

Abstract
The abundance of long noncoding RNAs (lncRNAs) and their wide range of functional roles in human cells are fast becoming realized. Importantly, lncRNAs have been identified as epigenetic modulators and consequently play a pivotal role in the regulation of gene expression. A human immunodeficiency virus-encoded antisense RNA transcript has recently been reported and we sought to characterize this RNA and determine its potential role in viral transcription regulation. The intrinsic properties of this human immunodeficiency virus-expressed lncRNA were characterized and the data presented here suggest that it functions as an epigenetic brake to modulate viral transcription. Suppression of this long antisense transcript with small single-stranded antisense RNAs resulted in the activation of viral gene expression. This lncRNA was found to localize to the 5′ long-term repeats (LTR) and to usurp components of endogenous cellular pathways that are involved in lncRNA directed epigenetic gene silencing. Collectively, we find that this viral expressed antisense lncRNA is involved in modulating human immunodeficiency virus gene expression and that this regulatory effect is due to an alteration in the epigenetic landscape at the viral promoter.
Introduction
Human immunodeficiency virus type-1 (HIV-1) is a lentivirus that causes a persistent infection, ultimately resulting in the demise of immune regulatory cells. In vivo, some HIV infections may undergo transcriptional shutdown leading to a state of viral latency in resting memory CD4+ cells (reviewed in ref. [1]). The underlying cellular mechanism(s) responsible for establishing and/or maintaining latency remain indistinct.2 It is known that the proviral genome functions analogously to host genes, interacting with chromosomal proteins and thus subject to the actions of the host epigenetic regulatory machinery (reviewed in ref. [3]). Notably, the reactivation of latent virus has been shown to involve the acetylation of histone tails specifically at the HIV promoter/LTR. Conversely, silent state chromatin marks to the viral promoter/LTR have been observed when the provirus is repressed.4 These insights suggest that epigenetic mechanisms are involved in viral latency. However, the mechanism by which these epigenetic marks are localized to particular targeted regions has remained unclear.
The complex paradigm of RNA-based gene regulation is slowly being revealed and there is a large body of evidence that infers a central role for noncoding RNAs (ncRNAs) in controlling gene expression by targeting various stages of epigenetic remodeling (reviewed in ref. [5]). We and others have observed that antisense long noncoding RNAs (lncRNAs) are able to guide epigenetic silencing complexes to targeted loci in the promoters of protein-coding genes resulting in gene silencing in human cells (reviewed in ref. [6]). It is thus becoming apparent that antisense ncRNAs are endogenous effector molecules capable of utilizing RNA-based transcriptional silencing pathways in human cells.7,8,9,10 This model lends itself to the possibility of disrupting lncRNA-mediated gene regulation. Indeed the direct targeting of antisense lncRNAs using small RNAs, functional in both the transcriptional11 and posttranscriptional gene silencing pathways,12,13 can lead to suppression of the targeted lncRNA. This suppression results in the derepression of the lncRNA targeted protein-coding gene and ultimately the activation of that gene.7,8,9,10
The number of functional lncRNAs being identified in human cells continues to rise and we are rapidly gaining an understanding of the disparate roles these transcripts play in different disease systems.14 Antisense ncRNAs emanating from the HIV genome during viral infection have been described15,16,17,18 and such transcripts are reportedly capable of suppressing HIV gene expression.17,18 As such we set out to further investigate the role of these HIV-encoded ncRNAs in the regulation of viral transcription and to gain insights into the mechanism by which this regulation occurs. We find here that the targeted suppression of HIV antisense ncRNAs by both posttranscriptional and transcriptional gene silencing of the ncRNA promoter can result in the activation of HIV gene expression and replication in both cell lines and primary human CD4+ T cells. Furthermore, we show that this ncRNA regulates viral gene expression by the endogenous RNA-directed epigenetic pathway involving DNA methyltransferase 3a (DNMT3a), histone deacetylase 1 (HDAC-1), and the polycomb protein enhancer of zeste homolog 2 (EZH2).
Results
Characterization of HIV-expressed antisense ncRNAs
Several independent studies have observed an antisense transcript emanating from the nef region of HIV (Figure 1a).15,16,18 In this study, we sought to verify and further characterize this HIV-expressed antisense transcript (Figure 1). The presence of this HIV-encoded antisense ncRNA was investigated in the ACH2 and J1.1 cell lines, which are chronically infected cell lines harboring the HIV-1 LAI strain and have served as in vitro culture models for latent HIV-1 infection. The antisense ncRNA was detected by directional reverse transcription (RT) and PCR using various primers (annotated in Figure 1a) and was clearly observed in both ACH2 (Figure 1b) and J1.1 cells (Figure 1c). The detection of the ncRNA by directional RT was specific as evidenced by the failure to detect the ncRNA when using a PCR primer set upstream from the RT primer (Figure 1d). The ncRNA transcript appears to span the majority of the viral genome (Figure 1e), and overlaps the 5′LTR (Figure 1f). Previous studies have found that some antisense ncRNAs involved in epigenetically modulating gene transcription lack polyadenylation.7 We find that the HIV-expressed antisense ncRNA characterized here also lacks a polyadenylated tail (Figure 1g), suggesting some level of nuclear retention8 and correlates with previous reports that HIV-expressed asRNAs are localized in the nuclei of infected cells.18 Taken together, these data verify the existence of an HIV-encoded antisense lncRNA and the properties thereof are consistent with previous observations of endogenous ncRNAs in human cells.19,20 The possibility therefore exists that this HIV-expressed long antisense transcript may be involved in the epigenetic regulation of HIV.
Figure 1.

Characterization of HIV-1-expressed antisense ncRNAs. (a) Schematic representation of the HIV-1 genome. The 5′LTR promoter is highlighted depicting the binding sites of the various directional RT (depicted by green arrows) and PCR (depicted by red arrows) primers used to detect the HIV-1 antisense ncRNA. (b) Detection of the HIV-1 antisense ncRNA in ACH2 cells. (c) Detection of the HIV-1 antisense ncRNA in J1.1 cells. The ncRNA was detected in (b) and (c) using directional RT primer 1 and the pol-specific PCR primer set 1. The HX10 molecular clone plasmid was used as a positive PCR control. (d) Specificity of ncRNA detection. The antisense ncRNA is undetectable with PCR primer set 4, which is upstream of the RT primer 1 site. (e) The antisense ncRNA spans the majority of the HIV genome. The HIV-encoded antisense ncRNA detected spans env, pol, and part of gag, significantly beyond the previously proposed antisense polyA signal16 as determined by RT with the gag-specific RT primer 1 and PCR with the env-specific primer set 2. (f) The HIV-encoded antisense ncRNA overlaps the 5′LTR. This panel shows detection of LTR overlapping antisense transcripts in ACH2 cells as determined by directional reverse transcription and PCR with the primers shown schematically. (g) The antisense ncRNA lacks a poly A tail. Directional RT was performed with the following primers: forward primer of primer set 3 (set 3 F) found within the LTR, a control oligo (dT) primer (dTT), and a combination of both set 3 F and dTT (mix), using total RNA or dTT-cleared RNA as a template. The ncRNA-specific RT primer was able to detect a product from the total and dTT-cleared RNA samples whereas the oligo (dT) control primer was only able to detect a primer in the total RNA as expected. This is consistent with previously published data showing that the antisense ncRNA is localized in the nucleus.18 HIV, human immunodeficiency virus; ncRNA, noncoding RNAs; RT, reverse transcription.
Activation of HIV gene expression by suppression of the HIV-expressed antisense lncRNA
Elucidation of the role of this HIV-encoded ncRNA and the molecular mechanism by which it acts could provide invaluable insight into a potential contributing component of viral latency. It has previously been reported that suppression of certain ncRNAs by RNA interference, either in vitro or in vivo, has resulted in derepression of the ncRNA targeted protein-coding gene and concomitant transcriptional activation.7,8,9,12 Similarly, we show that suppression of this HIV-expressed ncRNA by posttranscriptional silencing results in the activation of HIV (Figure 2). A small single-stranded antisense RNA targeted to the HIV-encoded antisense noncoding RNA transcript at the region overlapping the env gene (as154; Figure 2a) was cloned into an HIV-1 lentiviral vector (HIV7) containing the mycophenolic acid selectable IMPDH2 gene (Figure 2b). Activation of HIV gene expression and replication is shown in HIV-infected primary CD4+ T cells stably transduced with a lentiviral vector expressing the U6-driven as154 small RNA and is shown by quantitative PCR (qPCR) (Figure 2c) and p24 antigen enzyme-linked immunosorbent assay (Figure 2d). This activation correlates with a concomitant reduction of the HIV-expressed antisense lncRNA caused by posttranscriptional gene silencing (Figure 2e). Nuclear run-on assays show that the observed activation of HIV is transcriptional in nature (Figure 2f). These data showing knockdown of the HIV lncRNA and the consequent activation of HIV, suggest that the HIV antisense lncRNA plays a role in regulating HIV gene expression and replication. To determine possible changes in histone modifications in cells where the antisense lncRNA has been manipulated, chromatin immunoprecipitation (ChIP) analysis was performed at the viral 5′LTR using antibodies against the silent state histone modifications H3K27me3 and H3K9me2 (Figure 2g,h). In stable ACH2 cells expressing the small RNA as154 in which the HIV antisense lncRNA is downregulated, we observed a reduction in both H3K27me3 (Figure 2g) and H3K9me2 (Figure 2h) at the viral promoter. The HIV antisense lncRNA therefore appears to be responsible for regulating the epigenetic state at the viral promoter and subsequent viral gene transcription, similar to previous observations with endogenous lncRNAs.8
Figure 2.

The effects of a stably expressed antisense RNA targeted to the HIV-1-expressed antisense transcript on viral gene expression. (a) Schematic representation of the small antisense RNA (as154) target site within the HIV-1 expressed ncRNA transcript. (b) HIV-1 lentiviral vectors containing the MPA selectable IMPDH2 gene used to transduce and generate stable cell lines containing integrated vectors. The control vector is an empty HIV-1 lentiviral vector and the HIVas154 vector contains the activating U6-as154 expression cassette. (c,d) CD4+ T cells were transduced with the respective vectors (MOI: 20) and stable cells were infected with HIV-1 HX10 (MOI: 0.1). The cultures were assessed 72 hours later by (c) qRTPCR for viral expression and (d) by p24 antigen enzyme-linked immunosorbent assay. (e) The HIV-1 antisense RNA expression was characterized by directional RT and qPCR in the small RNA transduced CD4+ T cells as described in b and c. (f) Nuclear run-on analysis of the transduced HIV-1-infected CD4+ T cells. For c, e, and f, HIV gene expressing was measured by qPCR with primer set 5 within the RT region. For c–f, triplicate-treated cultures are shown as mean + SD and P values from a paired t-test. (g) ChIP analysis was performed to determine changes in histone modifications at the 5′LTR in cells expressing the small RNA as154. A reduction of H3K27me3 at the 5′LTR was observed in ACH2 cells transduced with a lentiviral vector expressing as154. (h) ChIP analysis also revealed a reduction of H3K9me2 at the viral 5′LTR in ACH2 cells transduced with a lentiviral vector expression as154. For g and h, enrichment at the 5′LTR was determined by qPCR with primer set 3, and n = 4 ChIP samples are shown as mean + SEM with P values from a paired t-test. ChIP, chromatin immunoprecipitation; HIV, human immunodeficiency virus; MOI, multiplicity of infection; MPA, mycophenolic acid; ncRNA, noncoding RNAs; RT, reverse transcription.
Susceptibility of HIV-expressed antisense ncRNA to transcriptional gene silencing
Next, we sought to determine the susceptibility of the HIV-encoded antisense lncRNA to silencing with small RNAs utilizing the transcriptional gene-silencing pathway. Target sites within the previously characterized promoter15 driving the HIV antisense lncRNA were determined by a recently published algorithm.11 Five single-stranded small antisense RNAs targeting the identified regions within the promoter (aspro1-5; Figure 3a) were cloned into lentiviral vectors (Figure 3b), stable cell cultures were generated, and their resulting activating properties determined.
Figure 3.

Transcriptional activation of HIV-1 by small antisense RNAs targeting of the asHIV promoter. (a) Schematic representation of the small antisense RNA target sites in the HIV-1 expressed ncRNA promoter.16 (b) HIV-1 lentiviral vectors containing the MPA selectable IMPDH2 gene used to transduce and generate stable cell lines containing integrated vectors. The control vector is an empty HIV-1 lentiviral vector and the HIVaspro vectors contain the U6-driven small RNAs targeted to the HIV-1 expressed ncRNA promoter, aspro1-5. (c) The effects of small antisense RNA promoter targeting on viral gene expression in vector transduced stable pools of J1.1 cells. (d) Activation of HIV-1 gene expression from nuclear run-on analysis in transduced J1.1 cells stably expressing small ncRNAs targeted to the asHIV promoter. (e) The effects of small RNA aspro5 on viral gene expression in primary CD4+ T cells. Stably-transduced aspro5-expressing CD4+ T cells were infected with HIV-1 NL4-3 (MOI: 0.1) and viral expression determined 72 hours later. (f) Specific inhibition of the HIV antisense ncRNA by small RNA aspro5. Transient cotransfection of cells with pME18S-asHIV18 expressing the full-length HIV-1 antisense ncRNA and pU6M2-aspro5 shows that aspro5 knocks down the antisense ncRNA in a dose-dependent manner. c and d represent data from stable cell populations. e and f show mean + SD of triplicate-treated cultures and P-values from a paired t-test. (g) ChIP analysis was performed to determine changes in histone modifications at the 5′LTR in cells expressing the small RNA aspro5. A reduction of H3K27me3 and H3K9me2 at the 5′LTR was observed in ACH2 cells transduced with a lentiviral vector expressing aspro5. For g, enrichment at the 5′LTR was determined by qPCR with primer set 3, and n = 4. ChIP samples are shown as mean + SEM with P-values from a paired t-test. ChIP, chromatin immunoprecipitation; HIV, human immunodeficiency virus; MOI, multiplicity of infection; MPA, mycophenolic acid ncRNA, noncoding RNAs.
We find that the targeted suppression of this HIV-expressed ncRNA with small RNAs utilizing the transcriptional silencing pathway also results in the activation of HIV expression (Figure 3c,d). Three of the small antisense RNAs (asRNAs) caused activation of viral gene expression in stable J1.1 cell populations expressing the asRNAs (Figure 3c) and nuclear run-on assays showed that the observed activation was at a transcriptional level (Figure 3d). One small asRNA targeted to the reported promoter for the HIV-expressed antisense ncRNA, aspro5, proved consistently effective at activating HIV expression across different cell lines. In addition, the level of activation by the aspro5 small asRNA was found to be at a similar level to that of a posttranscriptionally targeted small RNA, the as154 (Figure 2c,d). A lentiviral vector expressing the aspro5 small asRNA (Figure 3b) was then used to transduce primary CD4+ T cells. When these cells were infected with HIV those cultures stably expressing small asRNA aspro5 exhibited signficant viral activation relative to their control counterpart (Figure 3e). The aspro5-induced HIV activation correlated with the specific knockdown of the HIV antisense lncRNA in a dose-dependent manner in cells cotransfected with a plasmid expressing the HIV lncRNA and a plasmid expressing aspro5 (Figure 3f). These data suggest that the observed activation of viral gene expression is a consequence of aspro5-mediated inhibition of the HIV-expressed antisense lncRNA. To determine the effect of the small RNA aspro5-mediated inhibition of the HIV antisense lncRNA on the chromatin state at the viral promoter, ChIP analysis was performed at the 5′LTR (Figure 3g). In cells transduced with a lentiviral vector expressing aspro5, we observed a marked reduction in silent state histone modifications H3K27me3 and H3K9me2 compared to cell transduced with an empty vector (Figure 3g). Collectively, the observations presented in (Figures 2 and 3) suggest that this HIV-encoded lncRNA is involved in the epigenetic regulation of viral transcription.
Epigenetic-based mechanism of lncRNA-mediated HIV transcriptional regulation
To exclude the possibility of nonspecific gene knockdown effects or toxicities caused by the induction of a nonspecific interferon response following introduction of small activating RNAs into the cellular environment, IFN-β mRNA concentrations were measured in cells expressing as154 or aspro5 and in those cells treated with the dsRNA analogue poly (I:C). However, neither small RNA treated cell was observed to induce expression of IFN-β (Figure 4a). Endogenous mechanisms of ncRNA-based transcriptional regulation in human cells utilize various components of the epigenetic regulatory pathway. Recent findings have demonstrated that DNA methyltransferase 3A (DNMT3a) plays a pivotal role in this pathway through direct interactions with ncRNAs.8 Mechanistically ncRNAs are thought to direct DNMT3a and other proteins which form part of an epigenetic remodeling complex, to target loci ultimately resulting in stable epigenetic-based silencing of the ncRNA targeted locus (reviewed in ref. [21]). To determine whether the HIV antisense lncRNA described here directly interacts with DNMT3a, directional qRTPCR was performed on eluates from a DNMT3a immunoprecipitation (ChIP) for detection of the antisense ncRNA. We find here that the HIV antisense lncRNA associates with DNMT3a in both ACH2 and J1.1 cells (only Ach2 cells shown, Figure 4b). To determine whether this HIV-expressed lncRNA localizes to the HIV promoter, two synthetic biotinylated fragments of the HIV antisense lncRNA transcript, each 1,000 bp in length were generated by T7 transcription and transfected into ACH2 cells. One of these fragments (HIV asRNA LTR-gag) represented the 3′ end of the antisense lncRNA overlapping the 5′LTR region and the other fragment (HIV asRNA Nef) represented a fragment of the lncRNA, which overlapped the nef region. A pull-down of the biotinylated transcripts was performed 30 hours posttransfection and qPCR showed that the synthetic HIV antisense ncRNA fragment, representing the 3′ end of the endogenous antisense lncRNA, localized directly to the 5′LTR (Figure 4c). After independently showing that the HIV lncRNA interacts directly with DNMT3a (Figure 4b) and localizes to the 5′LTR (Figure 4c), we looked at the localization of both entities in a DNMT3a-ncRNA complex. Immunoprecipitation of DNMT3a followed by a biotin pull-down in cells transfected with synthetic biotinylated HIV antisense ncRNA transcripts revealed that the HIV lncRNA coimmunoprecipitated with DNMT3a specifically at the 5′LTR (Figure 4d) and were found by mass-spec analysis to be in complex with various histones and known lncRNA binding proteins, such as Nucleolin (Table 1).7 These observations suggest that the HIV antisense lncRNA is directly involved in localizing DNMT3a to the 5′LTR, leading to transcriptional regulation of the provirus, as has been observed previously with both genes and pseudogenes in human cells.8,10 Furthermore, ChIP analysis showed that the localization of DNMT3a to the HIV 5′LTR was diminished in ACH2 cells stably expressing the as154 or aspro5 small RNAs relative to an empty vector control (Figure 4e).
Figure 4.

Epigenetic-based mechanism of the HIV-expressed antisense lncRNA-mediated viral gene regulation. (a) IFN induction was assessed by measuring IFN-β mRNA concentration in total RNA extracted from cells expressing as154 or aspro5 and cells treated with poly (I:C) which served as a positive control. (b) DNMT3a interacts with the HIV-1 expressed antisense ncRNA. Immunoprecipitation of DNMT3a followed by directional RT with RT primer 4, a primer within the LTR, and specific for the antisense ncRNA, and then PCR with primer set 3, reveals interactions between DNMT3a and the HIV-1 expressed antisense ncRNA in ACH2 cells. (c) Localization of the HIV antisense ncRNA to the 5′LTR. A biotin pulldown was performed in cells transfected with two synthetic biotinylated fragments of the HIV antisense lncRNA, followed by qPCR of the DNA coprecipitated with the biotinylated RNA using LTR-specific primer set 3. A synthetic biotinylated antisense ncRNA fragment containing 5′LTR and Gag elements binds directly to the HIV 5′LTR in ACH2 cells whereas the control lambda and an antisense ncRNA fragment containing Nef and 3′ LTR content does not. (d) Localization of the ncRNA-DNMT3a complex to the 5′LTR. A ChIP assay for DNMT3a was performed in cells transfected with a T7-transcribed biotinylated HIV asRNA fragment followed by a biotin pulldown on the ChIP eluates. DNMT3a and the HIV antisense ncRNA appear to interact with each other and both localize to the 5′LTR. (e) The HIV antisense ncRNA guides DNMT3a localization to the 5′LTR. A ChIP of DNMT3a shows that DNMT3a localization to the 5′LTR/promoter is reduced in lentiviral transduced ACH2 cells stably expressing small asRNAs targeted to the HIV-expressed lncRNA compared to cells transduced with an empty vector (Control). (f) ChIP analysis of EZH2 at the 5′LTR and (g) ChIP analysis of HDAC-1 at the 5′LTR in lentiviral transduced ACH2 cells stably expressing the inhibitory small RNAs as154 and aspro5 compared to cells transduced with an empty vector (Control) show a reduction in these chromatin modifying proteins at the viral promoter when the HIV antisense lncRNA is inhibited. (h) The effects of a combination of 5-AC and TSA treatment (Treated) on aspro5-mediated activation. Stably-transduced Jurkat cells were infected with either HIV-1 HX10 or NL4-3 and treated with both 5-AC and TSA and HIV gene expression determined 72 hours after initial drug treatment. c, d, e, h show triplicate-treated cells as mean + SD and P-values from a paired t-test. f, g show the mean + SEM of n = 4 ChIP samples and P-values from a paired t-test. ChIP, chromatin immunoprecipitation; HIV, human immunodeficiency virus; lncRNA, long noncoding RNAs; RT, reverse transcription TSA, trichostatin A; 5-AC, 5-azacytidine.
Table 1. Proteins found by Mass-spec analysis to be associated with DNMT3a and the HIV-expressed antisense noncoding RNA at the HIV LTR/promoter.
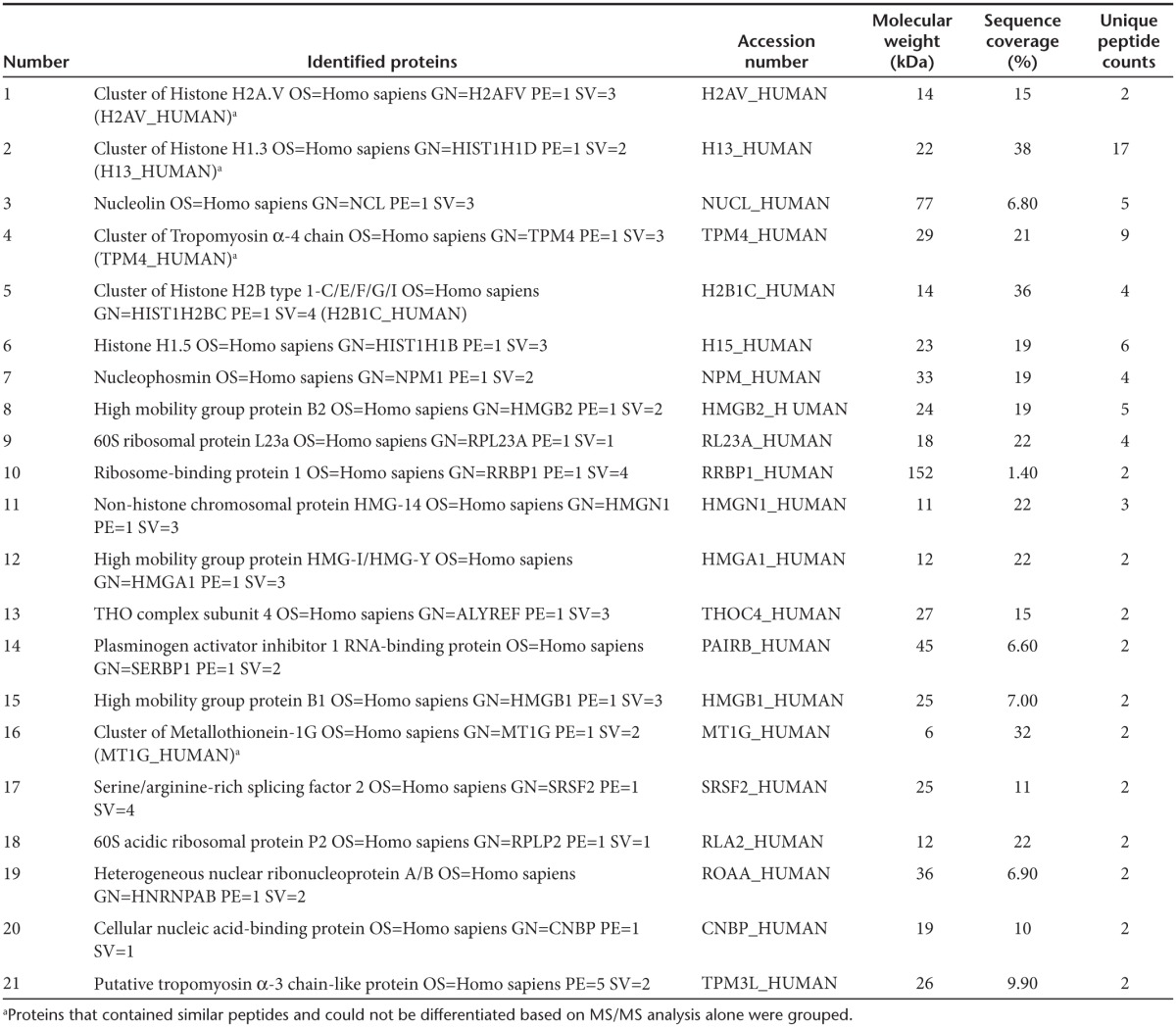
In addition to DNMT3a, the role of other chromatin modifying proteins such as EZH2 and HDAC-1 were investigated for their possible role in the HIV antisense lncRNA-mediated transcription regulation. ChIP analysis was performed at the 5′LTR of chronically infected ACH2 cells transduced with lentiviral vectors expressing the small inhibitory RNAs as154 or aspro5 (Figure 4f,g). A reduction in both EZH2 (Figure 4f) and HDAC-1 (Figure 4g) was observed at the viral promoter in cells in which the lncRNA had been manipulated by as154 or aspro5 when compared to cells transduced with an empty control vector. These data infer a vital role for the HIV-expressed lncRNA in recruiting and guiding a chromatin-remodeling complex consisting of proteins such as DNMT3a, EZH2, and HDAC-1 to the viral promoter. The presence of these proteins at the 5′LTR leads to changes in histone modifications and an altered chromatin state at the viral promoter resulting in reduced levels of viral gene expression.
Collectively, these data suggest that the HIV-expressed antisense lncRNA is involved in transcriptionally modulating expression of the virus, presumably through epigenetic-mediated mechanisms in the human cell, and that this pathway is susceptible to the regulatory effects of aspro5. To determine whether the aspro5 small asRNA is also acting via an epigenetic-based mechanism at the putative promoter of the HIV antisense lncRNA, cells stably expressing aspro5 were infected with either the CCR5 receptor tropic (HX10) or CXCR4 receptor tropic (NL4-3) HIV variants and subsequently treated with a combination of 5-azacytidine and trichostatin A. These compounds inhibit DNA methyltransferases and histone deacetylases, respectively, and hence actively suppress RNA-directed epigenetic-mediated gene silencing in human cells.22,23 The activating effect of the aspro5 in stably transduced cells expressing aspro5 and infected with HIV was reversed by treatment with 5-azacytidine and trichostatin A (Figure 4h), suggesting that aspro5 suppresses the expression of the HIV antisense lncRNA via an epigenetic-based mechanism.
Effects of aspro5 on viral latency
Several epigenetic marks have been associated with viral latency and we have shown that the HIV-expressed antisense lncRNA characterized above regulates HIV expression by an epigenetic-based mechanism. We therefore investigated the effects of aspro5-mediated repression of the HIV-encoded lncRNA on viral latency in an ex vivo memory CD4+ T-cell–based latency model.24,25,26 As previously described,24 naive CD4+T cells were isolated from a healthy donor and activated in nonpolarizing conditions for 3 days. At day 4 postactivation, cells were transduced with a control or aspro5-expressing lentiviral vector (Figure 3b) and kept in culture in the presence of IL-2. At day 5 postactivation, cells were infected with the replication-deficient HIV-1 NL4-3 (DHIV) and productive infection was monitored by analysis of green fluorescent protein (GFP) and p24 by flow cytometry at different time points. Marginal viral activation was observed at day 12 (Figure 5). Previous observations have demonstrated a requirement for active transcription in the initial establishment of ncRNA-directed gene silencing.27,28 Consistent with this mechanism, the activation of HIV mediated by the aspro5 appeared to require replication-competent virus, as replication defective, stably integrated variants in the resting memory CD4+ T-cell model were relatively resistant to this form of activation24,25 (Figure 5).
Figure 5.

Effects of aspro5 on latent viral replication in an ex vivo primary memory CD4+ T cell-based latency model.24,25 Cells were monitored for viral replication at different time points over 12 days and the effects of the aspro5 small asRNA were compared to an empty vector control.
Discussion
Recent advances in transcriptomic studies have revealed that a much larger fraction of the human genome is transcribed in both the sense and antisense directions than was previously envisioned.29,30,31 Examples of bidirectional transcription have demonstrated a role for these ncRNAs in the epigenetic regulation of gene expression.7,9,10,32,33 Here, we extend these observations to HIV and define a function for HIV-expressed antisense transcripts. The data presented here, along with previous observations,15,16,18 suggest that HIV expresses an endogenous antisense ncRNA that appears to be functional in the transcriptional and epigenetic regulation of the virus (Figure 6).
Figure 6.

Model for HIV-encoded antisense lncRNA-mediated regulation of viral transcription. (a) A long noncoding RNA (lncRNA) antisense to the viral genome is transcribed from a putative promoter within the nef gene. (b) The lncRNA recruits DNMT3a and possibly other chromatin remodeling proteins (enhancer of Zeste 2 (EZH2), histone deacetylase 1 (HDAC-1), G9a) and (c) guides these proteins to the viral promoter (5′LTR). (d) The localization of the chromatin-remodeling complex at the 5′LTR results in the formation of heterochromatin, which is characterized by H3K9 dimethylation, H3K27 trimethylation, and histone deacetylation. This alteration of the chromatin state at the viral promoter leads to transcriptional shutdown and an epigenetically silenced virus. (e) The HIV-encoded lncRNA may be inhibited by small single-stranded RNAs targeted to the promoter driving expression of the ncRNA via transcriptional gene silencing, or by small RNAs targeted to the ncRNA transcript via posttranscriptional gene silencing (shown). (f) The inhibition of the antisense lncRNA by small RNAs prevents the recruitment of chromatin remodeling proteins to the viral promoter (5′LTR). (g) The viral promoter remains in a euchromatin state, free of silent state epigenetic marks, and transcriptionally active ultimately resulting in elevated viral gene expression and replication. HIV, human immunodeficiency virus; lncRNA, long noncoding RNAs.
Noncoding RNAs have been shown to guide epigenetic complexes to targeted gene promoters resulting in transcriptional gene silencing of the targeted gene (reviewed in ref. [6]). Noncoding RNAs antisense to low-copy promoter-associated RNA are able to direct transcriptional silencing complexes containing HDAC-1, Ago-1, and DNMT3a to a targeted promoter leading to histone modifications and heterochromatin formation.10,27,34,35,36,37 These observations suggest that antisense ncRNAs are actively involved in the epigenetic regulation of gene expression. The evidence presented here and in previously published studies indicates that HIV-1 expresses a long ncRNA in the antisense orientation to the viral LTR and viral mRNA.15,16 Small RNAs targeting noncoding regions have previously been shown to induce epigenetic activation by targeting antisense lncRNAs.9,11,38,39 The data presented here demonstrate that HIV activation can be mediated by small RNAs targeted to the HIV-encoded antisense long ncRNA. Furthermore, we have shown that the localization of both DNMT3a and HDAC-1 to the viral LTR is reduced upon small RNA-mediated viral activation and in addition, we observe a loss of silent state histone modifications at the viral promoter when the HIV antisense lncRNA is inhibited. It is therefore likely that this viral-encoded lncRNA may guide epigenetic silencing complexes to the LTR and induce transcriptional shutdown. It appears therefore likely that HIV gene expression and replication may be regulated by a similar system to those endogenous cellular genes under antisense ncRNA-directed gene regulation and may also utilize a DNMT3a-mediated mechanism of action (reviewed in ref. [21]).
How and when HIV latency emerges has remained enigmatic but the process by which the virus undergoes latency appears to clearly involve epigenetic-based silencing mechanism.4 The activation of latent virus has been well characterized and requires the recruitment of histone acetyltransferases as well as other chromatin-remodeling proteins to the activated viral promoter/LTR.40,41 Conversely, enrichment of silent state chromatin marks (such as histone 3 lysine 9 trimethylation) and HDAC-1 have been observed at the LTR of transcriptionally inactive proviruses.42,43,44,45 These reports suggest a distinct role for chromatin remodeling, and thus the host epigenetic response, in viral latency. However, the endogenous factors responsible for guiding host epigenetic complexes to specific loci are currently unknown. Here we provide evidence of an HIV-encoded lncRNA that appears to regulate viral gene expression by inducing changes in chromatin structure at the LTR. It thus remains a possibility that this viral lncRNA may play a role in directing and guiding the establishment and/or maintenance of latency.
An alternative notion is that the HIV-expressed antisense transcript may align with a common theme of human cell specific integrating viral elements. Pseudogenes, some of which are essentially prehistoric viral entities, express antisense ncRNAs that are functional in the epigenetic regulation of gene expression7 and also function in a DNMT3a dependent manner.8 One similarity between HIV and endogenous pseudogenes is that both entities exhibit bidirectional transcription from their respective promoter elements. This suggests that these elements are important for epigenetic-based gene regulation, a concept that is supported by the observed action of aspro5 presented here (Figure 6).
While the essence of the endogenous mechanism by which HIV enters a state of latency remains to be fully determined, the data presented here suggest one mechanism by which HIV epigenetically regulates its own gene expression. We present here the possibility that the HIV-expressed antisense transcript is involved in the transcriptional shut down of the virus and that the inhibition of this transcript may lead to the activation of viral replication. An understanding of this underappreciated role for HIV-expressed antisense noncoding RNAs in the HIV viral lifecycle may prove useful in developing strategies to inhibit the emergence of viral latency.
Materials and Methods
Cell culture and viral infections. HEK293T cells were maintained in Dulbecco's modified Eagle's medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Life Technologies, San Diego, CA) and 50 μg/ml Pen/Strep (Mediatech) at 37 °C and 5% CO2. ACH2 and J1.1 cells are chronically infected cell lines harboring the HIV-1 LAI strain. ACH2, J1.1, and the J1.1 parental Jurkat cell lines were maintained in RPMI 1640 (Mediatech) supplemented with 10% fetal bovine serum and 50 μg/ml Pen/Strep at 37 °C and 5% CO2. Primary CD4+ T cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 50 μg/ml Pen/Strep, and 30 U/ml human rIL-2. Isolated CD4+ T cells were stimulated with anti-CD3/CD28 Dynabeads (Life Technologies). Cells were infected with the HIV-1 strains HX10 or NL4-3 at an multiplicity of infection of 0.1.
Lentiviral vectors and flow cytometry. Lentiviral vectors were produced by cotransfection of 10 µg of pRRE-GAG/Pol, 2.5 µg of pRSV-Rev, 5 µg of pMD2.G VSV-G, and 15 µg of the various HIV7-IGFP vectors into HEK 293T cells using calcium phosphate. High titer lentiviral vector stocks were prepared by ultracentrifugation on a 20% sucrose gradient in a SW32 Ti rotor (Beckman Coulter, Brea, CA) for 2 hours and 20 minutes at 19,400 rpm. The vectors were titered on HEK293T cells and analyzed for GFP expression by flow cytometry (FACSCalibur II, Becton, Dickinson and Company, East Rutherford, NJ) to determine infectious particles (IP) per milliliter. Jurkat, ACH2, and J1.1 cells were transduced at a multiplicity of infection of 1 IP/cell by spinoculation at 1,000×g for 30 minutes. Stimulated CD4+ cells were transduced at an multiplicity of infection of 20 IP/cell. Cells were placed under 2 µmol/l mycophenolic acid (Sigma-Aldrich, St Louis, MO) selection 72 hours posttransduction for 8 days; after which enrichment of GFP-positive cells was confirmed by flow cytometry. Cells were collected and fixed in 1% formaldehyde poststaining for all experiments. CD4 was stained using anti-CD4-APC (MHCD0405; Life Technologies). Stained/GFP-positive cells were analyzed using a BD FACSCalibur II.
RNA isolation and analysis. Total cellular RNA was isolated according to the manufacturer's instructions using the RNeasy Mini kit (Qiagen, Valencia, CA). RNA samples were DNase-treated using the Turbo DNA-free Kit (Life Technologies) according to the manufacturer's instructions. DNase-treated RNA samples were then standardized and reverse transcribed with Mu-MLV (Life Technologies) according to the manufacturer's instructions using a gene specific primer or an oligo-dT/random nonamer primer mix. The following gene specific primers were used: RT primer 1: 5′-cctagtataaacaatgagacaccaggg-3′; RT primer 2: 5′-cactcccaacgaagacaagata-3′; RT primer 3: 5′-ccctgtgagcctgcatggaatggatgaccc-3′; RT primer 4: 5′-actgctgacatcgagcttgctaca-3′. PCR was carried out using Kapa2G Fast HotStart ReadyMix (Kapa Biosystems, Cape Town, South Africa) according to the manufacturer's instructions. The following PCR primers were used: Primer set 1 F: 5′-tgagacaacatctgttgaggtggg-3′; Primer set 1 R: 5′-ggctgtactgtccatttatcagga-3′; Primer set 2 F: 5′-ttgttctgctgctgcacta-3′; Primer set 2 R: 5′-agctatgttccttgggttct-3′; Primer set 3 F: 5′-ctttccgctggggactttccagg-3′; Primer set 3 R: 5′-ccagagagacccagtacaggcaaaaagcag-3′; Primer set 4 F: 5′-ctttccgctggggactttccagg-3′; Primer set 4 R: 5′-ttcgctttcaggtccctgt-3′. Quantitative real-time PCR (qRT-PCR) was carried out using Kapa Sybr Fast universal qPCR mix (Kapa Biosystems, Woburn, MA) according to the manufacturer's instructions and an Eppendorf Mastercycler realplex (Eppendorf, Hamburg, Germany). Thermal cycling parameters started with 3 minutes at 95 °C, followed by 40 cycles of 95 °C for 3 seconds and 60 °C for 30 seconds. Specificity of the PCR products was verified by melting curve analysis. The following primers were used for qPCR: primer set 5 F: 5′-agggatggaaaggatcaccagcaa-3′; primer set 5 R: 5′-cccacctcaacagatgttgtctca-3′; β-actin F: 5′-aggtcatcaccattggcaatgag-3′; β-actin R: 5′-tctttgcggatgtccacgtca-3′. To determine induction of a nonspecific immune response, RNA was collected as described above 24 hours following treatment with 10 μg/ml poly(I:C; Sigma, MO, USA) and IFN-β induction was determined by qRT-PCR using the IFN-β specific primers: IFN-β F: 5′-tccaaattgctctcctgttgtgct-3′ and IFN-β R: 5′-ccacaggagcttctgacactgaaaa-3′.
p24 analysis. Samples were neutralized by lysis in Triton X-100 at a final concentration of 0.5%. Viral p24 analysis was performed by the Translational Virology Core at the University of California San Diego Center for AIDS Research using the PerkinElmer Extended Curve p24 enzyme-linked immunosorbent assay.
Immunoprecipitation analysis. ChIP analyses and RNA immunoprecipitation (RNA-IP) analyses were performed by standard protocols adapted for automation using the epMotion 5070 automated pipetting system (Eppendorf, Hamburg, Germany). ChIP eluates were purified using the MinElute PCR purification kit (Qiagen) and analyzed by qPCR. RNA-IP eluates were not treated with RNaseA. Eluates were DNase-treated, purified with acid phenol:chloroform (Life Technologies) and analyzed by directional RT and qPCR. Directional RT primer sequences are listed in the RNA isolation and analysis methods section above. qPCR to detect enrichment at the 5′LTR utilized primer set 3: primer set 3 F: 5′-ctttccgctggggactttccagg-3′; primer set 3 R: 5′-ccagagagacccagtacaggcaaaaagcag-3′. To determine nonspecific induction of the immune response.
Biotin pull-down assay. Fragments of the HIV antisense lncRNA ~1,000 bp long were generated by T7 transcription using the Ampliscribe T7-Flash Biotin–RNA Transcription Kit (Epicentre Biotechnologies, Madison, WI) according to the manufacturer's instruction. Templates for T7 transcription was prepared by PCR of pNL4-3 using primers: T7asHIV5′LTR F: 5′-cagtgaattgtaatacgactcactatagggtcctgtctgaag-3′ and asHIV5′LTR R: 5′-tggaagggctaatttggtcccaaaaaagac-3′ or PCR of pME18S-asHIV18 using primers: T7asHIV F: 5′-cagtgaattgtaatacgactcactatagggttctttgggagt-3′ and asHIV R: 5′-gaataacatgacctggatggagtggga-3′. Two fragments of the antisense lncRNA were generated, one fragment overlapping the 5′LTR and one fragment overlapping a region in the nef gene. Transcripts were transfected into ACH2 cells at a concentration of 50 nmol/l using the NEON Transfection System (Life Technologies). Thirty hours posttransfection, cells were cross-linked with formaldehyde at 1% for 10 minutes at room temperature followed by addition of glycine to a final concentration of 0.125 mol/l and a further incubation for 5 minutes at room temperature. Cells were then washed with phosphate-buffered saline supplemented with phenylmethylsulfonyl fluoride, aproteinin, and leupeptin and lysed with ChIP lysis buffer (50 mmol/l Hepes, 140 mmol/l NaCl, 1% Triton X, 0.1% nicotinamide adenine dinucleotide) on ice for 20 minutes. Chromatin was sheared by sonication. Cell lysates containing sheared chromatin, or ChIP eluates in the case of ChIP-biotin dual pull-down assays, were incubated with Dynabeads MyOne Streptavidin C1 (Life Technologies) prepared according to the manufacturer's instructions for 2 hours on a rotating platform. Beads were pulled down with a magnet for 3 minutes and washed with low salt immune complex wash buffer (0.1% sodium dodecyl sulfate; 1% Triton X-100; 2 mmol/l ethylenediaminetetraacetic acid (EDTA); 20 mmol/l Tris-HCl, pH 8.1; 150 mmol/l NaCl); High salt immune complex wash buffer (0.1% sodium dodecyl sulfate; 1% Triton X-100; 2 mmol/l EDTA; 20 mmol/l Tris-HCl, pH 8.1; 500 mmol/l NaCl); LiCl Immune complex wash buffer (0.25 mol/l LiCl; 1% NP40; 1% sodium deoxycholate; 1 mmol/l EDTA; 10 mmol/l Tris-HCl, pH 8.1); and Tris-EDTA (TE) buffer (10 mmol/l Tris-HCl; 1 mmol/l EDTA, pH 8.0). Each wash step was carried out for 3 minutes on a rotating platform. Streptavidin bead-biotinylated RNA–DNA complexes were resuspended in elution buffer (10 mmol/l Tris-HCl, pH 6.0; 1 mmol/l EDTA; and 2 mol/l NaCl) and heated at 70 °C for 10 minutes. The supernatants were used for mass spec. The triplicate sample elutes were then pooled together, speedvac'd, and subjected to LC/MS at University of New South Wales, Australia (BMSF Core facility, University of New South Wales, Canberra, Australia).
Electrospray tandem mass spectrometry. Mass Spectrometry was performed with the LTQ Fourier transform ion cyclotron resistance and the LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peak lists were generated using Mascot Daemon/extract_msn (Matrix Science, London, England, Thermo Fisher Scientific) using the default parameters, and submitted to the database search program Mascot (Matrix Science, UK; version 2.3.02; ref. [46]). Mascot was set up to search the Swiss-Prot database version_25_10_13 comprising 541,561 total entries (20,352 entries following taxonomy filter). Mascot was searched with taxonomy set to Homo sapiens, a peptide mass tolerance 4 ppm and fragment ion mass tolerance ± 0.4 Da. Oxidation of methionine and carbamidomethyl of cysteine were specified in Mascot as variable modifications. The enzyme specificity was set to trypsin with allowance for up to 1 missed cleavage site per peptide.
Criteria for protein identification: Scaffold (version Scaffold_4.2.1, Proteome Software, Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability by the Peptide Prophet algorithm47 with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 99.9% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.48 Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Proteins were annotated with gene ontology terms from National Center for Biotechnology Information (downloaded 19 December 2013).49 Those protein candidates from the anti-DNMT3a-asHIV IP were contrasted with control anti-DNMT3a-Lambda and no antibody-Lambda IPs. The top candidates not found in the controls are shown with the number of different peptide reads and percent coverage (Table 1).
Nuclear run-on assays. Approximately 4e6 cells were lysed with NP-40 lysis buffer (10 mmol/l Tris-HCl; 10 mmol/l NaCl; 3 mmol/l MgCl2; 0.5% NP-40). Nuclear lysates were incubated with transcription buffer (10 mmol/l Tris-HCl, pH 8.0; 2.5 mmol/l MgCl2; 150 mol/l KCl; 2 mmol/l dithiothreitol supplemented with 2 mmol/l ATP, CTP, and GTP; 1 mmol/l Biotin-16-AA-5′-UTP and 100 U RNase OUT) for 30 minutes at 37 °C. Samples were Dnase-treated (Turbo DNase, Life Technologies) and then incubated with nuclei lysis buffer (50 mmol/l Tris-HCl, pH 7.4; 5% sodium dodecyl sulfate; 0.125 mol/l EDTA) and proteinase K at 37 °C for 30 minutes. Transcribed RNA was purified by acid-phenol:chloroform extraction. Biotinylated RNA was captured by incubation for 2 hours with Dynabeads MyOne Streptavidin C1 (Life Technologies) prepared according the manufacturer's instruction. Captured RNA was washed three times with Bind/Wash Buffer (5 mmol/l Tris-HCl, pH 7.4; 0.5 mmol/l EDTA; 1 mmol/l NaCl) and analyzed by qRT-PCR.
5-Azacytidine and Trichostatin A treatment. Various vector stable transduced Jurkat cell lines were treated with 4 μmol/l 5-azacytidine and 0.5 μmol/l trichostatin A for 72 hours before analysis. Fresh 5-azacytidine was added to the cells every 24 hours.
Acknowledgments
The project was supported by NIHLB R01AI084406, NIAID R56 AI096861-01, PO1 AI099783-01, and ARC Future Fellowship FT130100572 to K.V.M. S.S. was supported by the Poliomyelitis Research Foundation James Gear International Fellowship and The National Research Foundation (South Africa) International Fellowship. The authors declare no conflict of interest.
References
- Persaud D, Zhou Y, Siliciano JM, Siliciano RF. Latency in human immunodeficiency virus type 1 infection: no easy answers. J Virol. 2003;77:1659–1665. doi: 10.1128/JVI.77.3.1659-1665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berges BK, Akkina SR, Remling L, Akkina R. Humanized Rag2(-/-)gammac(-/-) (RAG-hu) mice can sustain long-term chronic HIV-1 infection lasting more than a year. Virology. 2010;397:100–103. doi: 10.1016/j.virol.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok HP, Lever AM. Chromatin, gene silencing and HIV latency. Genome Biol. 2007;8:228. doi: 10.1186/gb-2007-8-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Kim KC, Roh TY, Park J, Jung KM, Lee JS, et al. Gene silencing in HIV-1 latency by polycomb repressive group. Virol J. 2011;8:179. doi: 10.1186/1743-422X-8-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV. The emerging role of RNA in the regulation of gene transcription in human cells. Semin Cell Dev Biol. 2011;22:351–358. doi: 10.1016/j.semcdb.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PG, Morris KV. RNA and transcriptional modulation of gene expression. Cell Cycle. 2008;7:602–607. doi: 10.4161/cc.7.5.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PG, Morris KV. Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcription. 2010;1:165–175. doi: 10.4161/trns.1.3.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson P, Ackley A, Vidarsdottir L, Lui WO, Corcoran M, Grandér D, et al. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat Struct Mol Biol. 2013;20:440–446. doi: 10.1038/nsmb.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV, Santoso S, Turner AM, Pastori C, Hawkins PG. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008;4:e1000258. doi: 10.1371/journal.pgen.1000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–206. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackley A, Lenox A, Stapleton K, Knowling S, Lu T, Kenny SS, et al. An algorithm for generating small RNAs capable of epigenetically mediating transcriptional gene silencing and activation in human cells. Mol Ther Nucleic Acids. 2013;2:e104. doi: 10.1038/mtna.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen MP, Lehtola T, Heinonen SE, Assefa GS, Korpisalo P, Girnary R, et al. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy. Circ Res. 2009;105:604–609. doi: 10.1161/CIRCRESAHA.109.200774. [DOI] [PubMed] [Google Scholar]
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- Landry S, Halin M, Lefort S, Audet B, Vaquero C, Mesnard JM, et al. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology. 2007;4:71. doi: 10.1186/1742-4690-4-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LB, Ambrus JL, Jr, Krawczyk KA, Sharma S, Brooks S, Hsiao CB, et al. Human Immunodeficiency Virus-Type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology. 2006;3:80. doi: 10.1186/1742-4690-3-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopman NC, Willemsen M, Liu YP, Bradley T, van Kampen A, Baas F, et al. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012;40:414–427. doi: 10.1093/nar/gkr719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi-Ishihara M, Yamagishi M, Hara T, Matsuda Y, Takahashi R, Miyake A, et al. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology. 2012;9:38. doi: 10.1186/1742-4690-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV. Non-coding RNAs, epigenetic memory and the passage of information to progeny. RNA Biol. 2009;6:242–247. doi: 10.4161/rna.6.3.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV. Long antisense non-coding RNAs function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics. 2009;4:296–301. doi: 10.4161/epi.4.5.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowling S, Morris KV. Epigenetic regulation of gene expression in human cells by noncoding RNAs. Prog Mol Biol Transl Sci. 2011;102:1–10. doi: 10.1016/B978-0-12-415795-8.00003-9. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA, et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem. 2008;283:23353–23363. doi: 10.1074/jbc.M709651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AM, De La Cruz J, Morris KV. Mobilization-competent Lentiviral Vector-mediated Sustained Transcriptional Modulation of HIV-1 Expression. Mol Ther. 2009;17:360–368. doi: 10.1038/mt.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods. 2011;53:54–61. doi: 10.1016/j.ymeth.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011;7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci USA. 2007;104:12422–12427. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli S, Pastori C, Magistri M, Carbone GM, Catapano CV. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. EMBO J. 2009;28:1708–1719. doi: 10.1038/emboj.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40 Database issue:D912–D917. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell. 2007;131:706–717. doi: 10.1016/j.cell.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Ebralidze AK, Guibal FC, Steidl U, Zhang P, Lee S, Bartholdy B, et al. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev. 2008;22:2085–2092. doi: 10.1101/gad.1654808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez S, Pisano DG, Serrano M. Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle. 2008;7:2601–2608. doi: 10.4161/cc.7.16.6541. [DOI] [PubMed] [Google Scholar]
- Hawkins PG, Santoso S, Adams C, Anest V, Morris KV. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res. 2009;37:2984–2995. doi: 10.1093/nar/gkp127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery L, Nakielny S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J Biol Chem. 2004;279:49479–49487. doi: 10.1074/jbc.M409070200. [DOI] [PubMed] [Google Scholar]
- Weinberg MS, Villeneuve LM, Ehsani A, Amarzguioui M, Aagaard L, Chen ZX, et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA. 2006;12:256–262. doi: 10.1261/rna.2235106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Younger ST, Nguyen NB, Hardy DB, Monia BP, Corey DR, et al. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol. 2008;15:842–848. doi: 10.1038/nsmb.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson A, Holloway A, Reeves R, Tremethick DJ. Recruitment of SWI/SNF to the human immunodeficiency virus type 1 promoter. Mol Cell Biol. 2004;24:389–397. doi: 10.1128/MCB.24.1.389-397.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007;81:10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, et al. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]