

Highlights
-
•
Mutation G805R is in the transmembrane domain of the LDLR.
-
•
A polar residue in the transmembrane domain induced metalloproteinase cleavage.
-
•
Mutation G805R caused reduced amounts of the precursor LDLR.
-
•
Reduced amounts of precursor LDLR led to reduced amounts of the mature LDLR.
-
•
Mutation G805R prevented γ-secretase cleavage within the transmembrane domain.
Abbreviations: DAPT, N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester; DiD, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate; LDL, low density lipoprotein; LDLR, low density lipoprotein receptor
Keywords: Endoplasmic reticulum, Familial hypercholesterolemia, LDL receptor, Metalloproteinase, Mutation, Transmembrane domain
Abstract
More than 1700 mutations in the low density lipoprotein receptor (LDLR) gene have been found to cause familial hypercholesterolemia (FH). These are commonly divided into five classes based upon their effects on the structure and function of the LDLR. However, little is known about the mechanism by which mutations in the transmembrane domain of the LDLR gene cause FH. We have studied how the transmembrane mutation G805R affects the function of the LDLR. Based upon Western blot analyses of transfected HepG2 cells, mutation G805R reduced the amounts of the 120 kDa precursor LDLR in the endoplasmic reticulum. This led to reduced amounts of the mature 160 kDa LDLR at the cell surface. However, significant amounts of a secreted 140 kDa G805R-LDLR ectodomain fragment was observed in the culture media. Treatment of the cells with the metalloproteinase inhibitor batimastat largely restored the amounts of the 120 and 160 kDa forms in cell lysates, and prevented secretion of the 140 kDa ectodomain fragment. Together, these data indicate that a metalloproteinase cleaved the ectodomain of the 120 kDa precursor G805R-LDLR in the endoplasmic reticulum. It was the presence of the polar Arg805 and not the lack of Gly805 which led to ectodomain cleavage. Arg805 also prevented γ-secretase cleavage within the transmembrane domain. It is conceivable that introducing a charged residue within the hydrophobic membrane lipid bilayer, results in less efficient incorporation of the 120 kDa G805R-LDLR in the endoplasmic reticulum membrane and makes it a substrate for metalloproteinase cleavage.
1. Introduction
The low density lipoprotein receptor (LDLR) binds low density lipoprotein (LDL) at the cell surface and internalizes LDL by receptor-mediated endocytosis [1]. Mutations in the LDLR gene which lead to defective LDLRs and disrupted clearance of LDL, cause familial hypercholesterolemia [1]. Typically, familial hypercholesterolemia heterozygotes have plasma LDL cholesterol levels in the range of 6–11 mmol/l, whereas homozygotes have plasma LDL cholesterol levels of approximately 20 mmol/l [1].
The LDLR is synthesized as a 860 amino acid protein. After the 21 amino acid signal peptide has been cleaved off, the mature 839 amino acid LDLR is inserted in the endoplasmic reticulum (ER) membrane and the ectodomain undergoes folding and glycosylation in the ER [1]. The properly folded LDLR exits the ER and the N-linked sugars are modified and the O-linked sugars are elongated in the Golgi apparatus. This makes the apparent molecular weight increase from 120 to 160 kDa [2]. After transport to the cell membrane, the LDLR becomes concentrated in clathrin-coated pits [3].
The LDLR has five functional domains [4]. The N-terminal ligand-binding domain consists of seven repeats of approximately 40 amino acids each. The next domain of approximately 400 amino acids has a high degree of homology with the precursor for the epidermal growth factor and contains a 280 amino acid β-propeller. The third domain consists of 58 amino acids immediately outside the cell membrane and is enriched in O-linked sugars. The transmembrane domain consists of 22 amino acids, and the 50 residue cytoplasmic domain contains the motifs required for concentrating the LDLR in clathrin-coated pits.
More than 1700 different mutations in the LDLR gene (www.ucl.ac.uk/ldlr) have been found to cause familial hypercholesterolemia and these may be classified into five classes based on their effects on the LDLR [5]. Class 1 mutations prevent the synthesis of immunodetectable LDLR. Class 2 mutations result in mutant LDLRs which are completely (Class 2a) or partially (Class 2b) retained in the ER. Class 3 mutations result in mutant LDLRs which are incorporated in the cell membrane, but are defective in binding LDL. Class 4 mutations result in mutant LDLRs which fail to concentrate in clathrin-coated pits. Class 5 mutations result in mutant LDLRs which fail to release LDL in the endosome, leading to intracellular degradation of the mutant LDLR. A suggested additional class of mutations results in mutant LDLRs which fail to undergo basolateral sorting in polarized cells [6].
The transmembrane domain of the LDLR is encoded by exon 16 and the 5′ part of exon 17 [7] and five of the reported mutations in this domain are missense mutations (www.ucl.ac.uk/ldlr). However, the mechanism by which mutations in this part of the gene affect the function of the LDLR, has not been characterized. In this study we have performed a series of studies to determine the mechanism by which mutation G805R (c.2413G > A, Ref. seq.: NM_000527.4) [8] in exon 17 of the LDLR gene causes familial hypercholesterolemia.
2. Results
2.1. Segregation analysis and bioinformatics analysis of mutation G805R
Mutation G805R is a rare mutation in Norway. Only two of a total of 1850 unrelated patients provided with a molecular genetic diagnosis of familial hypercholesterolemia, carry mutation G805R. Segregation analysis among family members of one of the index patients revealed that mutation G805R segregated with hypercholesterolemia through 14 meioses (Supplementary Fig. S1). The probability that this co-segregation occurred by chance is (1/2)14 = 0.006%. Mutation G805R was predicted to be pathogenic by the software programs PolyPhen2 (Score: “Probably Damaging”), SIFT (Score: “Not Tolerated”) and Mutation Taster (Score: “Disease Causing”). A multiple sequence alignment of the human LDLR (residues 764–839) and orthologs from 18 mammals and 8 additional vertebrate species found by searching the UniProt [9] and Ensembl [10] databases, is shown in Fig. 1. This alignment shows Gly805 to be highly conserved and there is a complete lack of residues with charged side chains in the transmembrane domain. Together, these data indicate that mutation G805R is pathogenic. The alignment also shows poor residue conservation in the ectodomain and in the transmembrane domain, but the latter is strongly enriched in hydrophobic residues. Three conserved positively charged residues of the cytoplasmic domain (Lys811, Arg814, and Lys816 in the human LDLR, Fig. 1) appear to be necessary for inserting the receptor with correct topology in the membrane, according to the positive-inside rule [11].
Fig. 1.
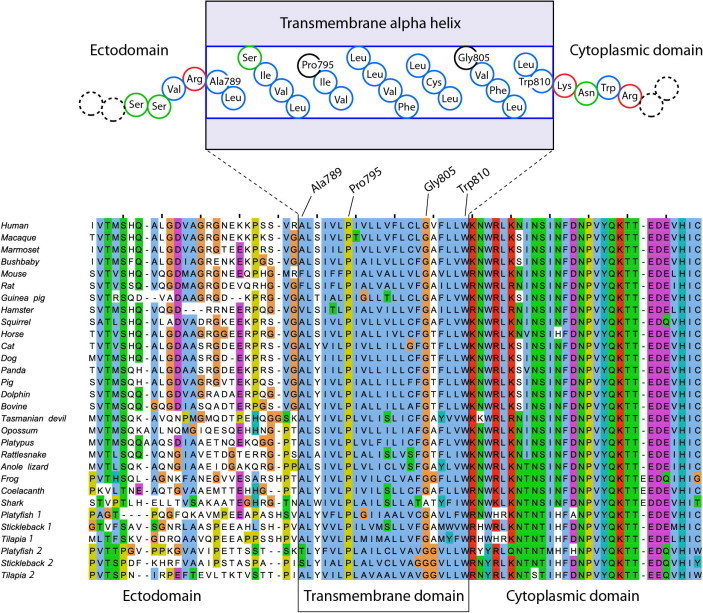
The LDLR transmembrane domain is devoid of residues with charged side chains. Multiple sequence alignment of the human LDLR (residues 764–839) and 29 vertebrate homologs shows a complete lack of charged residues in the transmembrane alpha helix passing through the membrane lipid bilayer. In the cartoon at the top, non-polar-aliphatic and aromatic residues are indicated by blue circles, the highly conserved Pro795 and Gly805 by black circles, basic residues by red circles, and polar, neutral residues by green circles. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.2. Characteristics of G805R-LDLR as determined by Western blot analysis
To determine how mutation G805R affects the 120 kDa precursor LDLR in the endoplasmic reticulum and/or the 160 kDa mature LDLR at the cell surface, HepG2 cells were transiently transfected with the G805R-LDLR plasmid. Because the wild-type (WT) LDLR has been found to undergo γ-secretase cleavage within the transmembrane domain [12], the cells were cultured in the presence or absence of the γ-secretase inhibitor N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester (DAPT) to study whether mutation G805R affects γ-secretase cleavage. γ-Secretase cleavage of the WT-LDLR is preceded by ectodomain cleavage to generate a residual C-terminal 17 kDa LDLR fragment, which is acted upon by γ-secretase to release the cytoplasmic domain [12]. The released cytoplasmic domain is unstable and has a short half-life which is typical of cytoplasmic domains released as part of regulated intramembrane proteolysis [13,14]. Thus, it is predominantly in the presence of DAPT which inhibits γ-secretase cleavage, that a 17 kDa C-terminal cleavage fragment of the LDLR can be detected by Western blot analysis [12].
Cell lysates of transfected HepG2 cells were subjected to Western blot analysis using an antibody against the C-terminal HA-tag. This antibody detects the 120 kDa precursor LDLR, the 160 kDa mature LDLR and a 17 kDa C-terminal cleavage fragment (Fig. 2). HepG2 cells transfected with the G805R-LDLR plasmid had markedly lower amounts of both the 120 kDa and the 160 kDa LDLR forms as compared to cells transfected with the WT-LDLR plasmid, of 21% (±5) and 25% (±6), respectively. However, the relative intensities of the bands representing the two LDLR forms, were similar to that of the WT-LDLR (Fig. 2). Because mutation G805R caused reduced amounts of both the precursor 120 kDa and mature 160 kDa G805R-LDLR and not a selective reduction of the mature 160 kDa form, it appears that this mutation primarily reduced the amount of the precursor form which led to reduced amounts of the mature form at the cell surface. Reduced amounts of the cell-surface G805R-LDLR were also demonstrated by confocal laser-scanning microscopy (Supplementary Fig. S2).
Fig. 2.
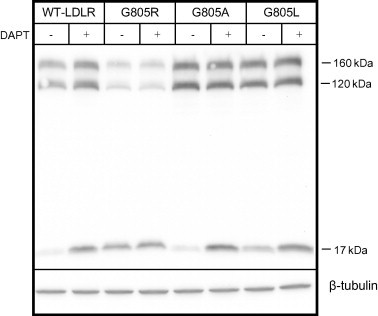
Effect of mutation G805R on the amount of precursor and mature G805R-LDLR. HepG2 cells were transiently transfected with the WT-LDLR plasmid or LDLR plasmids containing mutations G805R, G805A or G805L. The cells were cultured in the presence or absence of the γ-secretase inhibitor DAPT (10 μM). Cell lysates were subjected to Western blot analysis using an antibody against the C-terminal HA tag. The 160 kDa mature LDLR, the 120 kDa precursor LDLR and a C-terminal 17 kDa cleavage fragment are indicated. Beta-tubulin was used as a loading control. One representative of three separate experiments is shown.
When HepG2 cells transfected with the WT-LDLR plasmid were cultured in the absence of DAPT, virtually no 17 kDa C-terminal fragment was observed for the WT-LDLR (Fig. 2). However, for the G805R-LDLR a distinct band representing the 17 kDa C-terminal fragment was observed in the absence of DAPT and apparently no increased intensity of this band was observed in the presence of DAPT (Fig. 2). These findings indicate that mutation G805R by mimicking the effect of DAPT, prevents γ-secretase cleavage within the transmembrane domain.
2.3. Role of Gly805
To determine whether the reduced amounts of both the precursor and mature G805R-LDLR were due to the lack of Gly805 or to the presence of Arg805, the effect of mutations G805A and G805L on the amounts of LDLRs were studied. As determined by Western blot analysis of lysates from HepG2 cells transiently transfected with the G805A-LDLR plasmid or the G805L-LDLR plasmid, the amounts of the respective 120 kDa precursor LDLRs and the 160 kDa mature LDLRs were similar to that of the WT-LDLR (Fig. 2). Moreover, as determined by the increased amount of the respective 17 kDa C-terminal LDLR fragments in the presence of DAPT, these two mutant LDLRs were subjected to γ-secretase cleavage similarly to that of the WT-LDLR (Fig. 2). Thus, it is the presence of Arg805 within the transmembrane domain which reduces the amounts of the 120 kDa precursor form and prevents γ-secretase cleavage within the transmembrane domain.
2.4. Effect of helix-destabilizing residues on γ-secretase cleavage
γ-Secretase cleavage of transmembrane alpha helices rely on helix-destabilizing residues such as glycine, proline and serine, to promote local unfolding within the alpha helix [15]. To study the role of the highly conserved residues Pro795 and Gly805, as well as Ser791 within the transmembrane domain of the LDLR (Fig. 1), HepG2 cells were transiently transfected with mutant LDLR plasmids where these residues had been mutated to the alpha helix-stabilizing residue leucine. γ-Secretase cleavage was measured as the ability of DAPT to increase the amount of the 17 kDa C-terminal fragment. By transfecting plasmids containing one or more mutations as indicated in Fig. 3, it was evident that mutating individual residues only slightly reduced γ-secretase cleavage, whereas mutating the two highly conserved Pro795 and Gly805 almost completely reduced γ-secretase cleavage. Mutating all three residues appeared to completely prevent γ-secretase cleavage. Thus, helix-destabilizing residues are important for γ-secretase cleavage of the transmembrane domain of the LDLR.
Fig. 3.
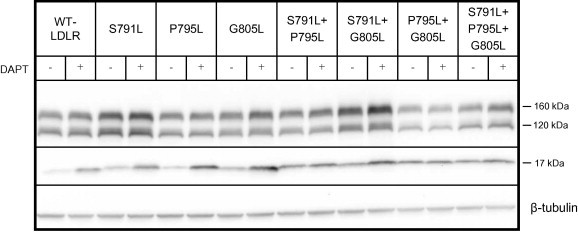
Effect of mutating the alpha helix-destabilizing residues Ser791, Pro795 and Gly805 on γ-secretase cleavage. HepG2 cells were transiently transfected with the WT-LDLR plasmid or plasmids containing one or more of the mutations S791L, P795L and G805R, as indicated. The cells were cultured in the presence or absence of the γ-secretase inhibitor DAPT (10 μM). Cell lysates were subjected to Western blot analysis using an antibody against the C-terminal HA tag. The 160 kDa mature LDLR, the 120 kDa precursor LDLR and a C-terminal 17 kDa cleavage fragment are indicated. Beta-tubulin was used as a loading control. One representative of three separate experiments is shown.
2.5. G805R-LDLR does not undergo proteasomal degradation
One mechanism for degradation of mutant proteins in the ER, is ER-associated degradation which involves ubiquitination of the cytoplasmic domain and subsequent proteasomal degradation [16]. To study whether the low amount of the precursor G805R-LDLR was due to proteasomal degradation, transfected HepG2 cells were cultured in the presence or absence of the proteasome inhibitor lactacystine. Whereas, lactacystine increased the amounts of the precursor and mature forms of the WT-LDLR by 61% (±10) and 18% (±16), which is consistent with previous reports [12,17], lactacystine increased the amount of the precursor G805R-LDLR by 13% (±23) and reduced the amount of the mature G805R-LDLR by 21% (±19) (Fig. 4). Thus, the low amounts of G805R-LDLR in cell lysates were apparently not due to proteasomal degradation of the precursor form of G805R-LDLR.
Fig. 4.
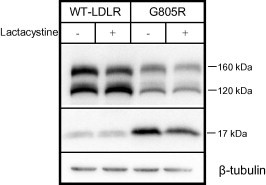
Effect of lactacystine on the amount of G805R-LDLR. HepG2 cells were transiently transfected with the WT-LDLR or the G805R-LDLR plasmids and the cells were cultured in the presence or absence of proteasome inhibitor lactacystine (10 μM). Cell lysates were subjected to Western blot analysis using an antibody against the C-terminal HA tag. The 160 kDa mature LDLR, the 120 kDa precursor LDLR and a C-terminal 17 kDa cleavage fragment are indicated. Beta-tubulin was used as a loading control. One representative of three separate experiments is shown.
2.6. The ectodomain of G805R-LDLR is cleaved by a metalloproteinase
In the culture medium of HepG2 cells transfected with the WT-LDLR plasmid, a 140 kDa N-terminal degradation product caused by ectodomain cleavage close to the cell membrane, can be observed (Fig. 5). When the cells were cultured in the presence of batimastat which inhibits a broad spectrum of metalloproteinases, no 140 kDa fragment was found in the culture medium (Fig. 5). These data are consistent with previous findings that metalloproteinase inhibitors prevent shedding of the 140 kDa ectodomain fragment at the cell surface [18]. Moreover, in the medium of cells transfected with the WT-LDLR plasmid, a 28 kDa fragment was observed (Fig. 5). This fragment represents the ligand-binding repeats 1–4 which is cleaved off from the 160 kDa mature LDLR at the cell surface [19]. The 140 kDa ectodomain fragment was also observed in the medium of cells transfected with the G805R-LDLR plasmid, but not when the cells were cultured in the presence of batimastat (Fig. 5). Thus, also G805R-LDLR is subjected to metalloproteinase cleavage.
Fig. 5.
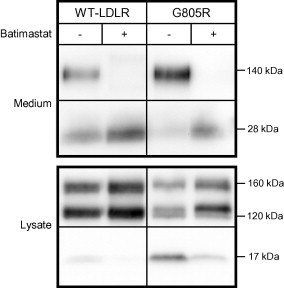
Effect of batimastat on the amount of G805R-LDLR. HepG2 cells were transiently transfected with the WT-LDLR or G805R-LDLR plasmids and the cells were cultured in the presence or absence of the metalloproteinase inhibitor batimastat (10 μM). Lysates and culture media were subjected to Western blot analysis. An antibody against the C-terminal HA tag was used to probe the lysates and an antibody against the ligand-binding domain was used to probe the media. In the media a 140 kDa ectdomain fragment and a 28 kDa fragment containing ligand-binding repeats 1–4 are shown. In the lysates the 160 kDa mature LDLR, the 120 kDa precursor LDLR and a C-terminal 17 kDa cleavage fragment are indicated. One representative of three separate experiments is shown.
Batimastat also affected the amounts of the LDLR in cell lysates. However, the effect of batimastat on G805R-LDLR was different from that on the WT-LDLR (Fig. 5). Whereas, batimastat increased the amounts of the 120 and 160 kDa forms of the WT-LDLR by 12% (±5) and 30% (±13), respectively, it markedly increased the amounts of the 120 and 160 kDa forms of the G805R-LDLR by 82% (±30) and 142% (±47), respectively (Fig. 5). Moreover, batimastat markedly reduced the amount of the residual 17 kDa C-terminal G805R-LDLR cleavage fragment by 86% (±8) (Fig. 5). Together, these data indicate that the reduced amounts of the 120 and 160 kDa forms of G805R-LDLR were caused by ectodomain cleavage of the 120 kDa form by a metalloproteinase.
Such a metalloproteinase-induced ectodomain cleavage could have taken place at the cell membrane, but this could not explain the reduced amounts of the 120 kDa precursor G805R-LDLR located in the ER. Thus, it is likely that metalloproteinase cleavage of G805R-LDLR predominantly takes place in the ER. The cleaved ectodomain is then released into the ER lumen for subsequent transport through the secretory pathway to appear in the media of transfected cells as a 140 kDa fragment after modification of sugars in the Golgi apparatus. Such an ectodomain cleavage of the G805R-LDLR in the ER and subsequent secretion of the released ectodomain, is reminiscent of that of transmembrane proteins undergoing rhomboid cleavage in the ER [20]. Thus, whereas similar amounts of the 140 kDa ectodomain fragment due to metalloproteinase cleavage is found in the culture media from cells expressing the WT-LDLR or the G805R-LDLR (Fig. 5), the underlying mechanism appears to be different. The WT-LDLR is predominantly cleaved at the cell surface, whereas the G805R-LDLR is predominantly cleaved in the ER.
2.7. Functionality of the G805R-LDLR at the cell membrane
As determined by Western blot analysis, a proportion of the 120 kDa G805R-LDLR escaped metalloproteinase cleavage to generate a mature 160 kDa G805R-LDLR located at the cell surface (Fig. 2 and Supplementary Fig. S2). Studies were therefore undertaken to determine the functionality of the 160 kDa G805R-LDLR at the cell surface. CHO T-REx cells stably transfected with the G805R-LDLR plasmid were used to determine the kinetics of internalization of DiD-labelled LDL. As can be seen from Fig. 6, cells expressing G805R-LDLR internalized DiD–LDL with high affinity and saturation kinetics similarly to that of cells expressing the WT-LDLR. This finding indicates that the proportion of G805R-LDLR which escapes metalloproteinase cleavage, functions normally.
Fig. 6.
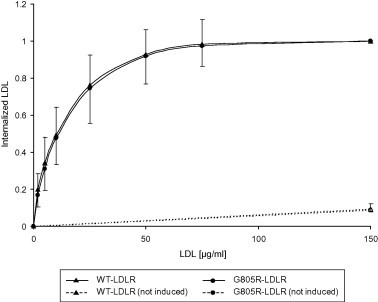
Internalization of DiD–LDL by the cell-surface G805R-LDLR. CHO T-REx cells stably transfected with the WT-LDLR plasmid or the G805R-LDLR plasmid were incubated with tetracycline (1 μg/ml) to induce the expression of the transgenes, and incubated with increasing concentrations of DiD–LDL for 2 h at 37 °C. The amount of internalized DiD–LDL was determined by flow cytometry. The amount of fluorescence detected after incubation with 150 μg/ml of DiD–LDL was given an arbitrary value of 1.0 for cells transfected with each of the plasmids. Mean (±SD) values from three separate experiments are shown. SD for the WT-LDLR is shown as upward symbols and SD for the G805R-LDLR is shown as downward symbols.
Studies were also performed to determine the stability of the mature 160 kDa G805R-LDLR at the cell surface. This was done by measuring the decay of the 160 kDa G805R-LDLR after removal of the transcription inducer tetracycline. These analyses revealed that the 160 kDa G805R-LDLR was slightly less stable than the WT-LDLR with half-lives of 13 and 16 h, respectively (Fig. 7, Quantified in Supplementary Fig. S3). A tendency for reduced stability of the G805R-LDLR at the cell surface, could possibly result from an increased propensity of the 160 kDa G805R-LDLR to undergo metalloproteinase cleavage also at the cell surface.
Fig. 7.

Stability of G805R-LDLR. CHO T-REx cells stably transfected with the WT-LDLR plasmid or the G805R-LDLR plasmid were incubated with tetracycline (tet) (1 μg/ml) to induce the expression of the transgenes. The media were removed and replaced with media without tetracycline and cultured for the indicated time intervals (h: hours), before Western blot analysis of cell lysates was performed using an antibody against the ligand-binding domain of the LDLR. Western blot of cells cultured in the absence of tetracycline to induce gene expression (No tet) was used as a control. One representative of three separate experiments is shown.
3. Discussion
In this study we have characterized the mechanism by which mutation G805R in the transmembrane domain of the LDLR gene causes familial hypercholesterolemia. As determined by Western blot analysis of lysates from transfected HepG2 cells, there were reduced amounts of the 120 kDa precursor G805R-LDLR and consequently reduced amounts of the 160 kDa mature cell-surface G805R-LDLR. The underlying mechanism for the reduced amounts of the 120 kDa precursor G805R-LDLR appeared to be that the ectodomain of G805R-LDLR was cleaved by a metalloproteinase in the ER. This cleavage generated an ectodomain fragment released into the ER lumen which appeared as a secreted 140 kDa fragment in the culture medium. It was the presence of Arg805 and not the lack of Gly805 which promoted metalloproteinase cleavage. Moreover, the proportion of G805R-LDLR which escaped ectodomain cleavage and was inserted in the cell membrane, functioned normally. Thus, mutation G805R causes familial hypercholesterolemia by reducing the number of cell-surface LDLRs without affecting their function.
The approximately 20–22 residues of the transmembrane alpha helix of single-pass membrane proteins are generally strongly enriched in aliphatic and other hydrophobic residues which stabilize the structure in the nonpolar interior of the membrane lipid bilayer. The sequence alignment in Fig. 1 shows relatively poor residue conservation in the LDLR transmembrane domain in vertebrates. However, there is clearly a selection against mutations that introduce polar and in particular charged residues in the transmembrane domain, with not one single charged residue among the more than 600 membrane-interacting residues. Thus, it is likely that mutation G805R results in a mutant LDLR that is less efficiently inserted into the ER membrane due to the energetic cost of dehydrating and transferring the charged guanidinium group of arginine into the nonpolar lipid bilayer. However, experimental studies have demonstrated that the energetic cost of inserting arginine residues in transmembrane helices, is rather low due to the arginine side chain “snorkelling” out of the lipid bilayer core [21]. Thus, a single arginine residue in a transmembrane alpha helix may lead to significant tilting of the helix as well as bilayer thinning as the guanidinium group is lifted towards the bilayer surface [22]. These changes may make the G805R-LDLR a substrate for metalloproteinase cleavage.
A likely metalloproteinase to act on G805R-LDLR in the ER is “a disintegrin and metalloproteinase” (ADAM) 17 which is induced by phorbol 12-myristate 13-acetate. ADAM17 is inhibited by batimastat and has been found to cause ectodomain shedding of a large number of transmembrane proteins [23]. ADAM17 has also been implicated by Begg et al. [19] as being the enzyme which cleaves the ectodomain of the WT-LDLR at the cell surface. However, silencing ADAM17 by the use of si-RNA caused only a marginal increase in the amount of the 120 kDa G805R-LDLR (data not shown). This indicates that also other metalloproteinases are involved. Also rhomboids such as the ER-resident rhomboid protease RHBDL4 have been shown to cleave transmembrane domains with helix-destabilizing residues [20,24,25].
Usually, ectodomain cleavage by a metalloproteinase as part of regulated intramembrane proteolysis, is followed by γ-secretase cleavage within the transmembrane domain to release the cytoplasmic domain [13]. However, as determined by our experiments where the transfected cells were cultured in the presence or absence of the γ-secretase inhibitor DAPT, mutation G805R prevented subsequent γ-secretase cleavage within the transmembrane domain. When Gly805 was substituted with Leu805 or Ala805, the transmembrane domain was subjected to γ-secretase cleavage similarly to that of the WT-LDLR. Thus, it was the presence of Arg805 within the transmembrane domain which prevented γ-secretase cleavage.
The near absolute conservation of residues Pro795 and Gly805 in the LDLR is a strong indication of the functional importance of these residues. They may possibly reduce the rigidity of the transmembrane helix and allow membrane-bound proteinases access to the LDLR peptide backbone. Proline is known as a helix breaker [26], while the lack of a side chain at glycine makes the conformation of the alpha helix backbone less stable at this residue. Our data show that these conserved residues are important for γ-secretase cleavage of the transmembrane alpha helix. However, whether γ-secretase cleavage of the LDLR plays any physiological role, remains to be determined.
There are other examples of monogenic diseases caused by mutations which lead to charged residues being introduced in the transmembrane domain of the respective proteins. One such example is mutation G308R within the transmembrane domain of fibroblast growth factor receptor 3 which accounts for the underlying mutation in 97% of achondroplasia cases [27]. Another example is mutation G163R in myelin protein zero which causes Charcot–Marie–Tooth disease Type 1b [28]. Such mutations which introduce hydrophilic residues within the hydrophobic lipid bilayer, could cause genetic diseases by affecting γ-secretase cleavage and prevent subsequent release of cytoplasmic domains for intracellular signalling, or by causing ectodomain shedding leading to reduced amounts of functioning protein.
In conclusion, the mechanism by which mutation G805R causes familial hypercholesterolemia, is through ectodomain cleavage of G805R-LDLR in the ER by a metalloproteinase. Because this mechanism is different from those underlying the previously reported classes of mutations in the LDLR gene, mutation G805R should be considered a member of a novel class of mutations causing familial hypercholesterolemia. Whether the other naturally occurring mutations in the transmembrane domain of the LDLR gene also induce ectodomain cleavage of the mutant LDLR, remains to be determined.
4. Materials and methods
4.1. Reagents
DAPT and lactacystine were purchased from Sigma–Aldrich Corp. (St. Louis, MO). Batimastat was from Calbiochem (Darmstadt, Germany). All other reagents were standard laboratory reagents.
4.2. Cell cultures
CHO T-REx cells (Invitrogen, Carlsbad, CA) express a tetracycline repressor which enables tetracycline-induced expression of genes cloned into plasmids containing the tetracycline operator 2 element. These cells were maintained in Ham’s F-12 medium (PAA Laboratories GmbH, Pasching, Austria) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml blasticidine and 100 μg/ml zeocin. HepG2 cells (European Collection of Cell Cultures, Wiltshire, UK) were maintained in Modified Eagle’s medium (PAA Laboratories GmbH, Pasching, Austria) containing 10% fetal bovine serum, 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin and non-essential amino acids, and were grown in collagen-coated plates (BD Biosciences, San Jose, CA).
4.3. Plasmids and transfections
Generation of plasmids pcDNA4-WT-LDLR and pcDNA4-WT-LDLR-HA, encoding the LDLR without or with a C-terminal HA tag, has previously been described [29]. pcDNA4-WT-LDLR was used as template to generate the mutant G805R-LDLR. The pcDNA4-WT-LDLR-HA plasmid was used as a template to generate G805R-LDLR-HA, G805L-LDLR-HA, G805A-LDLR-HA, S791L-LDLR-HA and P795L-LDLR-HA. Mutageneses were carried out using QuickChange II XL Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. The oligonucleotide sequences used for mutageneses are listed in Supplementary Table S1. HepG2 cells were transiently transfected using FuGENE HD (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s instructions. Stable transfections of CHO T-REx cells were performed as previously described [30]. The integrity of the plasmids and the transgenes of stably transfected cell lines was confirmed by DNA sequencing.
4.4. Western blot analyses
Western blot analyses of cell lysates and culture media were carried out as previously described [31]. Briefly, 24 h after transfection culture media and cell lysates were obtained and run on 4–20% Tris–HCl Criterion Precast Gels (Bio-Rad, Hercules, CA) and blotted onto Immuno-Blot PVDF Membranes (Bio-Rad, Hercules, CA). To detect the LDLR, the membranes were immunostained with a rabbit anti-HA antibody (Invitrogen, Carlsbad, CA) or a rabbit polyclonal anti-LDLR antibody which detects the linker region between ligand-binding repeats 4 and 5 (Fitzgerald Industries International, Concord, MA). Anti β-tubulin antibody was from USBiological (Salem, MA).
4.5. Flow cytometry
LDL (density 1.019–1.063 g/ml) was isolated by ultracentrifugation of human serum and labeled with fluorescent 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) (Sigma–Aldrich Corp., St. Louis, MO) as described by Pitas et al. [32]. A FACS Canto flow cytometer (BD Biosciences, San Diego, CA) was used to determine the amount of internalized DiD–LDL as previously described [33]. Briefly, stably transfected CHO T-REx were seeded out at 200,000 cells per well in 6-well plates, and incubated over night in culture media containing tetracycline (1 μg/ml) to induce gene expression. The cells were then incubated with DiD–LDL for 2 h at 37 °C before the amount of internalized DiD–LDL was measured by flow cytometry.
Appendix A. Supplementary data
References
- 1.Goldstein J.L., Hobbs H.H., Brown M.S. Familial hypercholesterolemia. In: Scriver C., Beaudet A.L., Sly W.S., Valle D., editors. The metabolic & molecular basis of inherited disease. McGraw-Hill; New York: 2001. pp. 2863–2914. [Google Scholar]
- 2.Tolleshaug H., Goldstein J.L., Schneider W.J., Brown M.S. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell. 1982;30:715–724. doi: 10.1016/0092-8674(82)90276-8. [DOI] [PubMed] [Google Scholar]
- 3.Brown M.S., Goldstein J.L. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 4.Südhof T.C., Goldstein J.L., Brown M.S., Russell D.W. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228:815–822. doi: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hobbs H.H., Russell D.W., Brown M.S., Goldstein J.L. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu. Rev. Genet. 1990;24:133–170. doi: 10.1146/annurev.ge.24.120190.001025. [DOI] [PubMed] [Google Scholar]
- 6.Koivisto U.M., Hubbard A.L., Mellman I. A novel cellular phenotype for familial hypercholesterolemia due to a defect in polarized targeting of LDL receptor. Cell. 2001;105:575–585. doi: 10.1016/s0092-8674(01)00371-3. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto T., Davis C.G., Brown M.S., Schneider W.J., Casey M.L., Goldstein J.L., Russell D.W. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 8.Leren T.P., Manshaus T., Skovholt U., Skodje T., Nossen I.E., Teie C., Sørensen S., Bakken K.S. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway. Semin. Vasc. Med. 2004;4:75–85. doi: 10.1055/s-2004-822989. [DOI] [PubMed] [Google Scholar]
- 9.UniProt Consortium Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White S.H., von Heijne G. The machinery of membrane protein assembly. Curr. Opin. Struct. Biol. 2004;14:397–404. doi: 10.1016/j.sbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Tveten K., Strøm T.B., Berge K.E., Leren T.P. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. J. Lipid. Res. 2013;54:1560–1566. doi: 10.1194/jlr.M034371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown M.S., Ye J., Rawson R.B., Goldstein J.L. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 14.Lal M., Caplan M. Regulated intramembrane proteolysis: signalling pathways and biological functions. Physiology. 2011;26:34–44. doi: 10.1152/physiol.00028.2010. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard S.J. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 16.Brodsky J.L. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura H., Tomoda H., Miura K., Takishima K., Omura S. Lactacystine increases LDL receptor level on HepG2 cells. Biochem. Biophys. Res. Commun. 1996;227:684–687. doi: 10.1006/bbrc.1996.1569. [DOI] [PubMed] [Google Scholar]
- 18.Begg M.J., Sturrock E.D., van der Westhuysen D.R. Soluble LDL-R are formed by cell surface cleavage in response to phorbol esters. Eur. J. Biochem. 2004;271:524–533. doi: 10.1046/j.1432-1033.2003.03953.x. [DOI] [PubMed] [Google Scholar]
- 19.Tveten K., Strøm T.B., Cameron J., Holla Ø.L., Berge K.E., Leren T.P. Characterization of a naturally occurring degradation product of the LDL receptor. Mol. Genet. Metab. 2012;105:149–154. doi: 10.1016/j.ymgme.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Lemberg M.K. Intramembrane proteolysis in regulated protein trafficking. Traffic. 2011;12:1109–1118. doi: 10.1111/j.1600-0854.2011.01219.x. [DOI] [PubMed] [Google Scholar]
- 21.Schow E.V., Freites J.A., Cheng P., Bernsel A., von Heijne G., White S.H., Tobias D.J. Arginine in membranes: the connection between molecular dynamics simulations and translocon-mediated insertion experiments. J. Membr. Biol. 2011;239:35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vostrikov V.V., Hall B.A., Greathouse D.V., Koeppe R.E., Sansom M.S.P. Changes in transmembrane helix alignment by arginine residues revealed by solid-state NMR experiments and coarse-gained MD simulations. J. Am. Chem. Soc. 2010;132:5803–5811. doi: 10.1021/ja100598e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber S., Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. doi: 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- 24.Fleig L., Bergbold N., Sahasrabudhe P., Geiger B., Kaltak L., Lemberg M.K. Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Cell. 2012;47:558–569. doi: 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Moin S.M., Urban S. Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. eLIFE. 2012;1:e00173. doi: 10.7554/eLife.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacArthur M., Thornton J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 27.Monsonego-Ornan E., Adar R., Feferman T., Segev O., Yayon A. The transmembrane mutation G308R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol. Cell. Biol. 2000;20:516–522. doi: 10.1128/mcb.20.2.516-522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eggers S.D.Z., Keswani S.C., Melli G., Cornblath D.R. Clinical and genetic description of a family with Charcot–Marie–Tooth disease type 1b from a transmembrane MPZ mutation. Muscle Nerve. 2004;29:867–869. doi: 10.1002/mus.20034. [DOI] [PubMed] [Google Scholar]
- 29.Sørensen S., Ranheim T., Bakken K.S., Leren T.P., Kulseth M.A. Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J. Biol. Chem. 2006;281:468–476. doi: 10.1074/jbc.M507071200. [DOI] [PubMed] [Google Scholar]
- 30.Tveten K., Holla Ø.L., Ranheim T., Berge K.E., Leren T.P., Kulseth M.A. 4-Phenylbutyrate restores the functionality of a misfolded mutant low-density lipoprotein receptor. FEBS J. 2007;274:1881–1893. doi: 10.1111/j.1742-4658.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- 31.Strøm T.B., Tveten K., Leren T.P. PCSK9 acts as a chaperone for the LDL receptor in the endoplasmic reticulum. Biochem. J. 2013 doi: 10.1042/BJ20130930. [DOI] [PubMed] [Google Scholar]
- 32.Pitas R.E., Innerarity T.L., Weinstein J.N., Mahley R.W. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis. 1981;1:177–185. doi: 10.1161/01.atv.1.3.177. [DOI] [PubMed] [Google Scholar]
- 33.Cameron J., Holla Ø.L., Ranheim T., Kulseth M.A., Berge K.E., Leren T.P. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum. Mol. Genet. 2006;15:1551–1558. doi: 10.1093/hmg/ddl077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.