–945. doi: 10.1111/j.1600-6143.2004.00427.x. [DOI] [PubMed] [Google Scholar]
- 46.Moss NG, Powell SL, Falk RJ. Intravenous cyclosporine activates afferent and efferent renal nerves and causes sodium retention in innervated kidneys in rats. Proc Natl Acad Sci USA. 1985;82(23):8222–8226. doi: 10.1073/pnas.82.23.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W, Li JL, Hosaka M, et al. Cyclosporine A-induced hypertension involves synapsin in renal sensory nerve endings. Proc Natl Acad Sci USA. 2000;97(17):9765–9770. doi: 10.1073/pnas.170160397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlaich MP, Grassi G. Sympathoexcitation in calcineurin inhibitor-induced hypertension: villain or innocent bystander? J Hypertens. 2010;28(9):1809–1810. doi: 10.1097/HJH.0b013e32833e0e74. [DOI] [PubMed] [Google Scholar]
- 49.Magina S, Santos J, Coroas A, et al. Salt sensitivity of blood pressure in patients with psoriasis on ciclosporin therapy. Br J Dermatol. 2005;152(4):773–776. doi: 10.1111/j.1365-2133.2005.06518.x. [DOI] [PubMed] [Google Scholar]
- 50.Damiano S, Scanni R, Ciarcia R, Florio S, Capasso G. Regulation of sodium transporters in the kidney during cyclosporine treatment. J Nephrol. 2010;23(Suppl 16):S191–S198. [PubMed] [Google Scholar]
- 51.Canzanello VJ, Textor SC, Taler SJ, et al. Renal sodium handling with cyclosporin A and FK506 after orthotopic liver transplantation. J Am Soc Nephrol. 1995;5(11):1910–1917. doi: 10.1681/ASN.V5111910. [DOI] [PubMed] [Google Scholar]
- 52.Esteva-Font C, Ars E, Guillen-Gomez E, et al. Ciclosporin-induced hypertension is associated with increased sodium transporter of the loop of Henle (NKCC2) Nephrol Dial Transplant. 2007;22(10):2810–2816. doi: 10.1093/ndt/gfm390. [DOI] [PubMed] [Google Scholar]
- 53.Mohebbi N, Mihailova M, Wagner CA. The calcineurin inhibitor FK506 (tacrolimus) is associated with transient metabolic acidosis and altered expression of renal acid-base transport proteins. Am J Physiol Renal Physiol. 2009;297(2):F499–F509. doi: 10.1152/ajprenal.90489.2008. [DOI] [PubMed] [Google Scholar]
- 54.Aker S, Heering P, Kinne-Saffran E, Deppe C, Grabensee B, Kinne RK. Different effects of cyclosporine a and FK506 on potassium transport systems in MDCK cells. Exp Nephrol. 2001;9(5):332–340. doi: 10.1159/000052629. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Zhang ZR, Chou CF, Liang YY, Gu Y, Ma HP. Cyclosporine stimulates the renal epithelial sodium channel by elevating cholesterol. Am J Physiol Renal Physiol. 2009;296(2):F284–F290. doi: 10.1152/ajprenal.90647.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoorn EJ, Walsh SB, McCormick JA, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17(10):1304–1309. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hadchouel J, Delaloy C, Fauré S, Achard JM, Jeunemaitre X. Familial hyperkalemic hypertension. J Am Soc Nephrol. 2006;17(1):208–217. doi: 10.1681/ASN.2005030314. [DOI] [PubMed] [Google Scholar]
- 58.Wilson FH, Disse-Nicodème S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 59.Colussi G, Bettinelli A, Tedeschi S, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007;2(3):454–460. doi: 10.2215/CJN.02950906. [DOI] [PubMed] [Google Scholar]
- 60.Melnikov S, Mayan H, Uchida S, Holtzman EJ, Farfel Z. Cyclosporine metabolic side effects: association with the WNK4 system. Eur J Clin Invest. 2011;41(10):1113–1120. doi: 10.1111/j.1365-2362.2011.02517.x. [DOI] [PubMed] [Google Scholar]
- 61.McCormick JA, Mutig K, Nelson JH, et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2010;14(3):352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calò LA. Vascular tone control in humans: insights from studies in Bartter’s/Gitelman’s syndromes. Kidney Int. 2006;69(6):963–966. doi: 10.1038/sj.ki.5000253. [DOI] [PubMed] [Google Scholar]
- 63.Paver WK, Pauline GJ. Hypertension and hyperpotassaemia without renal disease in a young male. Med J Aust. 1964;2:305–306. doi: 10.5694/j.1326-5377.1964.tb115766.x. [DOI] [PubMed] [Google Scholar]
- 64.Ellison DH, Brooks VL. Renal nerves, WNK4, glucocorticoids, and salt transport. Cell Metab. 2011;13(6):619–620. doi: 10.1016/j.cmet.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoorn EJ, Walsh SB, Unwin RJ, Ellison DH. Hypertension after kidney transplantation: calcineurin inhibitors increase salt-sensitivity. J Hypertens. 2012;30(4):832–833. doi: 10.1097/HJH.0b013e32835165e4. [DOI] [PubMed] [Google Scholar]