

Abstract
Cancer therapy using oncolytic viruses has gained interest in the last decade. Vesicular stomatitis virus is an attractive candidate for this alternative treatment approach. The importance of the immune response against tumor antigens in virotherapy efficacy is now well recognized, however, its relative contribution versus the intrinsic oncolytic capacity of viruses has been difficult to evaluate. To start addressing this question, we compared glycoprotein and matrix mutants of vesicular stomatitis virus (VSV), showing different oncolytic potentials for B16/B16gp33 melanoma tumor cells in vitro, with the wild-type virus in their ability to induce tumor-specific CD8+ T cell responses and control tumor progression in vivo. Despite the fact that wild-type and G mutants induced a stronger gp33-specific immune response compared to the MM51R mutant, all VSV strains showed a similar capacity to slow down tumor progression. The effectiveness of the matrix mutant treatment proved to be CD8+ dependent and directed against tumor antigens other than gp33 since adoptive transfer of isolated CD8+ T lymphocytes from treated B16gp33-bearing mice resulted in significant protection of naive mice against challenge with the parental tumor. Remarkably, the VSV matrix mutant induced the upregulation of major histocompatibility class-I antigen at the tumor cell surface thus favoring recognition by CD8+ T cells. These results demonstrate that VSV mutants induce an antitumor immune response using several mechanisms. A better understanding of these mechanisms will prove useful for the rational design of viruses with improved therapeutic efficacy.
Introduction
In recent years, oncolytic virotherapy has emerged as an alternative therapy against cancer showing great potential. Oncolytic tumor regression in vivo has been shown to be a multifactorial process involving viral gene expression and direct cell lysis, as well as the recruitment of immune effector cells directed against infected tumor cells.1,2,3 A well-characterized oncolytic virus being developed for such purpose is the prototypic Rhabdoviridae vesicular stomatitis virus (VSV). VSV possesses attractive intrinsic oncolytic properties as it replicates more efficiently in type-I interferon (IFN)-defective cells, a pathway frequently impaired during tumorigenesis.4 Several successful preclinical and clinical studies against prostate, breast, colorectal and liver cancers,5,6,7,8 as well as melanoma9 indicate that VSV oncolytic therapy is a promising alternative treatment against a number of cancer types.
In a recent clinical study, patients treated with oncolytic herpes virus were shown to harbor a very diverse tumor immune landscape.10 VSV treatment has also been shown to generate a variety of immune responses including tumor-specific CD8+ T cells that are induced following the release of tumor antigens by infected cells.2 Moreover, in models expressing exogenous antigens, VSV has been demonstrated to be a potent boost in a prime/boost oncolytic vaccination model.11 Other strategies that used irradiated tumor cells infected with VSV were also shown to provide some protection against tumor challenge.12 However, the tumor-specific immune response generated following VSV treatment is usually weak and leads only to a partial control of tumor growth. Hence, the causes for the high variability in the outcomes of VSV oncolytic therapy need to be better understood.13
Recently, our group has characterized various VSV glycoprotein (G) mutants.14 G mutants interfere with host cell metabolism by inhibiting cellular transcription and translation in a kinetic similar to the wild-type (WT) virus as opposed to the prototypic matrix (M) mutant (MM51R) that is slightly attenuated in vitro.14 G mutants proved to be especially cytolytic for B16 melanoma cells in vitro when compared to the MM51R mutant. One of the G mutants (G6R) also maintained the ability to induce type-I IFN in noncancerous cell lines at levels similar to the MM51R mutant suggesting that it could be a safe and potentially more effective alternative to MM51R. Furthermore, G mutants could still induce the translocation of calreticulin at the cell membrane following infection while the MM51R mutant had lost this ability.15 This endoplasmic reticulum–resident protein has been shown to function as a phagocytosis signal for dendritic cells16 and could potentially lead to the induction of immune-mediated cell death in vivo and subsequently to an increased antitumor immune response.
Given the differences in the oncolytic properties observed in vitro between G and M mutants of VSV, we sought to compare their immunomodulatory potential in vivo and correlate the antitumor immune response generated with survival in a B16/B16gp33 melanoma mouse model. Herein, we show that, while the MM51R mutant induced the weakest gp33-specific antitumoral CD8+ T cell immune response compared to WT or G mutants, it could nonetheless induce a functional antitumoral cytotoxic T lymphocyte (CTL) response that was efficient at controlling tumor progression. We found that this discrepancy was not the result of specific CD8+ T lymphocyte exhaustion since neither programmed cell death-1 (PD-1) nor programmed cell death 1 ligand-1 (PD-L1) blockade enhanced virotherapy in this system. However, we show that efficient targeting and lysis of tumor cells by CD8+ T cells likely reflected the remarkable ability of MM51R to upregulate major histocompatibility complex class-I (MHC-I) on tumor cells following infection.
Results
Wild-type and mutant VSV strains are similarly cleared from B16 tumors in vivo
Given that in vitro experiments had shown that VSV G mutants were as cytolytic as WT VSV for B16 melanoma cells whereas the MM51R mutant could less efficiently affect B16 metabolism,14 we first wanted to assess whether the different VSV mutants persisted in B16 tumors for different periods of time in vivo. In this experimental condition, infectious virus titers for all VSV strains studied declined with similar kinetics and all strains were cleared from the tumor site between 4 and 6 days postinfection indicating that the in vitro replication rates of VSV in B16 cells did not significantly affect viral clearance kinetics in vivo (Figure 1a). Due to the rapid elimination of infectious virus within the tumor tissue, three intratumoral infections were performed in every following treatment to induce local inflammation for a longer period of time. Despite this, no replicative virion could be detected at the tumor injection site 4 days after the last VSV dose neither for the WT nor the various mutants (data not shown).
Figure 1.
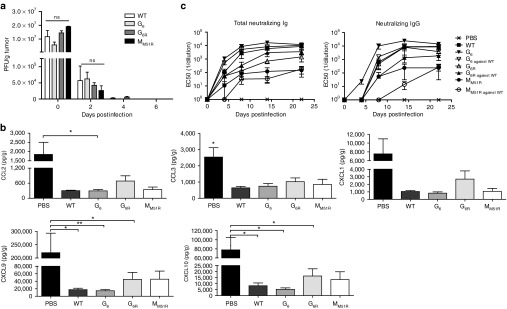
Rapid vesicular stomatitis virus (VSV) clearance from B16 melanoma tumors. (a) C57Bl/6 mice (n = 3 mice per group per time point) were injected subcutaneously with B16 cells and infected with a single 5 × 108 PFU intratumoral dose of either VSV WT or the mutants on day 7, harvested 15 minutes after injection (day 0) and on indicated days postinfection. Virus titers were determined using a standard plaque assay. Data are the mean ± SEM of three tumors. (b) C57Bl/6 mice (n = 9/group) were injected subcutaneously with B16 cells and infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7. On day 8, tumors were harvested, homogenized, and supernatants were analyzed for chemokine expression levels using a multiplex assay. Data show the combination of three independent experiments. (c) C57Bl/6 mice (n = 6) were injected subcutaneously with B16 cells and infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7, 9, and 11. Sera from the indicated days after infection were tested for total neutralizing immunoglobulins and IgGs using a plaque reduction assay. Indicated data points represent serum dilutions that reduced plaque formation by 50%. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
WT VSV and the MM51R mutant are actively being developed for clinical oncolytic applications. For obvious safety reasons, viral replication has to be restricted both in space and time, which however limits virus availability for oncolysis. For VSV, this is likely achieved through the induction of the innate antiviral immune response within infected tumor cells and through the rapid generation of neutralizing antibodies. Thus, we first compared the cytokine induction profile intratumorally following treatment with each VSV strain. Apart from five chemokines that were mainly downregulated following infection compared to nontreated animals (CXCL1, CXCL10, CCL3, CCL2, and CXCL9; Figure 1b), no other change in common inflammatory cytokine levels were observed (data not shown).
We next compared the kinetics of neutralizing antibody development against each mutant (Figure 1c). Total VSV-specific neutralizing immunoglobulins (mainly comprised of IgMs in the first few days) were detectable for all VSV strains by day 4 following the first VSV injection correlating with viral clearance kinetics. Class switching to neutralizing immunoglobulin G (IgGs) occurred by day 8. Of note, neutralizing IgGs were detected as early as day 4 after G6R treatment. The neutralizing antibody response reached its maximum level between 8 and 12 days postinfection. G6R and WT VSV induced the strongest and fastest humoral response, followed by G6, while the matrix mutant MM51R generated a weaker humoral response. We also analyzed the cross-neutralization ability of the antibodies elicited by the mutants to neutralize WT VSV. We found that even though the point mutations found in the glycoprotein of G6 and G6R mutants lie in close proximity to the dominant neutralizing B-cell epitope,17 cross-neutralization occurred for all mutants. Since no virus could be detected for any viral strain neither in the tumor nor in the spleen 4 days after the last injection, we conclude that the combined action of the innate antiviral immune response and neutralizing antibody response was sufficient to clear infection. These results suggest that all VSV mutants studied should be safe oncolytic agents because they are rapidly cleared in vivo, however this will likely limit the treatment efficacy window.
VSV treatment promotes immune cell infiltration
Since the generation of an efficient tumor-specific immune response is important for sustained tumor control, we characterized tumor infiltration by immune cells following VSV treatment. Using the B16 melanoma model expressing the surrogate tumor antigen gp33, we found that infection with VSV G or M mutants doubled the percentage of infiltrating immune cells while WT VSV increased their proportion by fivefold (Figure 2a). VSV treatment had little to no effect on the proportions of CD4+ T cells, B cells, natural killer (NK) cells, neutrophils, or macrophages (Figure 2b). However, treatment with every VSV mutant decreased the proportions of intratumoral regulatory T cells (Figure 2c). We also found a dramatic decrease in the numbers of intratumoral dendritic cells following infection. Furthermore, VSV treatment slightly increased myeloid-derived suppressor cells. Nevertheless, the main difference was found within the CD8+ T cell subset that doubled with treatment with WT, G6, and G6R mutants. Only G6 and G6R mutants significantly increased the CD8/CD4 ratio in the tumor (Figure 2d). Thus, VSV virotherapy induced immune cell infiltration in the tumor while G mutants favored an increased CD8/CD4 T cell ratio.
Figure 2.

Tumor infiltration by immune cells following vesicular stomatitis virus (VSV) treatment. C57Bl/6 mice (n = 9) were injected subcutaneously with B16gp33 cells and infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7, 9, and 11. On day 8 following the first VSV dose, tumor and spleen were harvested and immune cell subpopulations were characterized by flow cytometry. Mean ± SEM of three independent experiments showing total tumor infiltrating CD45+ cells (a) and subpopulation proportions of CD4 T cells (CD4+CD25−), regulatory T cells (CD4+CD25+), CD8 T cells, B cells (B220+), natural killer cells (NKp46+), myeloid-derived suppressor cells (MDSC) (Gr1+CD11b+), Neutrophils (Gr1+CD11b−), dendritic cells (Gr1−CD11b+CD11c+) and macrophages (Gr1−CD11b+F4/80+) were measured in the spleen and tumor tissue (b). Separate percentages of Tregs, CD8+ T cells, MDSC, and dendritic cells (c) as well as CD8/CD4 ratio (d) are also shown. *P < 0.05, **P < 0.01, ***P < 0.001.
VSV treatment induces a functional antitumor CTL response
To evaluate the local and systemic T cell activation induced by VSV oncolytic therapy, we compared the activation status of CD4+ and CD8+ T cells found in the tumor and spleen. Based on L-selectin (CD62L) and CD44 expression, we found no difference in the activation status of CD4+ T cells between treated and nontreated animals (data not shown) correlating with the weak CD4+ T cell infiltration observed (Figure 2b). However, a decrease in naive splenic and intratumoral CD8+ T cells was noted as well as a slight increase in effector CD8+ T cells following VSV infection (Figure 3a). As expected, at this early stage of the immune response, very few central memory T cells were found.
Figure 3.

Immune response induced following vesicular stomatitis virus (VSV) infection. B16gp33-bearing mice (n = 9) were infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7, 9, and 11. On day 8 following the first VSV dose, tumor and spleen were harvested and the activation profile of CD8+ T cells was analyzed by flow cytometry. (a) Naive (CD62L+CD44−), effector/effector memory (CD62L–) and central memory T cells (CD62L+CD44+) as well as (b) PD-1 expression on T cells in tumor and spleen after VSV treatments are shown. Data are the mean ± SEM of three independent experiments. (c) B16gp33-bearing mice (n = 9) were infected as above. On day 8 following the first VSV dose, splenocytes were isolated and stimulated ex vivo with VSV-N (RGYVYQGL) or gp33 (KAVYNFATM) peptides to analyze cytokine secretion and (d) VSV-N (RGYVYQGL), gp33 (KAVYNFATM), TRP-2 (VYDFFVWL), or gp100 (EGSRNQDWL) peptides for degranulation assay. Data are the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
We also characterized the expression of PD-1 at the cell surface. PD-1 is expressed upon T cell receptor activation, contributes to the contraction of the immune response but is also expressed by exhausted T cells. In contrast to the M mutant, treatment with WT VSV or G mutants induced a robust systemic activation of CD8+ T cells as shown by the high proportion of CD8+ PD-1+ T cells in the spleen (Figure 3b). However, PD-1 overexpression may also indicate the development of an exhausted phenotype. As for intratumoral CD8+ T lymphocytes, cells found locally were mainly PD-1 positive. These results suggest that VSV strains with an intact M protein induce the strongest T cell activation.
VSV treatment has been shown to induce limited γ-interferon (IFN-γ) secretion by adoptively transferred tumor-specific CD8+ T cells in draining lymph nodes18; hence, we characterized the systemic antitumor response of endogenous splenic CD8+ T cells. We first compared the intracellular cytokine expression profiles (TNF-α and IFN-γ) of CD8+ T cells. We found a robust antiviral CTL response against VSV independently of the mutant used (Figure 3c). Moreover, VSV strains containing an intact matrix protein could mount the strongest CD8+ T cell response against the gp33 surrogate tumor epitope when compared to nontreated animals. Furthermore, treatment with G6R induced a more polyfunctional antitumor CD8+ T cell response with more cells secreting both TNF-α and IFN-γ.
Effector CD8+ T cells kill cancer cells via degranulation of their cytolytic content. VSV-specific cytotoxic T cells were found in the spleen of treated animals, likely contributing to the elimination of infected cells (Figure 3d). VSV treatment additionally generated gp33-specific CTLs with the M mutant not reaching significance. In addition, CTLs against the self tumor-associated antigens tyrosinase-related protein (TRP)-2 and gp100 were also detectable in every case although not in proportions significantly above that of nontreated mice.
Levels of gp33-specific T cell responses do not correlate with prolonged mice survival
We then sought to determine if the ability of VSV mutants to induce a gp33-specific T cell response correlated with an improved therapeutic efficacy. Since expression of gp33 is likely to significantly increase B16 immunogenicity, we sought to determine the therapeutic potential of the various VSV mutants and to evaluate the predictive value of T cell responses directed toward surrogate tumor antigens against the more biologically relevant parental B16 tumor. Surprisingly, although the G6R mutant was the most potent in terms of inducing polyfunctional antitumor CD8+ T cells against gp33, it did not slow down tumor progression significantly more than other VSV mutants nor WT VSV (Figure 4). Oppositely, treatment with the M mutant, although it was the poorest at inducing gp33-specific T cell responses, prolonged mice survival to a similar degree as the WT or G mutants. Altogether, these results demonstrate that the capacity to induce a stronger immune response against a surrogate tumor antigen does not necessarily correlate with improved therapeutic efficacy.
Figure 4.
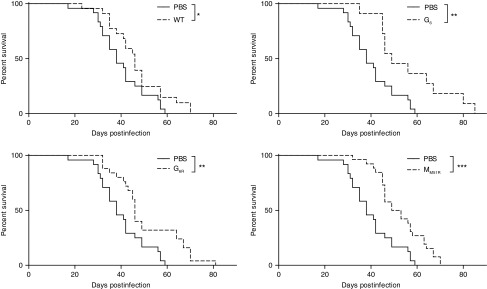
Vesicular stomatitis virus (VSV) therapy in the B16 tumor model. C57Bl/6 mice (n = 11–26) were injected subcutaneously with B16 cells and infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7, 9, and 11. Survival (tumors < 1.7 cm in any diameter) is shown. Data show three combined independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
The strength of the CD8+ T cell response against endogenous tumor antigens is VSV strain dependent
Since MM51R treatment led to prolonged survival rates despite a reduced CTL response against the surrogate tumor antigen gp33, we assessed whether CD8+ T lymphocytes directed against endogenous tumor antigens were involved in tumor control. To do so, we adoptively transferred purified CD8+ T cells, harvested 4 days after the last VSV treatment of B16gp33 melanoma-bearing mice, into naive mice. We then inoculated recipient mice with the parental B16 tumor cell line and followed tumor progression. We found that tumor-specific CD8+ T lymphocytes generated following treatment with VSV mutants could slow down tumor growth efficiently compared with CD8+ T cells taken from mock-infected animals (Figure 5a). However, tumor-specific CD8+ T cells generated by treatment with WT VSV were the poorest at controlling tumor progression even if a strong and functional gp33-specific immune response was induced. On the other hand, CD8+ T cells harvested from MM51R-treated mice efficiently controlled tumor burden despite the fact that this virus induced the lowest gp33-specific T cell response. Survival rates observed following transfer of CD8+ T cells from VSV-treated B16gp33-bearing mice (Figure 5b) correlated with those seen after treatment with the various VSV strains (Figure 4) indicating that the increase tumor control achieved by oncolytic VSV therapy is largely CD8+ T cell dependent. All together, these results show that the strength of the immune response induced against a surrogate tumor antigen does not adequately predict tumor control in vivo and suggest that the MM51R mutant may be able to induce CTL responses against a broader pool of endogenous tumor antigens using different mechanisms when compared to the WT virus or G mutants.
Figure 5.

Adoptive transfer of activated CD8+ T lymphocytes limits tumor progression. B16gp33-bearing mice (n = 10) were infected locally at the tumor site with 5.0 × 108 PFU of WT, mutant vesicular stomatitis virus (VSV) or mock infected on day 7, 9, and 11. (a) On day 8 following the first VSV dose, the spleen was harvested and CD8+ T cells purified. 5.0 × 106 cells were transferred intravenously into naive recipients 24 hours prior to parental B16 inoculation and tumor growth was measured. Data represent the mean ± SEM and is representative of two independent experiments. Overall survival (n = 20) was also assessed (b) and data are from two independent experiments combined. *P < 0.05, **P < 0.01.
VSV infection modulates the expression of cell-surface molecules
Tumor cells have been shown to express PD-L1 in certain conditions.19 We therefore characterized its expression and of its receptor PD-1 on B16gp33 cells upon VSV infection in vitro. Surprisingly, infection with each VSV strain induced the upregulation of PD-1 (Figure 6a) on B16gp33 cells while only the MM51R mutant led to PD-L1 upregulation (Figure 6b) compared to mock-infected cells, potentially providing a mechanistic explanation for the discrepancy observed with other VSV strains in terms of gp33-specific T cell response induction. We tested several other cell lines of mice and human origins and discovered that various transformed and nontransformed cell lines (human hepatocarcinoma HepG2 cells, L929 mouse fibroblasts, or Vero monkey kidney cells) upregulate PD-1 when infected with VSV (data not shown). While the biological significance of this observation remains unknown, PD-1 expression in response to VSV infection could potentially affect virotherapy efficacy.
Figure 6.
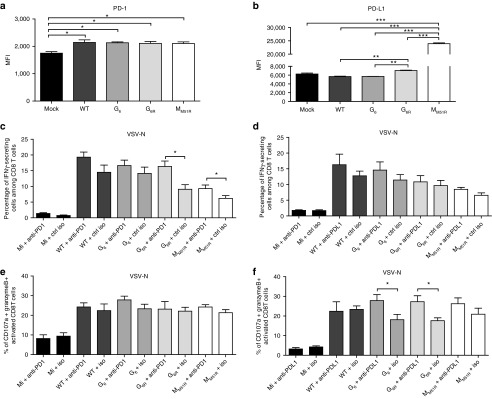
Expression of PD-1 and PD-L1 on B16gp33 cells following vesicular stomatitis virus (VSV) infection. B16gp33 cells were infected at a multiplicity of infection of 10 or mock infected. At 24 hours postinfection, cells were labeled to measure (a) PD-1 and (b) PD-L1 expression by flow cytometry. Data are the mean ± SEM and representative of two independent experiments in triplicates. B16gp33-bearing mice (n = 6–9) were also infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7, 9, and 11 and treated either with (c,e) anti-PD-1 (250 μg), (d,f) anti-PD-L1 (200 μg) or isotype control antibodies on day 6, 10, and 14. On day 8 following the first VSV dose, splenocytes were isolated and stimulated ex vivo with VSV-N (RGYVYQGL) to analyze cytokines secretion. Data show the combination of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Myeloid-derived suppressor cells present within the tumor microenvironment can also express PD-L1,20 which can negatively regulate T cell responses via interaction with PD-1 thus suppressing their effector functions.19,21 VSV treatment slightly increased the proportions of myeloid-derived suppressor cells (Figure 2c), while VSV infection modulated PD-1 and/or PD-L1 expression on tumor cells in vitro. Therefore, we sought to determine if PD-1 or PD-L1 blockade could enhance virotherapy in vivo. Based on the number of antigen-specific CD8+ T cells able to secrete IFN-γ and/or degranulate (CD107a and granzyme B expression), we found that PD-1 and PD-L1 blockade influenced only the VSV-specific T cell response (Figure 6c–f) and not the tumor-specific immune response (data not shown). While PD-1 blockade slightly increased anti-VSV CD8+ T cell functionality, anti-PD-L1 treatment increased VSV-specific CD8+ T cell cytotoxicity. The lack of effect on the antitumoral immune response observed following blockade of the PD-1 pathway is further correlated with the fact that VSV infection failed to modulate PD-1 or PD-L1 expression at the tumor cell surface in vivo (data not shown).
VSV matrix mutant upregulates the expression of cell-surface MHC-I on tumor cells
We then characterized the expression of MHC-I at the surface of B16 cells following VSV infection. As expected, MHC-I expression was downregulated upon in vitro infection with WT VSV or the G mutants (Figure 7a). Strikingly, the MM51R mutant tripled MHC-I surface expression on B16gp33 cells 24 hours postinfection. We confirmed this phenomenon in vivo (Figure 7b) and showed that T cell receptor-transgenic CD8+ T cells recognizing the gp33 epitope could be activated to a greater extent when incubated with dissociated B16gp33 tumor cells from MM51R-treated mice (Figure 7c). This suggests that the VSV matrix protein may limit MHC-I expression at the cell surface and that this effect is abolished by the M51R mutation allowing for its upregulation. This mechanism could be a contributing factor for the ability of MM51R treatment to induce an efficient antitumoral CD8+ CTL response.
Figure 7.

Expression of major histocompatibility complex class-I (MHC-I) on B16gp33 cells following vesicular stomatitis virus (VSV) infection. (a) B16gp33 cells were infected at an multiplicity of infection of 10 or mock-infected. At 24 hours postinfection, cells were labeled to measure MHC-I expression by flow cytometry. Data are the mean ± SEM and representative of two independent experiments in triplicates. (b) B16gp33-bearing mice (n = 4–5) were infected locally at the tumor site with 5.0 × 108 PFU of WT or mutant VSV on day 7. The following day, tumors were isolated and (b) labeled for the detection of MHC-I expression on CD45− cells or (c) cocultured with CFSE-labeled P14 transgenic splenocytes to analyze degranulation of CD8+CFSE+ cells. *P < 0.05, **P < 0.01, ***P < 0.001, CFSE, carboxyfluorescein succinimidyl ester.
Discussion
Oncolytic virotherapy has proven to be a promising cancer treatment in several successful clinical studies.8,22,23 VSV, one of the most studied prototypic oncolytic viruses, is now in clinical trials for liver cancer treatment in the United States (NCT01628640). However, a number of questions remain unanswered about the mechanisms that contribute to virotherapy efficacy. One such question is the relative contribution of the immune response versus the intrinsic oncolytic activity of a particular virus to the treatment outcome. To address this question and assess if a virus mutational status impacts the antitumoral immune response, we sought to determine the ability of WT, M, and G VSV mutants to induce tumor-specific immune responses and control tumor progression.
We found that the in vitro replication potential of the various VSV strains studied did not correlate with their respective capacity to persist in tumor tissue in vivo. Various pattern-recognition receptors have been shown to contribute to initial viral detection. Recognition of double-stranded RNA is achieved by the retinoic acid–inducible gene 1 and/or the melanoma differentiation–associated protein 5, leading to type-I IFN expression and the generation of an antiviral state through the expression of genes such as protein kinase R and the Mx GTPase. The VSV matrix protein was shown to block nucleocytoplasm mRNA export resulting, among other things, in the shut off of the IFN response and in apoptosis induction. MM51R induces type-I IFN production since the mutated protein can no longer interfere with this transport. We have previously shown that the G6R mutant induced significant type-I IFN secretion in L929-infected cells that was similar or even higher than the MM51R mutant even though it expresses a wild-type matrix protein.14 In vivo, IFN production can then prime an antiviral state in tumor-surrounding cells limiting efficient viral spread within the tumor tissue. Other studies have shown that the innate antiviral immune response induced against VSV plays a critical role in antitumor therapy in immunocompetent hosts.24 Indeed, in a B16-OVA mouse model, tumor regression using VSV intratumoral treatment was dependent on innate immune signaling through myeloid differentiation primary response gene 88 (MyD88) adaptor protein, type-III IFN-λ, CD8+ T cells, and NK cells, without requiring viral replication.1,25 Since in our study all VSV strains were cleared with the same kinetics, even those with a matrix protein able to block nucleocytoplasm mRNA transport, the antiviral type-I IFN pathway seems not the only mechanism involved in viral clearance.
Various tumor types constitutively produce cytokines and chemokines, thus favoring leukocyte migration into tumor tissue potentially facilitating the priming of the adaptive immune response. Paradoxically, however, immune infiltrates often do not offer protection against tumors but actually promote tumor growth by secreting reactive oxygen and nitrogen species, proteases, prostaglandins, and angiogenic growth factors.26 Specific chemokines, such as growth-related oncogene-α (CXCL1), directly induce the proliferation of melanoma cells.27 Stimulation of cancer stem cells has also been attributed to the increased secretion of CCL2 by cancer-associated fibroblasts when compared to normal cell-associated fibroblasts.28 CCL3 was associated with the maintenance of leukemia-initiating cells in chronic myeloid leukemia29 as well as the regulation of intratumoral leukocyte accumulation in a lung metastasis mouse model.30 Moreover, CXCL9 has proved to be highly secreted by tumor endothelial cells in melanoma metastases.31 In our model, VSV treatment led in all cases to a reduction of these chemokines. This may lead to two potential outcomes; limit suppressive myeloid cell migration into the tumor and limit tumor growth and metastasis, but could also prevent efficient recruitment of desirable immune cells. Therefore, the innate immune response to oncolytic viral treatment could in fact limit initial CTL priming.
Because point mutations in the G6 and G6R mutants are proximal to the dominant neutralizing VSV epitope,17 we also tested whether the development of the neutralizing antibody response against the G mutants was affected compared to the M mutant or WT VSV. G mutants induced a similar neutralizing antibody response with an efficient class switch to IgGs. Although viral spread through the blood stream following intratumoral injection is expected to be limited, antibodies could still contribute to a certain extent to viral clearance. Point mutations could have also modified the crossneutralization ability of the antibodies induced. However, mutations in G6 and G6R did not impair neutralization of the WT viral particle. Interestingly, we observed a reduced neutralizing antibody response after MM51R virus infection. This decreased B cell response along with the diminished CTL response observed against VSV MM51R may in fact allow for the establishment of a broader antitumoral immune response. While the WT, G6, and G6R mutants induce a stronger antiviral response, this response may limit the ability of VSV to induce a T cell response to tumor antigens.
Characterization of immune subpopulations within the tumor microenvironment following treatment showed a large proportion of T and B cells, as well as myeloid-derived suppressor cells. VSV administration also led to a decreased number of intratumoral DCs, as previously described,32 and increased CD8+ T cells. Therefore, treatment with VSV contributes to the establishment of an inflammatory milieu within the tumor that in turn leads to the recruitment of other immune mediators. All three viruses containing an intact matrix protein (WT, G6, and G6R) induced a better CTL response against the surrogate tumor antigen gp33 when compared to the matrix mutant and, although statistically significant, differences were only marginal and their biological relevance remains unclear. It is important to point out, however, that differences in the immune response induced by the various VSV strains do not correlate with a disparity in their individual replicative ability but appear to be strictly resulting from the respective mutation(s) they harbor.
To assess the overall antitumor immune response induced by each VSV strain, we also analyzed T cell responses against endogenous tumor-associated self-antigens. The group of melanoma differentiation antigens includes proteins such as MART-1, gp100, tyrosinase, gp75/TRP-1, and TRP-2 and are expressed by normal and malignant melanocytes both in human and mice.33 However, in contrast to the immunogenic surrogate tumor antigen gp33, gp100, and TRP-2 are also expressed by normal melanocytes. Therefore, most antigen-specific T cells directed against these self-antigens are deleted or tolerized explaining their poor immunogenicity.34 CTLs against these antigens were detectable in our model but in very low quantity when taken separately. However, in order to mount an efficient tumor-specific immune response, CTLs may be primed against various antigens, which could collectively confer protection.
In a previous study, we found that the G6 and G6R mutants were highly cytopathic for B16 cells in vitro compared to the M mutant.14 Here, we show that apart from inducing a strong antiviral CTL response, which may facilitate the concomitant uptake of infected tumor antigens, they also initiate a tumor-specific polyfunctional CTL response. Treatment with the MM51R mutant was the poorest at inducing a CTL response against the gp33 surrogate tumor antigen compared to nontreated animals. Nevertheless, treatment with all VSV strains significantly prolonged survival compared to nontreated mice. Unexpectedly, the increased functional anti-gp33 CD8+ T cell response induced by G6R treatment did not lead to a better tumor control, whereas the weaker anti-gp33 CD8+ T cell response elicited by the MM51R treatment did not worsen the survival. Since differences between mutants in both virus availability and robustness of the gp33-specific immune response did not correlate with survival, we assessed whether the CD8+ T cell response directed against endogenous tumor antigens was critical for tumor control. Adoptive transfer experiments revealed that T cells from WT-treated mice could not by themselves significantly control parental B16 tumor growth. However, CD8+ T cells harvested from G6R-treated animals that generated a polyfunctional gp33-specific immune response achieved tumor control as did CD8+ T cells from MM51R-treated mice indicating that these mutants could induce the broadest antitumoral immune response. We cannot exclude the possibility that the response against the immunodominant gp33 epitope could have limited the induction of a more robust response against more subdominant self tumor antigens expressed by B16 although we think this is unlikely since, at best, only 10 percent of the total CD8+ T cell response was directed against gp33 leaving sufficient naive CD8+ T cells in the repertoire to respond to other tumor epitopes. Taken together, these results indicate that induction of strong CTL responses against surrogate tumor antigens is not a good predictor of treatment efficacy suggesting that the overall response against a diverse tumor antigen repertoire is probably more important.
VSV treatment was also associated with a diminution of naive CD8+ T cells within the spleen and with the upregulation of PD-1 expression. Expectedly, T cells found in the tumor microenvironement were mainly of an activated phenotype since downregulation of L-selectin is required to cross the vascular endothelium and migrate into the tumor. We also found that the MM51R mutant induced much less PD-1 expression on CD8+ T cells in the spleen than the G mutants or WT VSV. Acute VSV infection does not usually lead to T cell exhaustion, but this immune dysfunction has been reported in cancer.35 However, it is more likely, here, that the strong VSV replication induced a robust T cell activation and PD-1 upregulation followed by a rapid contraction of the immune response; WT VSV potentially leading to an early contraction phase. On the opposite, the weaker cytopathic properties of the VSV M mutant could explain why fewer CD8+ T cells express PD-1 following infection. Surprisingly, in vitro infection of B16gp33 melanoma cells also induced the expression of PD-1. To explore if this was of biological relevance, we combined VSV treatment with PD-1 or PD-L1 blockade. We observed no significant improvement over VSV treatment alone. This result is compatible with our subsequent observation that VSV infection in vivo failed to modulate these markers.
We next examined other surface molecules that could be induced following VSV infection to explain the efficacy of the MM51R treatment despite the poor anti-gp33 response. Strikingly, VSV M mutant infection induced MHC-I upregulation at the tumor cell surface both in vitro and in vivo. The VSV matrix protein was shown to alter CD1d trafficking, a molecule structurally similar to MHC-I,36,37 inhibiting antigen presentation to, among other things, natural killer T cells.38 Furthermore, other viruses, like the poxvirus Orf or myxoma virus, have been found to interfere with antigen presentation by impairing MHC-I surface expression due to the disruption of the Golgi apparatus or the loss of β2-microglobulin, respectively.39,40 Thus, the VSV matrix protein could participate in the retention of MHC-I molecules within infected cells while the mutated protein may lack this ability. MHC-I upregulation at the tumor cell surface following MM51R treatment likely explains the significantly improved CD8+ T cell–dependent survival despite the poor gp33-specific CTL response induced by this mutant. This may result from an improved antigen presentation of multiple B16 antigens thus diluting a specific (gp33) response. One other possible explanation could come from the fact that the gp33 epitope is restricted to both H-2Db and H-2Kb in C57Bl/6 mice.41 The overexpression by MM51R may skew or alter MHC-I haplotype expression thus reducing gp33 presentation but still allowing for efficient presentation of endogenous cancer-related epitopes. More detailed analyses of the alteration in MHC-I expression induced following VSV infection will be required to further characterize this potential mechanism.
Altogether our results indicate that the induction of an antitumoral immune response is critical for efficient oncolytic virotherapy and that this can be achieved in different ways depending of the mutational status of a given virus. A better understanding of the mechanisms used by viruses to modulate the induction of the antitumoral immune response will prove extremely valuable for the design of more potent oncolytic viruses for clinical applications.
Materials and Methods
Cell lines and viruses. Mouse L929 fibroblasts, green monkey kidney Vero cells, and human HepG2 hepatocarcinoma cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. Murine B16 and B16gp33 melanoma cells were obtained from Dr A. Ochsenbein (Bern, Switzerland). The B16gp33 cells (H2-Db) were derived from B16.F10 cells transfected with a DNA minigene encoding the immunodominant CD8+ T cell epitope of the lymphocytic choriomeningitis virus (glycoprotein aa 33–41).42 Cells were grown in Dulbecco's modified Eagle medium (Life Technologies, Rockville, MD) supplemented with 10% fetal bovine serum and 200 μg/ml of G418 to select for retention of the gp33 minigene.
VSV G mutants, harboring solely mutation(s) in the envelope glycoprotein, were described previously.14,43 G6 and G6R are referred respectively as TP6 and TP6R1 in other studies.43,44 They were isolated from nonmutagenized VSV HR stocks by virtue of their small plaque-size phenotype on IFN-inducible cells but normal plaque size on Vero cells. HR (designated here as WT) is a heat-resistant variant of the San Juan isolate of the Indiana serotype. The MM51R mutant, originally named T102643 or AV1,45 is derived from mutagenized HR stocks and harbors the M51R substitution in its matrix protein. All viruses were propagated and titrated on Vero cells.
In vivo studies. All procedures were approved by the INRS Institutional Animal Care and Use Committee. To establish subcutaneous tumors, 5 × 105 B16 or B16gp33 cells in 100 µl phosphate-buffered saline (PBS) were injected into the flank of C57Bl/6 mice. They were infected locally at the tumor site with 5.0 × 108 plaque-forming unit (PFU) of WT or mutant VSV on day 7, 9, and 11. Mice were sacrificed 8 days following the first VSV injection for immune response analysis. For survival studies, mice were monitored at indicated time points and sacrificed when tumor burden exceeded 17 mm in diameter. For PD-1/PD-L1 blockade experiments, mice were infected as described above and treated i.p. either with anti-PD-1 antibody (clone RMP1-14; 250 μg) or anti-PD-L1 antibody (clone 10F.9G2; 200 μg) on day 6, 10, and 14. Control mice were injected with isotype control antibodies for each treatment groups (respectively clones 2A3 and LTF-2) (BioXCell, West Lebanon, NH). For the adoptive transfer experiment, splenocytes from control and treated mice were harvested at day 8 following the first VSV injection as described above. CD8+ T lymphocytes were isolated using a CD8 T cell enrichment kit (Stemcell, Vancouver, British Columbia, Canada) and 5.0 × 106 cells were injected intravenously into naive mice. Parental B16 cells were inoculated 24 hours later as described above and tumor growth was monitored.
Detection of infectious virus in vivo. Mice were injected into the flank with 5 × 105 B16 cells in 100 µl PBS. A single VSV dose of 5.0 × 108 PFU was given intratumorally 7 days after inoculation and tumors were harvested 15 minutes after infection (day 0) or every other day. Tumors were weighted, homogenized, and centrifuged to allow for the detection of virus in supernatants as determined by plaque assay on Vero cells as described previously.44
Quantification of intratumoral chemokines. Mice were injected into the flank with 5 × 105 B16 cells in 100 µl PBS. A single VSV dose was given intratumorally 7 days after inoculation and tumors were harvested 24 hours later. Tumors were then homogenized with an electric homogenizer (PolyTron homogenizer H3660-2A; Kinematica, Lucerne, Switzerland) and supernatants were assayed for CXCL1, CXCL10, CCL3, CCL2, and CXCL9 using a chemokine 5-plex bead immunoassay (Life Technologies) for the Luminex 100 system (Luminex Corporation, Austin, TX) according to the manufacturer's instructions.
Neutralizing antibody titration. Serum samples from mice bearing B16 tumors treated with VSV were collected at the indicated time points and tested for neutralizing activity using a plaque reduction assay as described previously.46 Briefly, serial twofold dilutions of 1/40 prediluted serum samples were mixed with equal volumes of VSV (using the same mutant injected for the oncolytic treatment) containing 5 × 103 PFU, and incubated for 90 minutes at 37 °C. A total of 100 μl of this solution was transferred onto Vero cell monolayers in 96-well plates and incubated for 1 hour at 37 °C. Monolayers were overlaid with medium containing 1% methylcellulose and incubated overnight at 37 °C. Then, the overlay was removed and the monolayer fixed and stained with 0.5% crystal violet dissolved in 5% formaldehyde, 50% ethanol, and 4.25% NaCl. The serum dilution reducing the number of plaques by 50% was taken as titer. To determine IgG titers, undiluted serum was treated with an equal volume of 280 mmol/l 2-ME for 1 hour at room temperature before samples were processed, as described above. For crossneutralization analysis, serum samples were mixed with VSV WT and processed as above.
Flow cytometry. Spleen and tumors were recovered from mice and dissociated to achieve single-cell suspensions using a nylon 100 μm cell strainer (BD Falcon, Mississauga, ON). Cells were washed, resuspended in phosphate-buffered saline containing 1% bovine serum albumin and 0.1% sodium azide (fluorescence-activated cell sorting (FACS) buffer), and incubated with directly conjugated primary antibodies for 30 minutes at 4 °C. Cells were then washed and resuspended in 200 µl FACS buffer containing 1% formaldehyde. Samples were acquired on a FACS Fortessa (BD Biosciences, San Diego, CA) and analyzed using the Flowjo software (Tree Star, Ashland, OR). Anti-CD45 PE/CF-594, anti-CD25 APC, and anti-NKp46 Alexa 700 were purchased from BD Biosciences. Anti-PD-1 fluorescein isothiocyanate (FITC), anti-CD4 APC/Cy7, anti-CD4 FITC, anti-CD8 phycoerythrin (PE)/Cy7, anti-CD62L Alexa 700, anti-CD44 PercP/Cy5.5, anti-B220 allophycocyanin (APC)/Cy7, anti-CD11b Pacific Blue, anti-Gr1 PE/Cy5, anti-CD11c FITC, anti-F4/80 APC, anti-PD-L1 Brilliant violet 421, and anti-CD86 PE were purchased from BioLegend (San Diego, CA). Purified anti-VSV-G from the monoclonal VSV-Indiana-specific IgG-secreting hybridoma VI1047 (kindly obtained from Dr R.M. Zinkernagel) was coupled to Alexa fluor 647 fluorochrome using the Alexa Fluor 647 Protein Labeling Kit (Life Technologies, Burlington, ON).
Intracellular staining assay. For IFN-γ and TNF-α intracellular staining, single-cell suspensions were prepared from spleen harvested 8 days after the first viral injection. Cytokine production, in response to viral or tumor antigens was measured by incubation with peptides (VSV-N; RGYVYQGL 5 µg/ml, gp33; KAVYNFATM 1 μg/ml, TRP-2; VYDFFVWL 5 µg/ml and gp100; EGSRNQDWL 5 µg/ml) in the presence of brefeldin A (10 μg/ml) and IL-2 (100 U/ml) for 5 hours. Cells were stained for surface markers, then fixed, and permeabilized for intracellular staining using fixation and permeabilization buffers from BioLegend according to the manufacturer's instructions. For granzyme B intracellular staining, cells were incubated with peptides in the presence of monensin A (20 μg/ml) and anti-CD107a FITC antibody for 5 hours. Cells were stained for surface markers, then fixed and permeabilized for intracellular staining. IFN-γ PE and TNF-α APC were obtained from BioLegend and granzyme B eFluor 450 was purchased from eBiosciences (San Diego, CA).
In vitro infections. B16gp33, HepG2, L929, and Vero cells were infected at a multiplicity of infection of 10 or mock infected. At 24-hour postinfection, cells were trypsinized, harvested, washed, and resuspended in FACS buffer. Cells were labeled with anti-PD-1 FITC, anti-PD-L1 brilliant violet 421, and anti-H2 PE (BioLegend) and analyzed by flow cytometry using a FACS Fortessa instrument (BD Biosciences) and the FlowJo software (Tree Star).
Tumor surface molecules staining. Mice were injected into the flank with 5 × 105 B16gp33 cells in 100 µl PBS. A single VSV dose was given intratumorally 7 days after inoculation and tumors were harvested 24 hours later. Tumors were then dissociated to achieve single-cell suspensions using a nylon 100 μm cell strainer (BD Falcon), washed, and resuspended in FACS buffer. Cells were labeled with anti-CD45 PE/CF594, anti-PD-1 FITC, anti-PD-L1 brilliant violet 421, and anti-H-2 PE (BioLegend) and analyzed by flow cytometry using a FACS Fortessa instrument (BD Biosciences) and the FlowJo software (Tree Star).
MHC-I recognition assay. Mice were injected into the flank with 5 × 105 B16gp33 cells in 100 µl PBS. A single VSV dose was given intratumorally 7 days after inoculation and tumors were harvested 24 hours later. Tumors were then dissociated to achieve single-cell suspensions using a nylon 100 μm cell strainer (BD Falcon), washed and coincubated with carboxyfluorescein diacetate succinimidyl ester–labeled P14 transgenic splenocytes (gp33-41-specific T cells) at a 1:1 ratio in the presence of monensin A (20 μg/ml) and APC-coupled anti-CD107a antibody for 5 hours. Cells were stained for surface markers and analyzed by flow cytometry using a FACS Fortessa instrument (BD Biosciences) and the FlowJo software (Tree Star).
Statistical analyses. Analysis of variance with Tukey post hoc testing was used for group comparisons and a Student's t-test was used for pair comparisons. P values of less than 0.05 were considered significant. Survival curves were plotted according to the Kaplan–Meier method, and statistical significance in the different treatment groups was compared using the log-rank test.
Acknowledgments
This work was supported by the Jeanne and J.-Louis Lévesque Chair in Immunovirology from the J.-Louis Lévesque Foundation and from a grant from the Canadian Institutes of Health Research (MOP-89797). V.J. and M.-P.L. acknowledge studentship support from the Fondation Armand-Frappier. P.L. holds a Fonds de recherche du Québec—Santé postdoctoral award and a Thomas F. Nealon, III Postdoctoral Research Fellowship from the American Liver foundation.
References
- Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, et al. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010;70:4539–4549. doi: 10.1158/0008-5472.CAN-09-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9:1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Puckett S, Lyles DS. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther. 2010;17:883–892. doi: 10.1038/cgt.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G, Xu S, Watanabe M, Jayakar HR, Whitt MA, Gingrich JR. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J Urol. 2010;183:1611–1618. doi: 10.1016/j.juro.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Huang TG, Ebert O, Shinozaki K, García-Sastre A, Woo SL. Oncolysis of hepatic metastasis of colorectal cancer by recombinant vesicular stomatitis virus in immune-competent mice. Mol Ther. 2003;8:434–440. doi: 10.1016/s1525-0016(03)00204-1. [DOI] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- Bridle BW, Boudreau JE, Lichty BD, Brunellière J, Stephenson K, Koshy S, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther. 2009;17:1814–1821. doi: 10.1038/mt.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay CG, Rintoul JL, Kus A, Paterson JM, Garcia V, Falls TJ, et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol Ther. 2012;20:1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelfanger DM, Offord CP, Dev J, Bajzer Z, Vile RG, Dingli D. Dynamics of melanoma tumor therapy with vesicular stomatitis virus: explaining the variability in outcomes using mathematical modeling. Gene Ther. 2012;19:543–549. doi: 10.1038/gt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelle V, Brassard F, Lapierre P, Lamarre A, Poliquin L. Mutations in the glycoprotein of vesicular stomatitis virus affect cytopathogenicity: potential for oncolytic virotherapy. J Virol. 2011;85:6513–6520. doi: 10.1128/JVI.02484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelle V, Poliquin L, Lamarre A. Vesicular stomatitis virus in the fight against cancer. Med Sci (Paris) 2013;29:175–182. doi: 10.1051/medsci/2013292015. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- Vandepol SB, Lefrancois L, Holland JJ. Sequences of the major antibody binding epitopes of the Indiana serotype of vesicular stomatitis virus. Virology. 1986;148:312–325. doi: 10.1016/0042-6822(86)90328-4. [DOI] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, et al. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum Gene Ther. 2011;22:1343–1353. doi: 10.1089/hum.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid T, Galanis E, Abbruzzese J, Sze D, Wein LM, Andrews J, et al. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): phase II viral, immunologic, and clinical endpoints. Cancer Res. 2002;62:6070–6079. [PubMed] [Google Scholar]
- Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18:1430–1439. doi: 10.1038/mt.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, et al. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol Ther. 2011;19:150–158. doi: 10.1038/mt.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichim CV. Revisiting immunosurveillance and immunostimulation: Implications for cancer immunotherapy. J Transl Med. 2005;3:8. doi: 10.1186/1479-5876-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond A, Thomas HG. Purification of melanoma growth stimulatory activity. J Cell Physiol. 1986;129:375–384. doi: 10.1002/jcp.1041290316. [DOI] [PubMed] [Google Scholar]
- Melgarejo E, Medina MA, Sánchez-Jiménez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. Int J Biochem Cell Biol. 2009;41:998–1001. doi: 10.1016/j.biocel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Baba T, Naka K, Morishita S, Komatsu N, Hirao A, Mukaida N. MIP-1α/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J Exp Med. 2013;210:2661–2673. doi: 10.1084/jem.20130112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol. 2008;181:6384–6393. doi: 10.4049/jimmunol.181.9.6384. [DOI] [PubMed] [Google Scholar]
- Amatschek S, Lucas R, Eger A, Pflueger M, Hundsberger H, Knoll C, et al. CXCL9 induces chemotaxis, chemorepulsion and endothelial barrier disruption through CXCR3-mediated activation of melanoma cells. Br J Cancer. 2011;104:469–479. doi: 10.1038/sj.bjc.6606056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveille S, Goulet ML, Lichty BD, Hiscott J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J Virol. 2011;85:12160–12169. doi: 10.1128/JVI.05703-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Fitzgerald EB, Southwood S, Sette A, Rosenberg SA, Kawakami Y. Identification of a shared HLA-A*0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2) Cancer Res. 1998;58:4895–4901. [PubMed] [Google Scholar]
- Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE. A molecular profile of T-cell exhaustion in cancer. Oncoimmunology. 2012;1:369–371. doi: 10.4161/onci.18342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabi F, Bradbury A. The CD1 system. Tissue Antigens. 1991;37:1–9. doi: 10.1111/j.1399-0039.1991.tb01836.x. [DOI] [PubMed] [Google Scholar]
- Brutkiewicz RR, Bennink JR, Yewdell JW, Bendelac A. TAP-independent, beta 2-microglobulin-dependent surface expression of functional mouse CD1.1. J Exp Med. 1995;182:1913–1919. doi: 10.1084/jem.182.6.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renukaradhya GJ, Khan MA, Shaji D, Brutkiewicz RR. Vesicular stomatitis virus matrix protein impairs CD1d-mediated antigen presentation through activation of the p38 MAPK pathway. J Virol. 2008;82:12535–12542. doi: 10.1128/JVI.00881-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga MC, Wang H, Barry M, McFadden G. Endosomal/lysosomal retention and degradation of major histocompatibility complex class I molecules is induced by myxoma virus. Virology. 1999;261:180–192. doi: 10.1006/viro.1999.9840. [DOI] [PubMed] [Google Scholar]
- Rohde J, Emschermann F, Knittler MR, Rziha HJ. Orf virus interferes with MHC class I surface expression by targeting vesicular transport and Golgi. BMC Vet Res. 2012;8:114. doi: 10.1186/1746-6148-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudrisier D, Oldstone MB, Gairin JE. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D(b) and H-2K(b) molecules. Virology. 1997;234:62–73. doi: 10.1006/viro.1997.8627. [DOI] [PubMed] [Google Scholar]
- Prévost-Blondel A, Zimmermann C, Stemmer C, Kulmburg P, Rosenthal FM, Pircher H. Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. J Immunol. 1998;161:2187–2194. [PubMed] [Google Scholar]
- Francoeur AM, Poliquin L, Stanners CP. The isolation of interferon-inducing mutants of vesicular stomatitis virus with altered viral P function for the inhibition of total protein synthesis. Virology. 1987;160:236–245. doi: 10.1016/0042-6822(87)90065-1. [DOI] [PubMed] [Google Scholar]
- Desforges M, Charron J, Bérard S, Beausoleil S, Stojdl DF, Despars G, et al. Different host-cell shutoff strategies related to the matrix protein lead to persistence of vesicular stomatitis virus mutants on fibroblast cells. Virus Res. 2001;76:87–102. doi: 10.1016/s0168-1702(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Charan S, Zinkernagel RM. Antibody mediated suppression of secondary IgM response in nude mice against vesicular stomatitis virus. J Immunol. 1986;136:3057–3061. [PubMed] [Google Scholar]
- Kalinke U, Bucher EM, Ernst B, Oxenius A, Roost HP, Geley S, et al. The role of somatic mutation in the generation of the protective humoral immune response against vesicular stomatitis virus. Immunity. 1996;5:639–652. doi: 10.1016/s1074-7613(00)80277-0. [DOI] [PubMed] [Google Scholar]