

Highlights
-
•
We generated pyrrole–imidazole (PI) polyamides that could bind to an E-box motif.
-
•
PI polyamide Myc-6 induces G1 arrest and apoptosis in human osteosarcoma MG63 cells.
-
•
Myc-6 represses tumor growth both in vitro and in vivo.
-
•
Myc-6 binds to the 5′-upstream region of noncoding RNA MALAT1 and reduces its expression.
-
•
Myc-6 exerts its tumor-suppressive ability through the down-regulation of MALAT1.
Abbreviation: PI polyamide, pyrrole–imidazole polyamide
Keywords: c-MYC, E-box, MALAT1, MG63 cells, Osteosarcoma, Pyrrole–imidazole polyamide
Abstract
Gene amplification and/or overexpression of the transcription factor c-MYC, which binds to the E-box sequence (5′-CACGTG-3′), has been observed in many human tumors. In this study, we have designed 5 pyrrole–imidazole (PI) polyamides recognizing E-box, and found that, among them, Myc-6 significantly suppresses malignant phenotypes of human osteosarcoma MG63 cells both in vitro and in vivo. Intriguingly, knockdown of the putative Myc-6 target MALAT1 encoding long noncoding RNA remarkably impaired cell growth of MG63 cells. Collectively, our present findings strongly suggest that Myc-6 exerts its tumor-suppressive ability at least in part through the specific down-regulation of MALAT1.
1. Introduction
Osteosarcoma is the most common primary malignant bone tumor in adolescents and young adults [1]. The curative approach to osteosarcoma includes surgery and chemotherapy. When surgery is not feasible, patients are sometimes subjected to radiotherapy. In the general protocol, 10–12 weeks of neoadjuvant chemotherapy followed by resection and adjuvant chemotherapy is performed to minimize trauma [2]. Although 10-year disease-free survival is around 60% of patients without metastasis, over 30% of the patients suffer metastatic disease at diagnosis [3]. Despite several improvements in the curative protocols, the actual survival trends of osteosarcoma have not been substantially improved over the past 20 years. To overcome the malignant osteosarcoma, the alternative treatments such as hormonal therapy, gene therapy and immune therapy are now under investigation, instead of the conventional chemotherapy, however, the efficacies of those alternative therapeutic strategies are limited and vary among patients [1]. Thus, the development of a novel approach should be required to improve the prognosis of the patients.
MYC family including c-MYC, N-MYC and L-MYC, has been shown to be one of the most frequently altered oncogenes involved in many malignancies. Indeed, the aberrant activation of MYC is detectable in approximately 70% of human cancers [4]. MYC acts as a nuclear sequence-specific transcription factor and exerts its transcriptional activity through the formation of the heterodimeric complex with MAX [5,6]. This MYC/MAX complex specifically binds to the consensus motif termed E-box (5′-CACGTG-3′) and transactivates its target genes implicated in the crucial cellular processes such as cell cycle regulation, proliferation, metabolism and mitochondrial biogenesis [5].
Amplification of c-MYC has been found in musculoskeletal neoplasms including osteosarcoma, chondrosarcoma and soft-tissue sarcoma [7–9]. It has been also shown that c-MYC is amplified in several osteosarcoma-derived cell lines, which are sometimes resistant to anti-cancer drugs such as doxorubicin and methotrexate [10]. Notably, knockdown of c-MYC resulted in a marked enhancement of sensitivity to cisplatin in osteosarcoma MG63 cells [11]. Since these findings indicate that the amplification of c-MYC plays a pivotal role in the regulation of osteosarcoma development and acquisition of chemo-resistance, molecule(s) targeting c-MYC and/or E-box might become a novel and attractive chemotherapeutic drug.
Pyrrole–imidazole (PI) polyamides, which have been developed from the DNA-binding antibiotics distamycin A, are small synthetic chemicals composed of the aromatic amino acids N-methylpyrrole (Py) and N-methylimidazole (Im) [12]. According to Dervan’s results, PI polyamides bound to the double-strand DNA in a sequence-specific manner and suppressed the transcription of their target genes. A combination of PI polyamides recognized the specific DNA base pairs, (Im/Py, Py/Im and Py/Py pairs bind to G–C, C–G, and A–T/T–A, respectively). A concatenation of these pairs made it possible to bind to a variety of specific DNA sequences. Since PI polyamides were efficiently delivered to cell nuclei without any specific drug-delivery systems, they might be powerful materials to develop gene-specific silencers [13]. Recently, we have generated various types of PI polyamides to down-regulate the expression of their target genes. For example, PI polyamide targeting the consensus activator protein-1 (AP-1)-binding site of the TGF-β1 promoter significantly inhibited its expression [14]. Additionally, PI polyamide recognizing AP-1-binding site within the promoter of matrix metalloproteinase (MMP)-9 gene markedly reduced its expression level, and thereby strongly inhibiting the migration and invasion of human breast cancer cells in vitro and also abrogating the metastasis in vivo [15].
In this study, we have generated 5 PI polyamides recognizing E-box and found that the PI polyamide termed Myc-6 suppresses the malignant phenotypes of human osteosarcoma MG63 cells at least in part through the specific down-regulation of MALAT1.
2. Materials and methods
2.1. Synthesis of PI polyamide targeting E-box
PI polyamides targeting E-box were designed as shown in Fig. 1A and synthesized as described previously [16]. All of the PI polyamides except for Myc-3 were dissolved in distilled water (at a final concentration of 1 mM) and small aliquots were stored at −20 °C. Myc-3 was dissolved in 50% DMSO and small aliquots were kept at −20 °C.
Fig. 1.
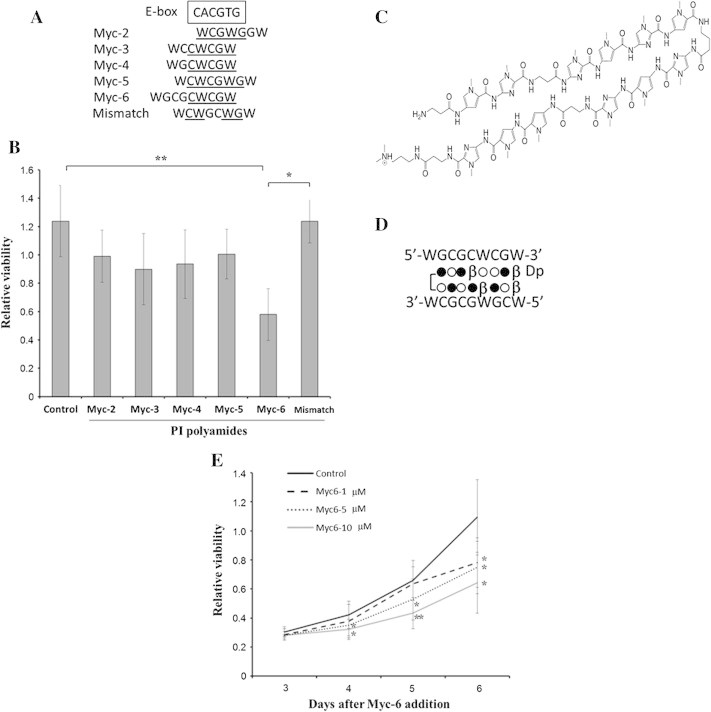
Myc-6 significantly represses cell viability of human osteosarcoma MG63 cells. (A) Sequence comparison between the putative targets of the indicated synthetic PI polyamides and the consensus E-box. Sequences corresponding to E-box are underlined. W indicates A or T. (B) WST-8 assay. MG63 cells were exposed to water (control), non-specific PI polyamide (mismatch) or with the indicated PI polyamides. Six days after the treatment, cell viability was examined by WST-8 assay. Data are expressed as the means ± SD. Differences were considered significant at p < 0.05. ∗p < 0.01;∗∗p < 0.001. (C) Chemical structure of Myc-6. (D) Schematic representation of DNA recognition by Myc-6. Open circle, filled circle, β and Dp indicate pyrrole, imidazole, β-alanine and dimethylpropanediamine, respectively. (E) Dose and time-dependency. Cell viability was measured by WST8 assay 3–6 days after addition of Myc-6. Differences between viability of cells treated with or without Myc-6 were considered significant at p < 0.05. ∗p < 0.05, ∗∗p < 0.01. Data were expressed as the means ± SD.
2.2. Cell culture and WST-8 assay
Human osteosarcoma MG63 cells were purchased from Health Science Resource Bank of Japan, and were grown in DMEM (Nakalai) supplemented with 10% of heat-inactivated fetal bovine serum (FBS), 100 μg/mL of streptomycin and 100 units/mL of penicillin. Cultures were maintained at 37 °C in a CO2 incubator with a controlled humidified atmosphere composed of 95% air and 5% CO2. Cell proliferation was assessed by WST-8 assay. Cells were seeded at a density of 200 cells/well in 96-well plate and allowed to attach overnight. Cells were then treated with the indicated concentrations of Myc-6. At the indicated time points after the treatment, medium was replaced with the fresh medium containing 1/10 volumes of WST-8 reagent, and cells were further incubated for additional 1 h followed by the measurement of absorbance at 450 nm using ARVO MX microplate reader (Perkin Elmer).
2.3. Detection of apoptosis
Apoptosis was detected by using Cell Meter™ Phosphatidiylserine Apoptosis assay kit (AAT Bioquest) following the manufacturer’s instructions. Briefly, cells were seeded at a density of 400 cells/well in 24-well glass bottom plate, and treated with the indicated concentrations of Myc-6. Following 2 days of the treatment, cells were washed in PBS once, mixed with 500 μl of assay buffer containing Apopxin Violet 500, and incubated for 15 min at room temperature. Cells were examined under a fluorescence microscope (Bravio 9000: KEYENCE) with 470ex/535em.
2.4. Immunoblotting
Cells were washed in PBS and lysed in RIPA buffer. Cell lysates (10 μg of protein) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto Immobilon-P membrane (Millipore). The membrane was blocked with Blocking-one (Nakalai) overnight at 4 °C, and then incubated with anti-caspase-3 (Cell Signaling Technology), anti-cleaved caspase-3 (5A1E: Cell Signaling Technology) or with anti-Actin (AC-15: Sigma) antibody for 1 h at room temperature, followed by the incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (GE Healthcare Life Sciences) for 1 h at room temperature. After washing in PBS containing 0.1% Tween 20, immuno-reactive bands were visualized by using ECL system (Amersham Biosciences) according to the manufacturer’s instructions.
2.5. Soft agar colony formation assay
For soft agar assay, cells (5000 cells/plate) were seeded in triplicate in 35-mm cell culture plates containing 0.35% agar and DMEM supplemented with 10% FBS. After 21 days, colonies were stained with crystal violet (WALDECK). Number of colonies with a diameter of > 100 μm was scored. The data shown were the average of 3 independent experiments.
2.6. Mouse xenograft model
Cells (3 × 106 cells) suspended in 200 μl of PBS were injected subcutaneously into the flank of 8-weeks-old male nude mice (n = 14). When the tumor volume reached to 200 mm3, the injection of water or Myc-6 (6 mg/kg body weight) was carried out intravenously and continued once weekly for a total of 4 treatments. Tumor volume was measured once weekly until 5 weeks after the first treatment. All experimental procedures performed on animals were approved by the Laboratory Animal Care Committee of Nihon University School of Medicine.
2.7. Microarray analysis
Cells were plated at a density of 2 × 105 cells/well in 6-well plate, and exposed to 10 μM of Myc-6. Twenty-four hours after the treatment, total RNA was prepared by using TRIZOL reagent (Invitrogen) followed by re-purification with RNAeasy kit (QIAGEN) according to the manufacturer’s protocols. Using Total RNA (100 ng), biotin-labeled cRNA probes were synthesized and hybridized to microarrays (Human Genome U133 Plus2.0: Affymetrix) following manufacturer’s instructions. The array data (CEL files) were quantified with the distribution free weighted method (DFW) [17] using statistical language R (http://www.r-project) [18] and Bioconductor (http://www.bioconductor.org/) [19]. To identify the differentially expressed genes, the rank products method [20] was applied to DFW quantified data. Probe sets with an FDR 0.05 or lower were regarded as having different expression levels between two groups.
2.8. Quantitative real-time PCR
Total RNA was extracted as described above. 500 ng of total RNA were reverse-transcribed using iScript (Bio-Rad). Quantitative real-time PCR was performed using SYBR® Premix ExTaq™ system (Takara). A mixture of cDNA derived from total RNA of MG63 cells treated with or without various amounts of Myc-6 was used as a reference. We then prepared dilution series of this cDNA mixture, and performed real-time PCR to obtain a standard curve for each gene. Three independent measurements were taken and the amounts were estimated by extrapolation from a standard curve. Expression values were normalized against the expression of β-2-microglobulin and used as an endogenous control. The sequences of the primers used in this study were listed in Supplementary Table 1.
2.9. siRNA-mediated silencing
For Knockdown of MALAT1, cells were plated at a density of 200 cells/well in a 96-well plate and allowed to attach overnight. Cells were then transiently transfected with control siRNA or with siRNA against MALAT1 by using LipofectAMINE RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions.
2.10. Statistical analysis
All values were expressed as the mean ± SE, and statistical significance was analyzed using the t-test. Significance was set at a p value of <0.05.
3. Results
3.1. Myc-6-mediated growth suppression of human osteosarcoma MG63 cells
We have designed and generated 5 pyrrole–imidazole (PI) polyamides targeting E-box to suppress malignant phenotypes of cancerous cells (Fig. 1A). Human osteosarcoma MG63 cells were treated with or without these PI polyamides. Six days after the treatment, their cell viability was examined by WST-8 assay. Among 5 PI polyamides examined, Myc-6 significantly inhibited MG63 cell growth and thus we have focused on Myc-6 (Fig. 1B). Schematic drawing of Myc-6 chemical structure and its binding to theoretical target sequence were shown in Fig. 1C and D, respectively. Additional WST assays clearly demonstrated that Myc-6 represses MG63 cell growth in a dose- and time-dependent manner (Fig. 1E).
3.2. MG63 cells undergo apoptosis in response to Myc-6
Since these results indicated that Myc-6 has an ability to prohibit MG63 cell growth, we asked whether Myc-6 could affect the cell cycle distribution. After 72 h of incubation with or without Myc-6, both floating and adherent cells were collected and their cell cycle distribution was examined by Tali image-based cytometer. As seen in Fig. 2A, a dose-dependent massive increase in number of cells with sub-G1 or G1 DNA content was detectable, and also number of cells bearing S phase DNA content was remarkably reduced in a dose-dependent fashion. These observations indicate that MG63 cells undergo apoptosis and/or G1 cell cycle arrest following Myc-6 exposure.
Fig. 2.
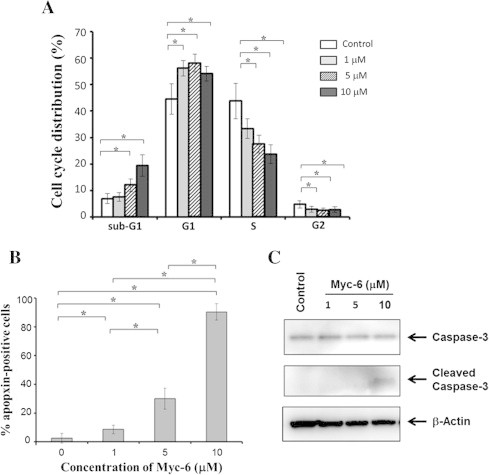
Myc-6-dependent induction of apoptosis. (A) Tali image-based cytometric analysis. MG63 cells were treated with the indicated concentrations of Myc-6 or left untreated. Following 72 h of Myc-6 exposure, the adherent and attached cells were collected and processed for Tali image-based cytometric analysis. Differences versus cells treated with or without Myc-6 were considered significant at p < 0.05. ∗p < 0.0001. The columns represent means ± SD. (B) Apopxin violet staining. Cells were treated as in (A). Forty-eight hours after the treatment, cells were incubated with apopxin violet 500 solution for 30 min, and then number of apopxin-positive cells was counted. ∗p < 0.01. (C) Cleavage of pro-apoptotic caspase-3. Cells were treated as in (A). Forty-eight hours after the treatment, cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies.
To adequately address this issue, MG63 cells treated with or without Myc-6 were examined by using apopxin violet which monitors the early phase of apoptosis. As shown in Fig. 2B, a dose-dependent marked increase in number of apopxin-positive cells was detectable in response to Myc-6. In contrast, number of apopxin-positive cells was less than 5% without Myc-6. Under the same experimental conditions, cell lysates were prepared and analyzed for the presence of cleaved pro-apoptotic caspase-3 by immunoblotting. As shown in Fig. 2C, the native caspase-3 was detected in cells regardless of Myc-6 treatment, whereas cleaved caspase-3 was detectable in cells exposed to 10 μM of Myc-6. Collectively, our present findings strongly suggest that Myc-6 has an ability to promote apoptosis in MG63 cells.
3.3. Administration of Myc-6 attenuates MG63-derived tumor growth in vivo
Considering that Myc-6 has a potential pro-apoptotic activity, it is likely that Myc-6 might exert tumor-suppressive ability in vitro and in vivo. To address this issue, MG63 cells were plated onto soft agar medium containing the indicated concentrations of Myc-6. Three weeks after the treatment, number of colonies with a diameter of > 100 μm was scored. The number of colonies was significantly decreased in response to 5 or 10 μM of Myc-6 as compared to that of control cells (Fig. 3A), suggesting that Myc-6 prohibits anchorage-independent growth of MG63 cells.
Fig. 3.
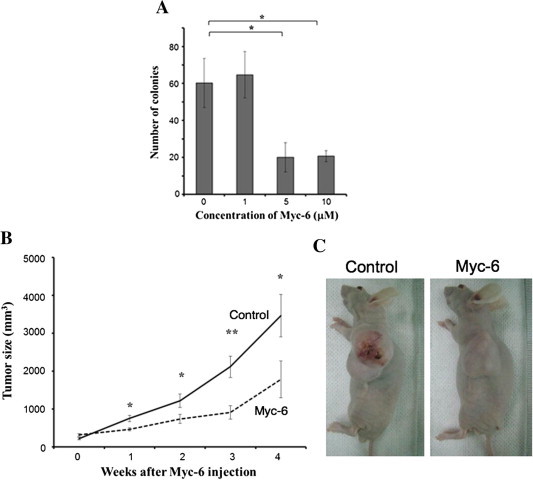
Tumor-suppressive activity of Myc-6 in vivo. (A) Anchorage-independent growth of MG63 cells. Cells were plated onto soft agar medium containing the indicated concentrations of Myc-6. Three weeks after Myc-6 treatment, number of colonies with a diameter of >100 μM was scored. Differences were considered significant at p < 0.05. ∗p < 0.01. The columns represent means ± SD. (B and C) Immuno-deficient nude mice bearing subcutaneously developed tumors derived from MG63 cells were injected intravenously with or without Myc-6 (6 μg/kg) once a week. At the indicated time periods after the first injection, tumor volume was measured. p < 0.05 was considered statistically significant. ∗p < 0.05; ∗∗p < 0.01 (B). Representative photographs of tumors were taken 4 weeks after Myc-6 injection (C).
To examine the possible effect of Myc-6 in vivo, we injected MG63 cells subcutaneously into each anterior flank region of athymic nude mice. When tumor volume reached to ∼200 mm3, mice were randomly divided into two groups. Experimental group received 6 mg/kg of Myc-6 intravenously once a week. Control group similarly received water. At the indicated time points after the first administration, tumor volume was measured. Four weeks after the treatment, mice were sacrificed. Consistent with the results obtained from soft agar colony formation assay, a remarkable reduction of tumor volume was observed in Myc-6-treated mice relative to that of control mice (Fig. 3B). The representative pictures were shown in Fig. 3C. These results strongly suggest that Myc-6 possesses tumor-suppressive activity in vivo.
3.4. MALAT1 is a putative Myc-6-target gene
To elucidate the precise molecular mechanism(s) behind Myc-6-mediated induction of apoptosis and suppression of tumor growth, we sought to identify a putative Myc-6-target gene(s) by using DNA microarray-based analysis. After the extensive screening, we have finally obtained 14 genes whose expression levels were significantly lower in Myc-6-treated MG63 cells relative to those in untreated cells (Table 1). The close inspection of their 5′-upstream regions (up to 1 kb) revealed that MALAT1, which encodes a long noncoding RNA [21], contains a theoretical Myc-6-target sequence including E-box-like motif (at positions −258 to −251) (Fig. 4A). To verify whether Myc-6 could specifically bind to this sequence, we have performed gel retardation assay. FITC-labeled double-strand oligonucleotide corresponding to the above-mentioned MALAT1 sequence or the unrelated sequence (Mismatch, Supplementary Table 2) was generated, incubated with or without Myc-6 and the reaction mixtures were separated by polyacrylamide gel electrophoresis. Unlike the non-specific oligonucleotide, a definite mobility shift of the specific oligonucleotide was observed in the presence of Myc-6 compared to that in the absence of Myc-6 (Fig. 4B). Furthermore, this mobility shift was clearly inhibited by the excess amount of specific oligonucleotide but not by non-specific one (Fig. 4C), indicating that Myc-6 specifically binds to its theoretical target site located within 5′-upstream region of MALAT1.
Table 1.
List of genes down-regulated by the treatment with Myc-6.
| FDR (%) | Gene symbol | Gene name |
|---|---|---|
| 0.00 | NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| 0.17 | DEPDC6 | DEP domain containing 6 |
| 0.14 | NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| 0.38 | COL14A1 | Collagen, type XIV, alpha 1 |
| 0.40 | MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| 1.00 | MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) |
| 1.23 | RNF145 | Ring finger protein 145 |
| 1.21 | MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| 1.20 | COL3A1 | Collagen, type III, alpha 1 |
| 1.19 | LAMA4 | Laminin, alpha 4 |
| 2.42 | NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| 2.65 | CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 |
| 2.95 | C5orf24 | Chromosome 5 open reading frame 24 |
| 3.67 | COL3A1 | Collagen, type III, alpha 1 |
| 4.04 | SFRP1 | Secreted frizzled-related protein 1 |
| 4.35 | ELK4 | ELK4, ETS-domain protein (SRF accessory protein 1) |
| 4.22 | BE466173 | RNA binding motif protein 39 |
| 4.86 | TAF15 | TAF15 RNA polymerase II, TATA box binding protein (TBP)-associated factor |
Fig. 4.
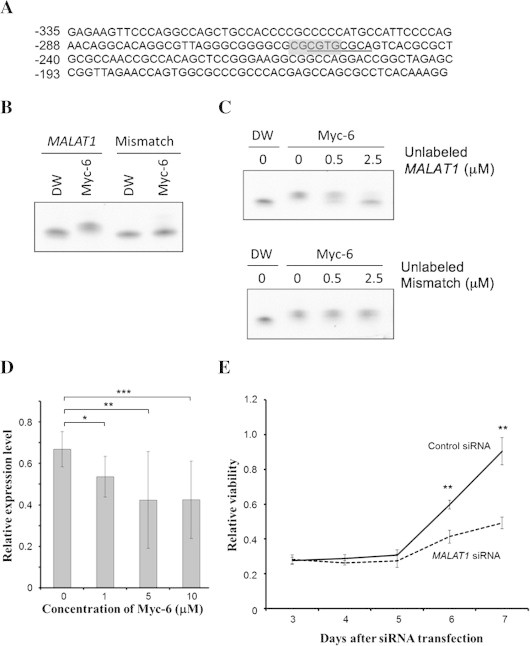
MALAT1 is one of possible target genes of Myc-6. (A) Sequence of 5′-upstream region of human MALAT1 gene. The positions relative to the first nucleotide of MALAT1 exon 1 (+1) are indicated. The putative Myc-6-target sequence is underlined, and E-box-like sequence is highlighted in gray. (B and C) Gel retardation assay. FITC-labeled specific (MALAT1) or non-specific (Mismatch) oligonucleotide (1 μM) was incubated with water or Myc-6 (5 μM) for 1 h at 37 °C. The reaction mixtures were separated by 4–20% gradient polyacrylamide gel electrophoresis and then analyzed by LAS4000 (FUJIFILM) (B). FITC-labeled specific oligonucleotide (0.5 μM) was incubated with Myc-6 (0.5 μM) in the presence or absence of the indicated amounts of unlabeled specific (upper) or non-specific (lower) oligonucleotides. The reaction mixtures were analyzed as in (B) (C). (D) Myc-6-mediated down-regulation of MALAT1. MG63 cells were treated with or without the indicated concentrations of Myc-6. Twenty-four hours after treatment, the expression level of MALAT1 was analyzed by real-time PCR. p < 0.05 was considered statistically significant. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. The columns represent means ± SD. (E) Knockdown of MALAT1 suppresses MG63 cell growth. MG63 cells were transfected with control siRNA or with siRNA against MALAT1. The cell viability was assessed by WST-8 assay. ∗∗p < 0.001.
We then investigated whether Myc-6 could reduce the expression level of MALAT1 by real-time PCR. Consistent with the results obtained from the microarray analysis, Myc-6 exposure led to a dose-dependent decrease in the expression level of MALAT1 in MG63 cells (Fig. 4D). Finally, we examined whether MALAT1 could be involved in the regulation of Myc-6-mediated growth suppression by employing siRNA-mediated knockdown of MALAT1. MG63 cells were transfected with control siRNA or with siRNA against MALAT1. As shown in Fig. 4E, a marked reduction in cell viability was observed in MALAT1-knockdown cells relative to that in control cells. Taken together, our present findings strongly suggest that E-box-targeting PI polyamide Myc-6 might exerts its growth- and tumor-suppressive activity at least in part through the specific down-regulation of MALAT1.
4. Discussion
In this study, we have found for the first time that Myc-6, a PI polyamide targeting E-box, efficiently suppresses malignant phenotypes of human osteosarcoma MG63 cells both in vitro and in vivo, which might be at least in part due to Myc-6-mediated specific down-regulation of MALAT1. As expected, MALAT1 promoter region possessed the theoretical Myc-6-binding sequence containing E-box-like motif. Thus, it is likely that PI polyamide-mediated specific targeting E-box might become an attractive strategy to develop novel anti-tumor drug(s) for the treatment of osteosarcoma.
The development of a small synthetic chemical compound(s), which could block oncogenic signaling pathway, would be ideal as a cancer therapeutic. Several challenges have persisted with respect to drug discovery, including virtual screening of large compound datasets based on computer-aided drug design. Among them, evidence is accumulating that PI polyamide-mediated gene silencing is an alternative and attractive approach to repress malignant phenotypes of tumors [15]. In the current study, we have synthesized 5 PI polyamides targeting E-box according to the general sequence recognition rule for PI polyamides [12]. Among them, Myc-6 displayed an ability to reduce the viability and anchorage-independent growth of MG63 cells in vitro. Moreover, administration of Myc-6 markedly suppressed MG63 cells-derived tumor growth in vivo. Of note, there were no significant changes in body weight between Myc-6-treated and untreated groups of healthy mice, indicating there exists no toxicity of the treatment (data not shown). Additionally, close inspection of their internal organs such as liver, kidney and lung revealed that no morphological changes can be found between them (data not shown). Collectively, it is likely that Myc-6 possesses potential tumor-suppressive function without any severe side effects. However, it should be noted that the remaining 4 PI polyamides examined appear to be biologically insufficient, as no remarkable reduction in cell viability was observed. Although the precise root of this problem has not yet been known, it might be due to their lack of sequence-specificity and/or efficient nuclear access. Further studies should be required to improve the molecular design of PI polyamides.
Another finding of the present study was that MALAT1 encoding a long noncoding RNA might be one of Myc-6-target genes. Consistent with our observations, it has been shown that MALAT1 is up-regulated in several human solid tumors and its aberrant higher expression level is tightly linked to tumor metastasis and recurrence [22]. Notably, forced expression of MALAT1 enhanced tumor formation in nude mice, while depletion of MALAT1 in cancerous cells reduced their tumorigenicity [23]. These findings suggest that MALAT1 has an oncogenic function, however, the precise molecular basis has been elusive. Recently Tripathi et al. described that depletion of MALAT1 leads to an induction of tumor suppressor p53 along with its targets such as p21WAF1 and GADD45, indicating that p53 and/or another p53 family members such as p73 might be a downstream mediator of MALAT1 [24]. Further experiments should be required to adequately address how MALAT1 could contribute to Myc-6-dependent tumor suppression.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This research was supported in part by MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2011-2015) to K.F., Y.T., N.F., T.U., M.S., H.N., and by JSPS KAKENHI Grant Number 23501313 to M.S., K.F., N.F. and U.T.
Appendix A. Supplementary data
List of primers used in real-time PCR.
List of oligoucleotides used in gel retardation assay.
References
- 1.Tan M.L., Choong P.F., Dass C.R. Osteosarcoma: conventional treatment vs. gene therapy. Cancer Biol. Ther. 2009;8:106–117. doi: 10.4161/cbt.8.2.7385. [DOI] [PubMed] [Google Scholar]
- 2.Gill J., Ahluwalia M.K., Geller D., Gorlick R. New targets and approaches in osteosarcoma. Pharmacol. Ther. 2013;137:89–99. doi: 10.1016/j.pharmthera.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Longhi A., Errani C., De P.M., Mercuri M., Bacci G. Primary bone osteosarcoma in the pediatric age: state of the art. Cancer Treat. Rev. 2006;32:423–436. doi: 10.1016/j.ctrv.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson J.A., Cleveland J.L. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 5.Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cascon A., Robledo M. MAX and MYC: a heritable breakup. Cancer Res. 2012;72:3119–3124. doi: 10.1158/0008-5472.CAN-11-3891. [DOI] [PubMed] [Google Scholar]
- 7.Barrios C., Castresana J.S., Ruiz J., Kreicbergs A. Amplification of c-myc oncogene and absence of c-Ha-ras point mutation in human bone sarcoma. J. Orthop. Res. 1993;11:556–563. doi: 10.1002/jor.1100110410. [DOI] [PubMed] [Google Scholar]
- 8.Ladanyi M., Park C.K., Lewis R., Jhanwar S.C., Healey J.H., Huvos A.G. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn. Mol. Pathol. 1993;2:163–167. [PubMed] [Google Scholar]
- 9.Barrios C., Castresana J.S., Falkmer U.G., Rosendahl I., Kreicbergs A. C-myc oncogene amplification and cytometric DNA ploidy pattern as prognostic factors in musculoskeletal neoplasms. Int. J. Cancer. 1994;58:781–786. doi: 10.1002/ijc.2910580605. [DOI] [PubMed] [Google Scholar]
- 10.Hattinger C.M., Stoico G., Michelacci F., Pasello M., Scionti I., Remondini D., Castellani G.C., Fanelli M., Scotlandi K., Picci P., Serra M. Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosomes. Cancer. 2009;48:289–309. doi: 10.1002/gcc.20640. [DOI] [PubMed] [Google Scholar]
- 11.Xie X.K., Yang D.S., Ye Z.M., Tao H.M. Enhancement effect of adenovirus-mediated antisense c-myc and caffeine on the cytotoxicity of cisplatin in osteosarcoma cell lines. Chemotherapy. 2009;55:433–440. doi: 10.1159/000265527. [DOI] [PubMed] [Google Scholar]
- 12.Dervan P.B., Edelson B.S. Recognition of the DNA minor groove by pyrrole–imidazole polyamides. Curr. Opin. Struct. Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 13.Murty M.S., Sugiyama H. Biology of N-methylpyrrole-N-methylimidazole hairpin polyamide. Biol. Pharm. Bull. 2004;27:468–474. doi: 10.1248/bpb.27.468. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda H., Fukuda N., Ueno T., Katakawa M., Wang X., Watanabe T., Matsui S., Aoyama T., Saito K., Bando T., Matsumoto Y., Nagase H., Matsumoto K., Sugiyama H. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole–imidazole polyamide targeting of the transforming growth factor-beta1 promoter. Kidney Int. 2011;79:46–56. doi: 10.1038/ki.2010.330. [DOI] [PubMed] [Google Scholar]
- 15.Wang X., Nagase H., Watanabe T., Nobusue H., Suzuki T., Asami Y., Shinojima Y., Kawashima H., Takagi K., Mishra R., Igarashi J., Kimura M., Takayama T., Fukuda N., Sugiyama H. Inhibition of MMP-9 transcription and suppression of tumor metastasis by pyrrole–imidazole polyamide. Cancer Sci. 2010;101:759–766. doi: 10.1111/j.1349-7006.2009.01435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bando T., Sugiyama H. Synthesis and biological properties of sequence-specific DNA-alkylating pyrrole–imidazole polyamides. Acc. Chem. Res. 2006;39:935–944. doi: 10.1021/ar030287f. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z., McGee M., Liu Q., Scheuermann R.H. A distribution free summarization method for Affymetrix GeneChip arrays. Bioinformatics. 2007;23:321–327. doi: 10.1093/bioinformatics/btl609. [DOI] [PubMed] [Google Scholar]
- 18.R development core team . R foundation for statistical computing; Vienna, Austria: 2006. R: A language and environment for statistical computing. [Google Scholar]
- 19.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A.J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J.Y., Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breitling R., Armengaud P., Amtmann A., Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. doi: 10.1016/j.febslet.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 21.Ji P., Diederichs S., Wang W., Boing S., Metzger R., Schneider P.M., Tidow N., Brandt B., Buerger H., Bulk E., Thomas M., Berdel W.E., Serve H., Muller-Tidow C. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 22.Gutschner T., Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703–719. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt L.H., Spieker T., Koschmieder S., Schaffers S., Humberg J., Jungen D., Bulk E., Hascher A., Wittmer D., Marra A., Hillejan L., Wiebe K., Berdel W.E., Wiewrodt R., Muller-Tidow C. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J. Thorac. Oncol. 2011;6:1984–1992. doi: 10.1097/JTO.0b013e3182307eac. [DOI] [PubMed] [Google Scholar]
- 24.Tripathi V., Shen Z., Chakraborty A., Giri S., Freier S.M., Wu X., Zhang Y., Gorospe M., Prasanth S.G., Lal A., Prasanth K.V. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:e1003368. doi: 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of primers used in real-time PCR.
List of oligoucleotides used in gel retardation assay.