

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and high-lethality fibrotic lung disease characterized by excessive fibroblast proliferation, extracellular matrix accumulation, and, ultimately, loss of lung function. Although dysregulation of some microRNAs (miRs) has been shown to play important roles in the pathophysiological processes of IPF, the role of miRs in fibrotic lung diseases is not well understood. In this study, we found downregulation of miR-26a in the lungs of mice with experimental pulmonary fibrosis and in IPF, which resulted in posttranscriptional derepression of connective tissue growth factor (CTGF), and induced collagen production. More importantly, inhibition of miR-26a in the lungs caused pulmonary fibrosis in vivo, whereas overexpression of miR-26a repressed transforming growth factor (TGF)-β1-induced fibrogenesis in MRC-5 cells and attenuated experimental pulmonary fibrosis in mice. Our study showed that miR-26a was downregulated by TGF-β1-mediated phosphorylation of Smad3. Moreover, miR-26a inhibited the nuclear translocation of p-Smad3 through directly targeting Smad4, which determines the nuclear translocation of p-Smad2/Smad3. Taken together, our experiments demonstrated the antifibrotic effects of miR-26a in fibrotic lung diseases and suggested a new strategy for the prevention and treatment of IPF using miR-26a. The current study also uncovered a novel positive feedback loop between miR-26a and p-Smad3, which is involved in pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and high-lethality fibrotic lung disease, which is characterized by alveolar epithelial cell injury, inflammation, cell accumulation, aberrant proliferation of fibroblasts, and exaggerated deposition of extracellular matrix (ECM) in the lung parenchyma, which finally lead to the damage of lung function.1,2 Currently, there is no effective treatment for IPF, and the overall prognosis is dismal, with a median survival of 3–5 years after diagnosis.3 Therefore, understanding the mechanisms of lung fibrosis is of great value for therapy of IPF.
MicroRNAs (miRs) comprise a class of evolutionarily conserved, noncoding small RNAs, 19–22 nucleotides in length, which bind to the 3′-untranslated region (UTR) of targets and repress the translation of target genes or degradation of target messenger RNAs (mRNAs).4 Increasing evidence supports the fact that miRs play indispensable roles in the progress of many diseases, such as diabetes, cancer, and cardiovascular disease.5,6,7 Some miRs have been reported to be related to pulmonary fibrosis. Liu et al.8 found that miR-21 is highly upregulated in the lungs of mice with bleomycin (BLM)-induced lung fibrosis and in the lungs of patients with IPF. In addition, it mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. Cushing et al.9 reported downregulation and an antifibrotic role of miR-29 in IPF. However, we have just begun to understand the role of miRs in lung fibrosis, and many mysterious insights remained yet to be uncovered. Continuous revelation of the roles of specific miRs involved in the pathogenesis of lung fibrosis should help us develop new therapeutic strategies for IPF and other interstitial lung diseases.
It has reported that miR-26a is downregulated in canine atrial fibrillation and rat cardiac fibrosis.10,11 Moreover, knockdown of miR-26a promotes the proliferation and differentiation of fibroblasts into myofibroblasts in vitro. Such studies indicate that miR-26a plays a protective role in the process of fibrosis. However, the role of miR-26a in the process of pulmonary fibrosis has not been studied in detail.
Our study aimed to reveal the role and mechanisms of miR-26a in the progress of pulmonary fibrosis. Our data confirmed that miR-26a was downregulated in mice with experimental pulmonary fibrosis and in IPF, and it restored fibrogenesis in vitro and in vivo. Moreover, we uncovered a reciprocal loop between miR-26a and p-Smad3, which was activated under the condition of external stimuli and exacerbated pulmonary fibrosis. Our results suggest that miR-26a plays a pivotal role in pulmonary fibrosis.
Results
Deregulation of miR-26a and TGF-β pathway in lungs of mice with experimental pulmonary fibrosis
To detect whether miR-26a is involved in the pathogenesis of pulmonary fibrosis, we first developed a mouse model of experimental pulmonary fibrosis by intratracheal injection of BLM. Four weeks after injection, Masson's staining showed markedly increased collagen deposition in BLM-treated mice compared with the level in the saline group (Figure 1a), with significantly upregulated hydroxyproline content (Figure 1b). Meanwhile, the mRNA expression of collagen I, collagen III, and matrix metalloproteinase-2/9 (MMP-2 and MMP-9) was strikingly elevated in lungs of mice with experimental pulmonary fibrosis (Figure 1c).
Figure 1.

Downregulation of miR-26a in the lungs of mice with experimental pulmonary fibrosis and in IPF patients. (a) Hematoxylin–eosin (HE) and Masson Trichrome staining of mouse lung sections showing interstitial fibrosis with collagen deposition 28 days after injection of bleomycin (BLM) (10 × 20; original magnification ×200). (b) Increased hydroxyproline content in the lungs of mice treated with BLM compared with the content in lungs of control group mice. (c) Increased collagen (Col) I, Col III, matrix metalloproteinase (MMP)-2, and MMP-9 mRNA expression in the BLM-treated mice compared with expression in control animals, measured by real-time PCR. (d) Western blot analysis of proteins in the TGF-β signaling pathway in BLM-treated mice as compared with the same in saline animals. (e) Real-time PCR analysis of expression deregulation of miRs in the lungs of mice with experimental pulmonary fibrosis. (f) Downregulation of miR-26a in IPF patients. Both the PCR and protein assessments are shown after normalization compared with internal controls. Mean ± SEM; n = 5 mice in each group; *P < 0.05; **P < 0.01 versus saline or normal group. CTGF, connective tissue growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
TGF-β signaling pathway plays an important role in the pathogenesis of fibrotic response.12 In our study, western blot showed that the expression of several proteins belonging to the TGF-β pathway was significantly upregulated in BLM-treated mice compared with that in saline mice (Figure 1d), indicating that TGF-β signaling pathway was activated in our model of experimental pulmonary fibrosis. We then carried out real-time polymerase chain reaction (PCR) on some selected miRs that have been reported to take a part in pulmonary fibrosis or cardiac fibrosis, to see whether they were deregulated in lungs of mice with experimental pulmonary fibrosis. As shown in Figure 1e, miR-21, miR-133, and miR-590 were upregulated, whereas miR-26a, miR-29, and miR-30 were downregulated in mice with experimental pulmonary fibrosis compared with the expression levels in control mice. Further study confirmed that miR-26a was downregulated in the lungs of IPF patients compared with the level in the lungs of normal people (Figure 1f).
To determine the potential role of miR-26a in the process of pulmonary fibrosis, we administered, intratracheally, injection of antagomiR-26a on days 1, 2, 8, 9, 15, and 16. On the 21st day after treatment, we evaluated collagen deposition in the lungs. As illustrated in Figure 2a, miR-26a was significantly inhibited by antagomiR-26a, whereas antagomiR-negative control (NC) was not. And, antagomiR-26a caused a marked collagen deposition, which was similar to that caused by BLM (Figure 2b–e). At the same time, transfection of the miR-26a inhibitor (AMO-26a) induced significant upregulation of col1a1 and col3a1 in MRC-5 cells (Figure 2f). These results suggest that both in vivo and in vitro inhibition of miR-26a in the lung activates TGF-β pathway and causes pulmonary fibrosis in mice.
Figure 2.
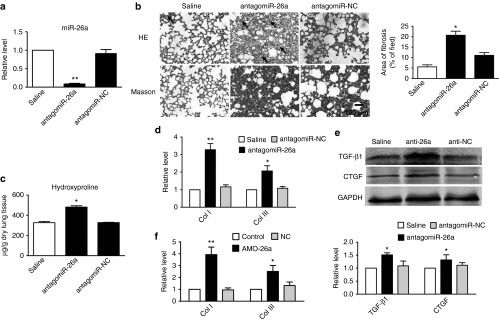
Inhibition of miR-26a caused pulmonary fibrosis in mice. (a) Real-time PCR analysis of miR-26a expression in mice treated with antagomiR-26a as compared with saline group. (b) Hematoxylin–eosin (HE) and Masson Trichrome staining of mouse lung sections showing interstitial fibrosis with collagen deposition 21 days after intratracheal injection of antagomiR-26a (original magnification ×200). (c) Increased hydroxyproline content in mice treated with antagomiR-26a. (d) Real-time PCR quantification of relative levels of collagen I (Col I) and Col III mRNAs in antagomiR-26a-treated and saline-treated mice. (e) Western blot analysis of TGF-β1 and CTGF proteins in antagomiR-26a-treated mice as compared with control animals. (f) Real-time PCR analysis of expression of Col I and Col III mRNAs in MRC-5 cells. Both the PCR and protein assessments are shown after normalization compared with internal controls. Mean ± SEM; n = 4; *P < 0.05, **P < 0.01 versus saline or control group. CTGF, connective tissue growth factor; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
miR-26a attenuates TGF-β1-induced fibrogenesis in cultured MRC-5 cells
Pulmonary fibrosis is characterized by abnormal deposition of ECM mainly consisting of collagen. We therefore transfected miR-26a into cultured MRC-5 human fetal lung fibroblasts to assess the effects of miR-26a on collagen production. As shown in Figure 3a, miR-26a abolished TGF-β1-induced secretion of collagen in MRC-5 cells whereas the NC failed to do so. Moreover, miR-26a reversed the upregulation of mRNA levels of Col I and Col III induced by TGF-β1 in MRC-5 cells (Figure 3b,c).
Figure 3.

miR-26a attenuates TGF-β1-induced fibrogenesis in cultured MRC-5 cells. miR-26a suppresses TGF-β1-induced increase in (a) collagen content, mRNA levels of (b) collagenase (Col) I and (c) Col III in cultured MRC-5 cells, whereas the CTGF mask alleviates this antifibrotic effect of miR-26a. n = 5, *P < 0.05, **P < 0.01 versus control, respectively; #P < 0.05 versus TGF-β1; &P < 0.05 versus TGF-β1+miR-26a. (d) 5-ethynyl-2′-deoxyuridine (EdU) fluorescence staining detects the newly synthesized DNA. (e) Western blot shows that miR-26a inhibited the TGF-β1-induced differentiation in MRC-5 cells. (f) Fluorescence-labeled smooth muscle α-actin (α-SMA) protein was visualized by fluorescence microscopy. miR-26a suppressed TGF-β1-induced expression of α-SMA in MRC-5 cells. α-SMA was stained in red and nuclei in blue (original magnification ×200). Both the PCR and protein assessments are shown after normalization compared with internal controls. n = 3, **P < 0.01 versus control, #P < 0.05 versus TGF-β1. SMA, smooth muscle α-actin. CTGF, connective tissue growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
The development of pulmonary fibrosis is associated with fibroblast proliferation, differentiation into pathological myofibroblasts, and increased collagen content. As shown in Supplementary Figure S1a,b, miR-26a inhibited the elevated cell viability of fibroblasts induced by 10% serum. Moreover, miR-26a alleviated the TGF-β1-induced proliferation of MRC-5 cells (Figure 3d). The data from Figure 3e showed that miR-26a inhibited the TGF-β1-induced upregulation of Smad4, whereas miR-26a had no significant effects on p-Smad3. In addition, western blotting and immunofluorescence assay showed that overexpression of miR-26a eliminated the TGF-β1-induced upregulation of smooth muscle α-actin, a marker of pathological myofibroblasts in lung fibroblasts, suggesting that miR-26a diminished the differentiation of myofibroblasts (Figure 3e,f).
miR-26a directly regulates the expression of CTGF in MRC-5 cells
Several studies have indicated that CTGF is critical for fibrotic response, including increased proliferation and differentiation of fibroblasts and the synthesis of collagen.13,14 We assumed that miR-26a inhibited pulmonary fibrosis by regulating CTGF. The results from our western blot analysis indeed lent evidence in support of this hypothesis: miR-26a mitigated the TGF-β1-induced upregulation of CTGF (Figure 4a).
Figure 4.

miR-26a posttranscriptionally regulates CTGF. (a) miR-26a reduced TGF-β1-induced upregulation of CTGF protein in MRC-5 cells. **P < 0.01 versus control, #P < 0.05 versus TGF-β1. (b) Sequence alignment showing miR-26a:CTGF complementarity for mouse, rat, and human genes. The matched base pairs are outlined by dashed red rectangles. The Genbank accession numbers of the genes are indicated in the brackets, and the positions of the target sites are numbered. (c) Compared with control, transfection of miR-26a with the luciferase reporter gene vector containing the wild-type 3′-untranslated region (UTR) of CTGF resulted in a significant decrease of luciferase activity. Coapplication of miR-26a with AMO alleviated the reduction of luciferase activity, whereas NC showed no effects. miR-26a has no effect on the mut-CTGF construct. (d) Compared with control, transfection of miR-26a resulted in a significant decrease of CTGF. Coapplication of miR-26a with AMO alleviated the reduction of CTGF, whereas NC showed no effects. (e) Real-time PCR shows that miR-26a had no effects on CTGF mRNA level. AMO: miR-26a inhibitor; NC: negative control. Both the PCR and protein assessments are shown after normalization compared with internal controls. Mean ± SEM; n = 4, represents four independent experiments under each condition; *P < 0.05, **P < 0.01 versus control; #P < 0.05 versus miR-26a; &P < 0.05 versus mut-CTGF. CTGF, connective tissue growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
By aligning miR-26a with the 3′-UTR sequence of the CTGF gene, we found a motif matching the seed sites of miR-26a (Figure 4b), suggesting CTGF to be a target of miR-26a. Our subsequent experiments validated this targeting relationship. As depicted in Figure 4c, cotransfection of miR-26a (20 nmol/l) with the luciferase expression vector carrying the target fragments caused a significant reduction of luciferase activities compared with the results of transfection with luciferase vector alone. The reduction of luciferase activities was efficiently reversed by the miR-26a inhibitor AMO-26a (10 nmol/l). In addition, we used a CTGF 3′-UTR construct with mutation on the predicted miR-26a binding sites. Thus, miR-26a failed to diminish the mutated luciferase activities, whereas the mutated miR-26a could do it (Figure 4c). This was further verified by the data illustrated in Figure 4d, CTGF expression was significantly downregulated in MRC-5 cells after transfection with miR-26a, which was reversed by the miR-26a antisense AMO-26a (miR-26a inhibitor). Real-time PCR showed that miR-26a had no effect on CTGF mRNA levels (Figure 4e).
To further confirm whether the antifibrotic effects of miR-26a are in fact mediated through CTGF, we introduced the miR-mask oligodeoxyribonucleotide to block the regulatory effect of miR-26a on CTGF. The miR-mask oligodeoxyribonucleotide techniques have been validated in masking the binding sites for miRs of interest in 3′-UTR of target mRNA.15,16 As shown in Figure 3a–c, miR-26a failed to weaken the fibrogenesis induced by TGF-β1 in MRC-5 cells when we cotransfected the CTGF mask, indicating that the effects of miR-26a are mediated through CTGF.
Enhancement of miR-26a expression attenuates experimental pulmonary fibrosis
To evaluate the antifibrotic effects of miR-26a in a mouse model of pulmonary fibrosis, we delivered miR-26a into mice through intratracheal injection of agomiR-26a once a day for 3 days before BLM administration (Figure 5a, subpart (A)). MiR in the form of agomiR has been previously shown to be highly efficient for sustained miR overexpression under both in vitro and in vivo conditions.17,18 At 28 days after treatment, we evaluated collagen deposition in the lungs. As shown in Figure 5b,c, agomiR-26a significantly alleviated the exaggerated deposition of collagen and depressed the boundary of fibrosis induced by BLM, whereas the negative control agomiR (agomiR-NC) had no effects on BLM-induced lung fibrosis. It was further confirmed that agomiR-26a attenuated increased hydroxyproline content and the mRNA expressions of collagen I, collagen III, MMP-2, and MMP-9 in lungs of mice treated with BLM (Figure 5d,e). Meanwhile, miR-26a reversed the BLM-induced upregulation of CTGF and col1a2 at the protein level (Figure 5f).
Figure 5.

miR-26a prevents experimental pulmonary fibrosis in mice. (a) Schematic representation of protocol used to study the ability of miR-26a to prevent (subpart (A)) and to attenuate (subparts (B,C)) experimental pulmonary fibrosis. Mice were pretreated with agomiR-26a for 3 days followed by injection of bleomycin (BLM) for 28 days. miR-26a significantly alleviated (b) collagen deposition and (c) area of fibrosis induced by BLM, whereas agomiR-NC had no effects. miR-26a markedly decreased (d) hydroxyproline content and (e) mRNAs of collagenase (Col) I, Col III, matrix metalloproteinase (MMP)-2, and MMP-9 in the lungs of mice treated with BLM. (f) miR-26a inhibited BLM-induced upregulation of Col1a2, Smad4, and CTGF protein levels. Both the PCR and protein assessments are shown after normalization compared with internal controls. n = 3, *P < 0.05, **P < 0.01 versus saline; #P < 0.05 versus BLM. NC, negative control. CTGF, connective tissue growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
To explore whether miR-26a has therapeutic potential in the treatment of lung fibrosis, we performed intratracheal injection of agomiR-26a once a day from day 7 to day 9 after instillation of BLM (Figure 5a, subpart (B)), the time point when fibrogenesis occurs,19,20 and analyzed the extent of fibrosis 28 days after administration of BLM. Under this condition, agomiR-26a attenuated collagen deposition and hydroxyproline content, as well as collagen and CTGF, at mRNA and protein levels in BLM-treated lungs (Figure 6a–e). We further administered agomiR-26a from day 14 to day 16 after infusion of BLM and then detected the extent of fibrosis (Figure 5a, subpart (C)). It was still able to attenuate lung fibrosis, although the effects were less effective than those in mice that were treated with agomiR-26a at earlier time points.
Figure 6.

Enhancement of miR-26a expression attenuates experimental pulmonary fibrosis. Administration of agomiR-miR-26a after 7 or 14 days infusion of bleomycin (BLM) attenuated (a) collagen deposition and (b) area of fibrosis, whereas agomiR-NC had no effects. miR-26a markedly decreased (c) hydroxyproline content and (d) mRNAs of collagenase (Col) I, Col III, matrix metalloproteinase (MMP)-2, and MMP-9 mRNAs in the lungs of mice treated with BLM. (e) miR-26a inhibited BLM-induced upregulation of Col1a2, Smad4, and CTGF protein levels. Both the PCR and protein assessments are shown after normalization compared with internal controls. n = 3, *P < 0.05, **P < 0.01 versus saline; #P < 0.05, ##P < 0.01 versus BLM. NC, negative control. CTGF, connective tissue growth factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
The positive feedback loop between miR-26a and p-Smad3 is involved in lung fibrosis
Analysis of the putative promoter sequence by the Genomatix algorithm revealed a binding site for Smad3 in the region upstream of the miR-26a gene. We then evaluated the effect of Smad3 on miR-26a through activation of Smad3 with recombinant TGF-β1, and real-time PCR showed that miR-26a expression was significantly repressed by TGF-β1 in MRC-5 cells (Figure 7a). Meanwhile, knockdown of Smad3 using a short hairpin RNA (shRNA) against Smad3 (pRetroSuper-GFP (green fluorescent protein) Smad3, sh-Smad3)21 decreased CTGF, a direct target of miR-26a (Figure 7b), and increased miR-26a, whereas the empty vector had no effect (Figure 7a), indicating that Smad3 negatively regulates transcription of miR-26a. Furthermore, we transfected the sh-Smad3 to inhibit the expression of Smad3 in MRC-5 cells before treating with TGF-β1, and sh-Smad3 could reverse the downregulation of miR-26a induced by TGF-β1 (Figure 7a), suggesting that the effects of TGF-β on miR-26a are in fact mediated by Smad3.
Figure 7.

A positive feedback loop between miR-26a and Smad3 is involved in the lung fibrosis. (a) Real-time PCR confirmed the effect of Smad3 on expression of miR-26a. (b) Inhibition of Smad3 with sh-Smad3 induced downregulation of CTGF in MRC-5 cells. (c) Sequence alignment showing the miR-26a:Smad4 complementarity for mouse, rat, and human genes. The matched base pairs are marked with dashed red rectangles. The Genbank accession numbers of the genes are indicated in the brackets, and the positions of the target sites are numbered. (d) Compared with control, transfection of miR-26a with the luciferase reporter gene vector containing the wild-type 3′-UTR of Smad4 resulted in a significant decrease of luciferase activity. Coapplication of miR-26a with AMO alleviated the reduction of luciferase activity, whereas NC showed no effects. miR-26a has no effect on the mut-Smad4 construct. (e) Compared with control, transfection of miR-26a resulted in a significant decrease of Smad4. Coapplication of miR-26a with AMO alleviated the reduction of Smad4, whereas NC showed no effects. (f) Real-time PCR shows that miR-26a had no effects on the expression of Smad4 mRNA. n = 4, represents three independent experiments under each condition. (g) Western blot and (h) immunofluorescence cell staining (original magnification ×400) analysis of nuclear translocation of p-Smad3 in the MRC-5 cells transfected with miR-26a or AMO. Both the PCR and protein assessments are shown after normalization compared with internal controls. n = 3, *P < 0.05, **P < 0.01 versus control; #P < 0.05, ##P < 0.01 versus miR-26a; &P < 0.05 versus mut-Smad4. NC, negative control. AMO: miR-26a inhibitor; CTGF, connective tissue growth factor; DAPI, 4',6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR, microRNA; PCR, polymerase chain reaction; TGF, transforming growth factor.
As illustrated in Figure 7c, TargetScan miR database predicted that Smad4, a co-Smad that plays an essential role in nuclear translocation of p-Smad2/3, contains a putative binding site for miR-26a, suggesting that it is a potential target for miR-26a. Consistent with this prediction, our results confirmed that miR-26a posttranscriptionally repressed Smad4 expression by binding to its 3′-UTR (Figure 7d–f). In addition, treatment with agomiR-26a prevented Smad4 upregulation in BLM-treated lungs (Figures 5f and 6e). On the basis of these data, we assumed that miR-26a exerts a regulatory effect on phosphorylated Smad3 (p-Smad3) in the nucleus. Indeed, p-Smad3 in the nucleus was markedly decreased in cells treated with miR-26a, and inhibition of miR-26a could facilitate translocation of p-Smad3 (Figure 7g,h). Moreover, miR-26a had no effect on the expression of total p-Smad3 (T-p-Smad3) (Figure 7g). These results indicated that TGF-β1 modulated the expression of miR-26a by regulating Smad3, and miR-26a inhibited the nuclear translocation of p-Smad3 to block the signaling events downstream of TGF-β1, which finally alleviated collagen deposition and reduced the lung fibrosis.
Discussion
In this study, we evaluated the antifibrotic role and the mechanisms of miR-26a action in governing pulmonary fibrosis (Figure 8). Our results showed downregulation of miR-26a in the lungs of mice with experimental pulmonary fibrosis and in IPF patients, which resulted in posttranscriptional derepression of CTGF and promoted the differentiation into pathological myofibroblasts and induced collagen production in cultured MRC-5 cells. Overexpression of miR-26a repressed TGF-β1-induced fibrogenesis in MRC-5 cells and attenuated pulmonary fibrosis in mice. In addition, miR-26a expression was downregulated by TGF-β1 induced activation of Smad3. Moreover, miR-26a inhibited the nuclear translocation of p-Smad3 by directly targeting Smad4, which is essential in the process of nuclear translocation of p-Smad2/Smad3. Thus, our study uncovered a novel positive feedback loop between miR-26a and p-Smad3, which may be involved in pulmonary fibrosis. These findings indicate that miR-26a is a strong antifibrotic miR in the lung and may be considered a new target for the prevention and treatment of IPF.
Figure 8.

Proposed model for the role of miR-26a in pulmonary fibrosis. In response to stimuli, the TGF-β1 signaling pathway is activated, which in turn downregulates expression of miR-26a by stimulating phosphorylation and nuclear translocation of Smad3 in lung fibroblasts. Reduction of miR-26a results in increase in CTGF, leading to production of collagens and pulmonary fibrosis. Moreover, miR-26a downregulation results in upregulation of Smad4, which increases nuclear translocation of p-Smad3 and further suppresses expression of miR-26a. Once the miR-26a/p-Smad3 loop is activated, it is perpetuated on its own and finally aggravates collagen deposition and other extracellular matrix proteins involved in fibrosis. CTGF, connective tissue growth factor; miR, microRNA; TGF, transforming growth factor.
A growing body of evidence has proven that miRs play important roles in the initiation and progress of pulmonary fibrosis and may be new therapeutic targets for fibrogenic lung diseases. For example, Kusum et al.1 found that let-7 inhibition in the lungs caused epithelial–mesenchymal transition, thickening of alveolar septa, and induction of pulmonary fibrosis. Liu et al.8 showed that miR-21 is upregulated in the lungs of mice with BLM-induced fibrosis and also in the lungs of patients with IPF and that administration of antisense miR-21 diminished the severity of experimental lung fibrosis in mice. In addition, miR-200, miR-31, and miR-29 have been shown to play an antifibrotic role in lungs.22,23,24
miR-26a has been reported to play important roles in the regulation of many diseases. Salvatori et al.22 found that miR-26a targets E2F7-induced cell proliferation and inhibits monocytic differentiation in acute myeloid leukemia cells. Suh et al.25 showed that miR-26a promotes apoptosis of hypoxic rat neonatal cardiomyocytes by repressing glycogen synthase kinase (GSK)-3β protein expression. Recently, Masahide et al.26 confirmed that miR-26a controls the proliferation and differentiation of cardiac fibroblasts and represses activation of atrial fibroblasts by downregulating transient receptor potential cation channel (TRPC) 3 protein. Wei et al.10 showed that miR-26a inhibits angiotensin II–stimulated collagen I and CTGF gene expression in cardiac fibroblasts. Previous studies using miR microarray showed that miR-26a was downregulated in the lungs of mice with experimental pulmonary fibrosis and in IPF patients.1,8 However, there has not been any evidence for the antifibrotic role of miR-26a in lung fibrosis. The current study thus is the first to uncover miR-26a as an antifibrotic miR in pulmonary fibrosis.
Pulmonary fibrosis is accompanied by aberrant proliferation of fibroblasts and increased deposition of ECM.27,28,29 In response to injury, lung fibroblasts undergo phenotypic modulation to become smooth muscle α-actin-positive myofibroblasts, a critical component both in the repair process following lung injury and in secretion of collagen. The presence of myofibroblasts and collagen at the site of injury ensures adequate scar formation to maintain the structural integrity and function of the alveoli.30 Transforming growth factor-β (TGF-β) pathway is an important mediator of fibrogenic response, including lung fibrosis, and can induce differentiation of pulmonary fibroblasts into myofibroblasts and can cause active synthesis of ECM proteins.31,32 In the pathological process associated with fibrosis, TGF-β1 interacts with a complex of transmembrane serine/threonine kinase receptors (TGF-βR I/TGF-βR II) and leads to phosphorylation of transcription factors Smad2/Smad3, which form a complex with Smad4. This heteromeric complex translocates into the nucleus and modulates other profibrotic genes, promoting proliferation and differentiation of fibroblasts to myofibroblasts and inducing deposition of collagen.33 CTGF, a major profibrotic molecule downstream of the TGF-β signaling pathway, is considered a key factor in differentiation of fibroblasts into myofibroblasts and in ECM synthesis.34 Our results showed that miR-26a repressed pulmonary fibrosis by inhibiting proliferation, differentiation, and collagen secretion in lung fibroblasts by targeting CTGF, which may be a molecular mechanism for miR-26a in lung fibrosis. Moreover, TargetScan database predicts Col1a2 as a potential target of miR-26a, and Wei et al.10 have verified that miR-26a posttranscriptionally regulates Col1a2 in cardiac fibroblasts. Thus, we assumed that miR-26a may be able to fulfill its antifibrotic role by directly regulating collagen synthesis apart from targeting CTGF in pulmonary fibrosis, which warrants further studies in detail.
In addition, Davis et al.35 showed that TGF-β1 promotes a rapid increase in the expression of mature miR-21 through a posttranscriptional step and promotes the expression of primary transcripts of miR-21 (pri-miR-21) through Smad activation. Other researchers confirmed Smad3 plays an important role in regulating expression of many miRs, such as inhibiting expression of let-7d and miR-29, in addition to increasing expression of transcripts such as miR-21, both of which contribute to the pathogenesis of lung fibrosis.1,23,36 Our data showed that TGF-β1 regulated miR-26a expression in a Smad3-dependent manner. However, whether Smad3 directly transactivates expression of miR-26a or not is unknown.
Some studies found that the downregulation of target imposed by miRs is quantitatively modest; even an overexpressed miR typically downregulates most of its endogenous targets by less than 50%, but most proteins should remain effective at levels greater than this degree of inhibition.4 Some miRs exert their effects through modulation of dozens of targets by relatively subtle changes in expression. Thus, the summation of small changes in multiple mRNAs may be responsible for the phenotypic effects of miRs.37 In this study, we uncovered a novel positive feedback loop between p-Smad3 and miR-26a during pulmonary fibrogenesis. We confirmed the Smad3-modulated expression of miR-26a on the one hand and miR-26a-regulated expression of Smad4 and nuclear translocation of p-Smad3 on the other hand. In response to injury, activation of p-Smad3 suppressed expression of miR-26a to relieve repression of CTGF, which in turn increased the production of collagen. On the other hand, downregulation of miR-26a is expected to downplay the positive feedback mechanism and enhance p-Smad3-mediated lung fibrosis. Once the loop is activated, it will run over and over again, and finally induce collagen deposition and lung fibrosis. This miR-26a/p-Smad3 positive feedback mechanism identified in this study may be a potential target for the prevention or therapy of pulmonary fibrosis.
In summary, our study showed that CTGF is one of the targets for miR-26a. MiR-26a attenuated fibrogenesis under both in vitro and in vivo settings. Moreover, we uncovered a positive feedback loop between miR-26a and p-Smad3, which was shown to be involved in the progress of IPF. Our study therefore has elucidated the action and mechanism of miR-26a in IPF and has indicated that miR-26a may be a novel target for treatment of lung disease–associated fibrogenesis.
Materials and Methods
IPF tissues. IPF lung tissue samples were obtained from surgical remnants of biopsies or lungs explanted from patients with IPF who underwent pulmonary transplantation, and control samples were obtained from resected tissues from patients with lung cancer through the Second Affiliated Hospital of the Harbin Medical University under the procedures approved by the Ethnic Committee for Use of Human Samples of Harbin Medical University (Harbin, China).
Experimental pulmonary fibrosis model and treatment. Use of animals (8-week-old C57BL/6 mice; 20–30 g) in this work was in accordance with the regulations of the Ethics Committees of Harbin Medical University and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). The mice anesthetized with pentobarbital (40 mg/kg, i.v.) were randomly divided into two groups: saline and bleomycin (BLM, Sigma-Aldrich, St Louis, MO). To induce pulmonary fibrosis, BLM was injected intratracheally at a dose of 1.5 U/kg body weight for 4 weeks. Control groups received the same volume of sterile saline only. All surgical procedures were performed under sterile conditions. The cholesterol-conjugated miR-26a mimics/antisense and negative control (agomiR -26a/antagomiR-26a and agomiR-NC/antagomiR-NC, respectively) were purchased from RiboBio (Guangzhou, China). Mice were treated either with agomiR-26a/antagomiR-26a (10 mg/kg body weight in 50 μl) or with agomiR-NC/antagomiR-NC by intratracheal injection.
Masson's trichrome staining. The lungs of mice in the BLM or saline group were quickly dissected and immersed in 4% neutral buffered formalin for 24 hours and stained with Masson's Trichrome. Fibrosis tissue was quantified with Image Pro Plus 6.0. Three areas were analyzed in each slide, and each area was divided into 100 squares. The number of collagen points (blue stain) was scored as 1 (present) or 0 (absent). Results are shown as the percentage of area occupied by fibrosis to the total area.
Collagen content assay. Total collagen content was measured by Sircol Collagen Assay Kit (Biocolor, County Antrim, UK) according to the manufacturer's instruction. Each sample was incubated at 4 °C with stirring for 24 hours. After centrifugation, 100 μl of each supernatant was assayed. One milliliter of Sircol dye reagent bound to collagen was added to each sample, and the solution was mixed at 4 °C for 30 minutes. After centrifugation, the pellet was suspended in 700 μl of the alkali reagent included in the kit and read at 540 nm with a spectrophotometer. Collagen standard solution was used to construct a standard curve. Total collagen content was also determined by assay of cell protein content. The amount of protein was colorimetrically assessed at 562 nm with BCA protein assay kit (Pierce, Rockford, IL).
Measurement of hydroxyproline. Hydroxyproline assay was performed to determine matrix protein within the lung tissues by Hydroxyproline Assay Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. Data are expressed as micrograms of hydroxyproline per milligram dry weight of lung tissue.
Real-time reverse transcriptase-PCR. Total RNA samples were extracted from lung tissues of mice and IPF patients, or from MRC-5 cells, using Trizol (Invitrogen, Carlsbad, CA). As delineated in the work of Yang et al.,38 the relative expression levels of mRNAs and miRs were quantified by the mirVana qRT-PCR miR Detection Kit in conjunction with real-time reverse transcriptase (RT)-PCR with SYBR Green I and TaqMan microRNA assays (Applied Biosystems, Foster City, CA), respectively. After circle reaction, the threshold cycle (Ct) was determined, and relative mRNA and miR levels were calculated based on the Ct values and normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase or U6 in each sample.
Cell culture and treatment. MRC-5 human fetal lung fibroblasts were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China). The cells were grown in 25 cm2 cell culture flasks with Dulbecco's Modified Eagle Medium (Hyclone) containing 10% fetal bovine serum (Hyclone, Logan, UT), 2 mmol/l L-glutamate, 100 U/ml penicillin G, and 100 U/ml streptomycin at 37 °C in 5% CO2, 95% air. The cells were maintained at subconfluent densities and used when growing to passages 3 or 4. After starvation in serum-free medium for 24 hours, MRC-5 cells were administered recombinant human TGF-β1 (Sigma-Aldrich) for 24 hours.
Transfection procedures. For transfection, cells were washed with serum-free medium once and then incubated with 4 ml serum-free medium for 4–6 hours. The miR (or the sh-Smad3 plasmid) and lipofectamine 2000 (Invitrogen) were separately mixed with 500 μl of Opti-MEM I Reduced Serum Medium (Gibco, Grand Island, NY) for 5 minutes. Then, the two mixtures were combined and incubated at room temperature for 20 minutes. The lipofectamine: miRs (or small interfering RNA) mixture was added to the cells and incubated at 37 °C for 6 hours. The miR-26a, AMO, and mut-miR-26a were purchased from RiboBio (Guangzhou, China). The CTGF mask was synthesized by Invitrogen. Subsequently, 5 ml of fresh medium containing 10% fetal bovine serum was added to the flasks, and the cells were maintained in culture until the following experiments.
Immunofluorescence cell staining. Cultured MRC-5 cells on sterile glass cover slips were washed briefly with cold phosphate-buffered saline 3 times and fixed with 4% paraformaldehyde for 15 minutes. Then, the cell membrane was penetrated by Triton X-100 for 1 hour and blocked by normal goat serum for 1 hour, at 37 °C. The cells were incubated with anti-smooth muscle α-actin or anti-p-Smad3 antibody (Abcam; Cell Signaling Technology, Beverly, MA) overnight at 4 °C and subsequently incubated with fluorescein isothiocyanate–conjugated goat anti-mouse or goat anti-rabbit antibody for 1.5 hours. The cells were washed with phosphate-buffered saline, and nuclei were stained with 4',6-diamidino-2-phenylindole (Roche Molecular Biochemicals, Basel, Switzerland) for 5 minutes at room temperature. Immunofluorescence was analyzed under a fluorescence microscope (Nikon 80i, Tokyo, Japan).
5-Ethynyl-2′-deoxyuridinefluorescence staining. 5-Ethynyl-2′-deoxyuridine fluorescence staining was used to detect the newly synthesized DNA in MRC-5 cells after the indicated treatment. All steps followed the manufacturer′s instructions of Cell-Light EdU DNA Cell Proliferation Kit (RiboBio).
Western blotting. For western blot analysis, total protein samples were extracted from tissues or cells using the procedure previously described.36 Tissues or cells were lysed with radio immunoprecipitation assay lysis buffer (Beyotime, Jiangsu, China). Proteins (60 μg) were fractionated on a 10% or 15% sodium dodecyl sulfate–polyacrylamide gel. After electrophoretically transferring to a pure nitrocellulose blotting membrane (Pall Life Science, Ann Arbor, MI), the blots were probed with primary antibodies, with glyceraldehyde-3-phosphate dehydrogenase (anti-glyceraldehyde-3-phosphate dehydrogenase antibody from Kangchen, Shanghai, China) as an internal control. Primary antibodies against TGF-β1, Smad3, Smad4, and p-Smad3 were rabbit polyclonal antibodies purchased from Cell Signaling Technology (Beverly, MA). Primary antibodies against CTGF or Col1a2 were rabbit or goat polyclonal antibodies and purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Immunoreactivity was detected using Odyssey Infrared Imaging System (Gene Company, Hongkong, China). The bands were quantified by measuring the band intensity for each group.
Luciferase reporter assays. CTGF/Smad4 3′-UTR containing the conserved miR-26a binding sites and their mutated 3′-UTRs were synthesized by Invitrogen and amplified by PCR. The PCR fragment was subcloned into the SacI and HindIII sites downstream of the luciferase gene in pMIR-Report (Promega, Madison, WI). The luciferase vector (0.1 μg) containing the 3′-UTR was cotransfected with miR-26a mimics into HEK-293 cells using lipofectamine 2000 (Invitrogen). As an internal control, 10 ng of Renilla luciferase reporters were also included. After 48-hour transfection, the cells were collected, and dual luciferase activities were measured by a luminometer according to the manufacturer's instructions.
Statistical analysis. All data are presented as mean ± SEM. One-way analysis of variance followed by Dunnett's posthoc test was used for multiple comparisons. A two-tailed value of P < 0.05 was considered a statistically significant difference. Data were analyzed using GraphPad Prism 5.0 and SPSS 14.0.
SUPPLEMENTARY MATERIAL Figure S1. Effect of miR-26a on cell viability of MRC-5 cells.
Acknowledgments
We thank Joan Massague for pRetroSuper-GFP Smad3 plasmid. This study was supported by National Basic Research Program of China (973 program, 2013CB531104); the Major Program of National Natural Science Foundation of China (grant 81230081); the Funds for Creative Research Groups of the National Natural Science Foundation of China (grant 81121003), the National Natural Science Foundation of China (31171094, 31300943); and the Postgraduate Research Innovation Fund of Heilongjiang (YJSCX2012-243HLJ). The authors declared no conflict of interest.
Supplementary Material
Effect of miR-26a on cell viability of MRC-5 cells.
References
- Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Slutsky AS. Idiopathic pulmonary fibrosis–new insights. N Engl J Med. 2007;356:1370–1372. doi: 10.1056/NEJMcibr070490. [DOI] [PubMed] [Google Scholar]
- Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–263. doi: 10.1038/nrm2868. [DOI] [PubMed] [Google Scholar]
- Hu X, Guo J, Zheng L, Li C, Zheng TM, Tanyi JL, et al. The heterochronic microRNA let-7 inhibits cell motility by regulating the genes in the actin cytoskeleton pathway in breast cancer. Mol Cancer Res. 2013;11:240–250. doi: 10.1158/1541-7786.MCR-12-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunwald E. Cardiovascular science: opportunities for translating research into improved care. J Clin Invest. 2013;123:6–10. doi: 10.1172/JCI67541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–2387. doi: 10.1161/CIRCULATIONAHA.110.958967. [DOI] [PubMed] [Google Scholar]
- Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Kim I-K, Kumar S, Jayasinghe S, Hong NY, Castoldi G, et al. NF-kappaB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. 2012;228:1433–1442. doi: 10.1002/jcp.24296. [DOI] [PubMed] [Google Scholar]
- Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123:1939–1951. doi: 10.1172/JCI62185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation. 2012;126:840–850. doi: 10.1161/CIRCULATIONAHA.112.094524. [DOI] [PubMed] [Google Scholar]
- Daniels A, van Bilsen M, Goldschmeding R, van der Vusse GJ, van Nieuwenhoven FA. Connective tissue growth factor and cardiac fibrosis. Acta Physiol (Oxf) 2009;195:321–338. doi: 10.1111/j.1748-1716.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465–472. doi: 10.1093/cvr/cvp130. [DOI] [PubMed] [Google Scholar]
- Wang Z, Luo X, Lu Y, Yang B. miRNAs at the heart of the matter. J Mol Med (Berl) 2008;86:771–783. doi: 10.1007/s00109-008-0341-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Li D, Liu X, Lin L, Hou J, Li N, Wang C, et al. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J Biol Chem. 2011;286:36677–36685. doi: 10.1074/jbc.M111.270561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–243. doi: 10.1016/j.ccr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, et al. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massagué J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell. 2006;125:929–941. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Yang S, Xie N, Cui H, Banerjee S, Abraham E, Thannickal VJ, et al. miR-31 is a negative regulator of fibrogenesis and pulmonary fibrosis. FASEB J. 2012;26:3790–3799. doi: 10.1096/fj.11-202366. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, et al. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther. 2012;20:1251–1260. doi: 10.1038/mt.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Banerjee S, de Freitas A, Sanders YY, Ding Q, Matalon S, et al. Participation of miR-200 in pulmonary fibrosis. Am J Pathol. 2012;180:484–493. doi: 10.1016/j.ajpath.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Choi E, Cha MJ, Song BW, Ham O, Lee SY, et al. Up-regulation of miR-26a promotes apoptosis of hypoxic rat neonatal cardiomyocytes by repressing GSK-3β protein expression. Biochem Biophys Res Commun. 2012;423:404–410. doi: 10.1016/j.bbrc.2012.05.138. [DOI] [PubMed] [Google Scholar]
- Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Lee CG, Cho S, Homer RJ, Elias JA. Genetic control of transforming growth factor-beta1-induced emphysema and fibrosis in the murine lung. Proc Am Thorac Soc. 2006;3:476–477. doi: 10.1513/pats.200603-040MS. [DOI] [PubMed] [Google Scholar]
- Cutroneo KR, White SL, Phan SH, Ehrlich HP. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J Cell Physiol. 2007;211:585–589. doi: 10.1002/jcp.20972. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Faherty N, Curran SP, O'Donovan H, Martin F, Godson C, Brazil DP, et al. CCN2/CTGF increases expression of miR-302 microRNAs, which target the TGFβ type II receptor with implications for nephropathic cell phenotypes. J Cell Sci. 2012;125 Pt 23:5621–5629. doi: 10.1242/jcs.105528. [DOI] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X, et al. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol. 2012;44:2152–2160. doi: 10.1016/j.biocel.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol. 2009;21:461–469. doi: 10.1016/j.ceb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of miR-26a on cell viability of MRC-5 cells.