

Abstract
Neuroglobin (NGB) is considered as an endogenous neuroprotective molecule against stroke, since the protein alleviates the adverse effects of hypoxic and ischemic insults. We previously demonstrated the functional link between NGB and mitochondria since it is required for respiratory chain function. Thus, here, we evaluated the relevance of this effect in the Harlequin (Hq) mouse strain, which exhibits retinal ganglion cell (RGC) loss and optic atrophy due to a respiratory chain complex I (CI) defect. A twofold decrease of NGB amounts was observed in Hq retinas. We constructed a recombinant adeno-associated virus which combines to the mouse NGB open reading frame, its 5′ and 3′UTR, for guarantying mRNA stability and translation capacity. The vector was administrated intravitreally to Hq mice and NGB expression was stable for up to 7 months without negative effect on retinal architecture or function. On the contrary, RGCs and their axons were substantially preserved from degeneration; consequently, CI activity in optic nerves was protected conferring improvements in vision. Hence, we established that NGB prevents respiratory chain impairment, therefore, protecting visual function otherwise compromised by mitochondrial energetic failure.
Introduction
Neuroglobin (NGB) was identified in vertebrates as a member of the globin superfamily, the protein is highly abundant in different brain regions and in the eye.1,2 NGB is now considered as a powerful neuroprotectant,3,4 and NGB expression is correlated with the pathogenesis of glaucoma and Alzheimer,5,6 and in experimental models of these disabling diseases, NGB overexpression appeared to prevent neuronal damage.7 Moreover, the evidence linking NGB and mitochondrial function has increased in the last years.8 Recently, we established that NGB localizes to the mitochondria throughout the rat retina. Further, NGB expression knockdown in retinal ganglion cell (RGC) provokes cellular degeneration and respiratory chain complex I (CI) and III defects in optic nerves resulting in visual function impairment.9 Likewise, we extensively studied the Harlequin (Hq) mouse phenotype that is primarily attributed to apoptosis-inducing factor (AIF) depletion.10 This phenotype involves most of the human neurodegenerative mitochondriopathies hallmarks, with age-dependent degeneration of retina, optic nerve, cerebellum, and cortical regions. Tissue deterioration is associated with a profound respiratory chain CI (RCCI) defect and a low content of some RCCI subunits in brain and retinas.11,12 AIF is a flavoprotein which localizes to the mitochondrial intermembrane space, with its N-terminus anchored to the inner membrane. In AIF-deficient ES cells, the abundance and activity of CI and complex III were reduced11 though the precise role of AIF within respiratory chain complexes is not yet elucidated.13 A functional overlap can exist between AIF and NGB proteins,13,14 but how AIF and NGB oxidoreductase activities could interact for preserving RCCI activity remains a significant question.
We demonstrated that 6–9-month-old Hq mice exhibit RGC loss which correlated with the disappearance of optic fibers and a severe defect of RCCI in optic nerves. RGC physiopathology and onset were an incentive to envisage a gene therapy in Hq eyes before RGC disappearance. The intravitreal administration of adeno-associated virus serotype 2 (AAV2)/2-AIF in animals aged between 4 and 6 weeks was successful in preventing RGC loss and preserving RCCI activity in optic nerves.15 Interestingly, in retinas and optic nerves from Hq mice, an approximate twofold decrease in the steady-state levels of NGB mRNA and protein was observed relative to controls. Hence, Hq mouse eye constitutes a suitable tissue for deciphering NGB mitochondrial function and testing our hypothesis that the protein could protect retinal cell integrity within a context of mitochondrial impairment. We constructed a recombinant AAV2 which combines to the mouse NGB open reading frame (ORF), its full-length 5′ and 3′UTRs, to ensure NGB mRNA stability and translation efficacy by mimicking physiological conditions.16 We demonstrated the long-term morphological and functional improvement of visual system in Hq mice following the intravitreal administration of AAV2/2-NGB. NGB expression driven by the vector in Hq retinas confers: (i) RGC viability improvement, (ii) RCCI activity protection in optic nerves, and (iii) visual function preservation. Since Hq mice replicate the main features of mitochondrial deficiency in humans,12 our data strengthen the hypothesis that NGB is a promising candidate for developing neuroprotective strategies aimed at preserving mitochondrial function in an array of ophthalmic conditions and neurodegenerative diseases.
Results
NGB expression in control and Hq mouse retinas
To examine the subcellular distribution of the NGB in adult mouse retinas, we prepared mitochondrial enriched fractions by differential centrifugation and performed western blot analysis (Figure 1a). Three proteins with apparent molecular masses of about 17, 19, and 21 kDa were revealed in homogenates and mitochondrial fractions. The abundance of NGB is very high in the mitochondrial fraction as observed for ATP synthase-β (a subunit of respiratory chain complex V). Interestingly, only very weak NGB signals were detected in the pellet (nuclei and unbroken cells) and the high speed supernatant. Most of the NGB was found in the mitochondria-enriched fraction. The presence of NGB within the mitochondria was confirmed by subjecting this fraction to a mild proteolysis with proteinase K. The three forms of NGB and the ATP synthase-β gave strong signals indicating that these proteins were inside the organelle. Next, mitochondria-enriched fractions were treated with proteinase K in the presence of Triton X-100; the detergent disrupts mitochondrial membranes giving access to the protease which can then digest mitochondrial membrane–enclosed proteins. ATP synthase-β and NGB protein signals were considerably diminished, confirming their localization inside the organelle in a protease-sensitive form (Figure 1a). Therefore, NGB localizes to the mitochondria of mouse retinas as we previously showed in rats.9
Figure 1.

Neuroglobin (NGB) subcellular localization and distribution in mouse retinas. (a) Western blot detection of NGB and ATP synthase-β proteins in the different subcellular fractions from mouse retinas. Homogenates, high-speed supernatant obtained after mitochondrial centrifugation, pellet containing nuclei and unbroken cells, and mitochondrial fractions were subjected to western blot analysis after electrophoresis of 30 μg per sample. To confirm the presence of the NGB inside the mitochondria, the enriched mitochondrial fractions were treated with 150 µg/ml of proteinase K (PK) in the presence or absence of 1% Triton X100 at 4 °C for 30 minutes. The western blots were examined with different antibodies (Supplementary Table S1). The “PageRuler Plus Prestained Protein Ladder” allowed the estimation of apparent molecular mass of each signal; in the right margin is annotated the markers with similar electrophoretic properties. (b) Immunofluorescence analysis of retinal flat mounts from adult mice immunostained for NGB and BRN3A proteins. BRN3A labeling was exclusively nuclear (green). The prominent NGB labeling (red) was punctuated in the cytoplasm. Some NGB-positive cells did not show BRN3A labeling, they might be displaced amacrine cells or astrocytes which reside in the ganglion cell layer (white arrowheads). The scale bar represents 50 µm for all panels. (c) Retinal flat mounts from adult mice were double labeled for NGB (red) and the mitochondrial protein NDUFB6, a respiratory chain complex I subunit (green). The yellow-orange pixels show that NGB and NDUFB6 were in close apposition within numerous cells (white arrowheads). The scale bar represents 50 µm for all panels.
To investigate, the abundance of NGB in RGCs, flat mounted retinas from 6-month-old mice were immunostained for NGB, BRN3A, and the mitochondrial NDUFB6 protein, a CI subunit (Figure 1b,c). BRN3A is a nuclear factor exclusively expressed by most of the RGCs in rodent retinas.17 All the BRN3A-positive cells showed an intense NGB labeling as punctuate dots in the cytoplasm. In addition, some NGB-positive cells were not immunostained for BRN3A; they may correspond to displaced amacrine cells or astrocytes (Figure 1b, white arrowheads). When antibodies against NGB and NDUFB6 were combined, the majority of cells exhibited similar labeling patterns indicating some extent of colocalization between the two proteins (Figure 1c; white arrowheads).
NGB distribution in other cell populations was analyzed in radial cryosections of retinas immunostained for NGB in control and Hq mice aged 6 months (Figure 2a). In normal mice, it appears that the protein was particularly abundant in the ganglion cell layer (GCL) (Figure 2a, white arrowheads) and the inner segments of photoreceptors, while very weak labeling with the NGB antibody was observed in photoreceptor outer segments. The inner and outer plexiform layers showed strong to moderate punctuate labeling; at the junction between the outer nuclear layer and the outer plexiform layer, the staining presumably represents signal from the mitochondria in the synaptic terminals of photoreceptors and in horizontal cells.18,19 The inner nuclear layer is usually divided in three regions: distal, middle, and proximal; NGB immunostaining is strong in the three levels, especially in the distal region, some of the fluorescent cells being located at the very inner margin of the inner nuclear layer, which could be amacrine cells as previously described.19 Overall, NGB labeling in retinal neurons was consistent with the known abundance of mitochondrial proteins in the different retinal compartments.18
Figure 2.

Cellular distribution and relative amounts of neuroglobin (NGB) in retinas from Harlequin (Hq) and control retinas. (a) The cellular distribution of NGB was examined by indirect immunofluorescence in retinal sections from 6-month-old control and Hq mice using a specific antibody against NGB (green) (Supplementary Table S1), nuclei were staining with 4′, 6-diamidino-2-phenylindole (blue). Scale bars are equivalent to 50 μm for both panels. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; IS, inner segments of photoreceptors; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, outer segment of photoreceptors. (b) RT-qPCR assays were performed using total RNAs from retinas (isolated from mice 6-month old) to determine the steady-state levels of NGB mRNA. RNAs purified from 32 control and 38 Hq retinas were evaluated. Primers used for the NGB mRNA are shown in Supplementary Table S2. (c) Western blot detection of NGB, apoptosis-inducing factor, and ATP synthase-β and β-Actin proteins was performed with 30 µg of whole protein extractions from Hq and control retinas (mice were euthanized at 6 months of age). Specific antibodies against NGB, apoptosis-inducing factor, ATP synthase-β, and β-actin recognized proteins of ~17, ~65, ~57, and ~42 kDa apparent molecular masses respectively as expected from their theoretical molecular weight estimations. In the right, the membrane before incubation with antibodies and the prestained markers sharing similar electrophoretic properties to proteins evaluated were shown. Protein extractions were performed from a single retina per sample; in these conditions it was difficult to reveal the 19 and 21 kDa bands with the antibody against NGB, while they were easily noticeable when biochemical extractions were performed from 36 pooled retinas to obtain “enriched mitochondrial fractions” (Figure 1a).
On the other hand, retinal sections from Hq mice showed a considerable reduction in the thickness of all the neuron layers with a significant diminution of the overall NGB immunostaining (Figure 2a, white arrowheads). To further characterize the reduction of NGB amount in Hq retinas, NGB mRNA steady-state levels in retinas from Hq mice were determined by real-time quantitative polymerase chain reaction (RT-qPCR). Total RNA preparations from 38 Hq and 32 control retinas from 6–8-month-old mice were examined. NGB mRNA was 45% less abundant in Hq retina than in controls (P < 0.0001) (Figure 2b). Whole protein extracts from Hq and control retinas were subjected to western blotting analysis using identical protein amounts purified from single retinas to corroborate these data. The specific antibody against NGB recognized an intense band of ~17 kDa in the protein extracts from control retinas (Figure 2c). In these samples, we were unable to reveal the 19 and 21 kDa bands, while they were noticeable when biochemical extractions were performed from 36 pooled retinas to obtain “enriched mitochondrial fractions” both in the homogenates and the mitochondrial fractions (Figure 1a). In Hq retinas, NGB signal was strongly reduced compared to control retinas; as expected, AIF was undetectable while ATP synthase-β and β-Actin amounts remained almost unchanged in control and Hq retinas (Figure 2c). These results confirm the overall reduction of NGB in Hq retinas relative to age-matched controls, both at the mRNA and protein levels.
Design of a gene therapy using the NGB gene for preventing retinal damage in Hq mice
The evaluation of optic nerves (longitudinal and transverse sections) from control and Hq mice of different ages using transmission electron microscopy allowed us to establish that very few ultrastructural differences were noticed between the two mice at the age of 6 weeks (Supplementary Figure S1). In 6-week-old Hq mice, the only difference relative to age-matched controls was the presence of few swollen axons. Between 3 and 6 months, severe alterations reflecting axonal degeneration were noticed in Hq mice: many axons appeared shrunken, cristae within mitochondria disappeared, and large vacuoles could be seen between the axolemmas and myelin sheaths. Moreover, invasion by astrocytic processes and phagocytosing cells disorganized the nerve structure. Ultimately, in older Hq mice (12–15 months), very few axons remained within the optic nerve and the axonal pathology shares some similarities with Wallerian degeneration.20 In an attempt to prevent RGC and optic nerve degeneration in Hq mice, a recombinant AAV2/2 encompassing the mouse NGB ORF associated with the full-length 5′ and 3′ UTRs of the gene was constructed to ensure mRNA stability and translation capacity16 (Figure 3a). Subsequently, a single intravitreal injection with the AAV2/2-NGB vector (2 × 109 VG per eye) was performed in 4–6-week-old Hq mice. Overall, 45 mice were euthanized 6–7 months after vector administration and extensively evaluated to determine the impact of NGB overexpression on RGC and respiratory chain integrity. Since NGB sequences were inserted into the pAAV-hrGFP; the Green Fluorescent Protein. GFP was used for assessing the efficacy of cell transduction. The reconstruction of whole retinal sections from injected eyes clearly shows a homogenous and intense GFP immunoreactivity restricted to the GCL; all the surface of the retina gave a positive signal with the antibody in this layer. In contrast, the untreated eye did not display GFP immunoreactivity (Figure 3b). Consequently, the GFP labeling obtained confirms the high efficiency of vector transduction. Since a 3xFLAG sequence is appended in frame to the C-terminal extremity of the NGB protein (Figure 3a), retinal sections from untreated and treated eyes were subjected to a double labeling experiment, with a mixture of anti-FLAG and anti-NGB antibodies (Figure 4a). The high-magnification confocal images of retinas revealed in the GCL of treated eyes several cells presenting a staining pattern of NGB similar to the one evidenced in Figure 2a for control mice: intense, located close to the nuclei, and in some positive cells, showing a punctuate aspect. The FLAG antibody labeled also many cells in the GCL from treated eyes; the labeling pattern was similar to the NGB one and some extent of overlapping was observed, as expected (Figure 4a). Next, we determined the abundance of the transduced NGB mRNA by real-time quantitative PCR in retinas from 15 Hq mice subjected to vector administration (Figure 4b). The steady-state levels of the transduced NGB mRNA contributed to an approximate threefold increase of the overall amount of NGB mRNA relative to the one measured in control retinas, while no change was evidenced in the relative steady-state levels of AIF1 mRNA between retinas from treated and untreated eyes (P = 0.33; Figure 4b). To confirm the NGB expression incremental after AAV2/2-NGB administration, the steady-state levels of the protein in retinas were evaluated for 6 treated eyes (about 8 weeks after the treatment), 8 untreated eyes, and compared to those found in 12 age-matched controls (Figure 4c). It appears unambiguously, as shown in Figure 2c, that the amount of NGB was decreased in retinas from Hq-untreated eyes; the estimation after normalization against the β-actin signal indicates a 54% reduction relative to age-matched controls (Figure 4c). In contrast, NGB amount increased significantly in retinas from Hq-treated eyes since it attained 90% of the value measured in control retinas (Figure 4c). Recombinant AAV2/2 administration in the vitreous body of rodents is known to transduce almost exclusively cells within the GCL as shown in Figure 3; besides, the proportion of these cells represent no more than 2% of the whole retinal cell population in rodents.21,22 Therefore, NGB protein levels in cells that expressed the transgene in the GCL of Hq-treated eyes were much higher than the one in untreated Hq retinas. Hence, AAV2/2-NGB administration to Hq eyes led to NGB overexpression essentially in resident GCL without noticeable adverse effects on mouse eyes up to 7 months.
Figure 3.

AAV2/2-Neuroglobin (NGB) vector generation and administration in Harlequin (Hq) eyes. (a) Physical map of the AAV2/2-NGB vector genome (7255 bp), encompassing mouse NGB sequences inserted into the pAAV-IRES-hrGFP plasmid: the NGB ORF (453 bp), encoding 151 amino acids (CDS), is in frame with three FLAG epitopes and transcribed under the control of the Cytomegalovirus promoter and the β-globin intron. The construction contains both untranslated regions at the 5′ (279 bp) and the 3′ (895 bp) ends of the mouse NGB mRNA (NM_022414.2). The plasmid possesses also a cassette allowing the expression of the recombinant humanized green fluorescent protein translated from the encephalomyocarditis virus internal ribosome entry site. (b) Transduction efficiency was evaluated by immunostaining for Green Fluorescent Protein (GFP) antibodies of retinal sections from Hq mice euthanized 6 months after a single intravitreal injection of AAV2/2-NGB (2 × 109 VG in one eye). A strong and homogenous labeling for GFP (green) was restricted to the GCL of the AAV2/2-NGB treated eye, while no GFP labeling was evidenced in the retina from the untreated eye. Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole (blue). Scale bars are equivalent to 0.5 mm and 50 μm (in the magnification). GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Figure 4.

Neuroglobin (NGB) transgene expression in Harlequin (Hq)-treated eyes. (a) AAV2/2-NGB transduction efficiency was further evaluated by immunostaining for a FLAG antibody which recognized the NGB protein synthesized from the vector. A strong labeling for FLAG (green) was restricted to the GCL of AAV2/2-NGB treated retinas; additionally, a high extent of fluorescent signal overlapping was observed in all the cells between antibodies against NGB (red) and FLAG. Scale bar is 50 μm for both panels. (b) RT-qPCR assays were performed with total RNAs extracted from 18 control retinas and 15 pairs of retinas isolated from Hq mice in which one eye was subjected to intravitreal injection of AAV2/2-NGB. Histograms show the steady-state levels of NGB mRNA and AIF1 mRNAs relative to the value assessed in RNAs from control retinas. Primers used are shown in Supplementary Table S2. (c) Representative western blots from retinas of 7-month-old Hq in which eyes were subjected to AAV2/2-NGB treatment reveals an increase of NGB signal relative to Hq-untreated retinas. The membrane before incubation with the antibodies and the prestained markers sharing similar electrophoretic properties to proteins evaluated were shown. Histogram (right panel) shows the relative amount of the NGB protein in retinas from untreated Hq (n = 8), treated Hq (n = 6), and age-matched controls (n = 12). The densities of NGB signals were normalized to β-actin signals.
NGB overexpression limits gliosis reaction in retinas from Hq mice
One prominent feature of the progressive retinal degeneration in Hq mice is glial cell activation, hence glial fibrillary acidic protein (GFAP) showed a significant increase, which begins in mice aged about 4 months.15 To determine whether NGB overexpression could prevent the active growth of Müller cell processes, retinal sections from treated and untreated eyes were subjected to immunochemistry for GFAP and NGB. The reconstruction of whole retinal sections confirmed the extensive glial response in the untreated eye of a 7-month-old Hq mice relative to both its contralateral eye in which the AAV2/2-NGB vector was injected and to an age-matched control (Figure 5a). A higher magnification of independent retinal sections confirms that in control retinas, GFAP immunofluorescence was confined exclusively to the GCL, corresponding to the end-feet of Müller cells and astrocytes resident in this cellular layer (Figure 5b). At 7 months of age, GFAP immunoreactivity was markedly increased in Hq mouse retinas and was not just restricted to the GCL since intense GFAP-stained Müller cell processes extended into the outer nuclear layer of the retina from the untreated eye (Figure 5b, white arrowheads in the middle panel). The steady-state levels of the GFAP mRNA also increased 6.4-fold relative to age-matched controls, confirming that an extensive glial response occurred in Hq retinas concomitantly with RGC degeneration (Figure 5c). In the retinal section from the AAV2/2-NGB-treated eye, NGB labeling was noticeably enhanced in the GCL substantiating both the efficacious vector transduction (Figures 3b and 4a) and the strong accumulation of both NGB mRNA and protein (Figure 4b,c). In opposition, GFAP immunoreactivity was distinctly less increased relative to contralateral untreated eyes, resulting in the limited labeling of the end-feet of Müller cells (Figure 5b). Accordingly, the GFAP mRNA abundance in treated retinas diminished by 19% compared to untreated retinas (n = 10, P = 0.002; Figure 5b). Therefore, NGB overexpression hinders GFAP upregulation in Müller glia; this subtle change may be beneficial for RGC survival in AAV2/2-NGB-treated eyes.
Figure 5.

Glial fibrillary acidic protein (GFAP) expression in retinas from Harlequin and control eyes. (a) Immunostaining for GFAP (red) and a reconstruction of whole retinal sections is shown in the low magnification images from a 7-month-old control and both eyes of an age-matched Hq mouse in which one eye was subjected to AAV2/2-NGB treatment. Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI) (blue); scale bar are equivalent to 0.5 mm. (b) In control retinas, GFAP expression was restricted to the GCL (red), while in retinas from untreated Hq mice, GFAP-labeled glial Müller cell processes that extended into the ONL (white arrowheads) and NGB signal (red) was weak in all the cell layers. Cell nuclei counterstained with DAPI are also shown (blue); scale bars are equivalent to 50 µm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. (c) RT-qPCR assays were performed with RNAs purified from 15 control retinas and from 10 pairs of retinas isolated from Hq mice in which one eye was subjected to intravitreal injection with AAV2/2-NGB. Relative GFAP mRNA variations are represented relative to the value assessed in RNAs from control retinas. Primers used for the GFAP gene are shown in Supplementary Table S2.
RGC loss in Hq mice is prevented by the intravitreal administration of AAV2/2-NGB vector
To determine the impact of NGB overexpression on RGC integrity, we proceeded to the estimation of RGC soma number in whole retinal sections from Hq-treated and Hq-untreated eyes subjected to immunostaining for BRN3A. We compared RGC numbers (revealed by BRN3A labeling) and the total number of cells in the GCL (revealed by 4′, 6-diamidino-2-phenylindole staining) in retinas from control mice (n = 26) and Hq mice in which one eye was subjected to AAV2/2-NGB administration (n = 15). As compared to controls, the number of RGCs were significantly reduced in retinas from Hq-untreated eyes (149 ± 10 versus 247 ± 6 in control retinas; P < 0.0001); a 40% diminution of the total amount of BRN3A-positive cells in the GCL in accordance with our previous study15 (Figure 6a). Consequently, the number of 4′, 6-diamidino-2-phenylindole-stained nuclei in the GCL was 18% lower than in retinas from controls (P = 0.01; Figure 6b). By contrast, RGC number in AAV2/2-NGB-treated eyes (188 ± 11) was 27% higher than in contralateral untreated eyes (P < 0.0001; Figure 6a) and attained 76% of the value measured in control retinas. Besides, the overall amount of cells in the GCL in treated retinas appeared increased relative to untreated eyes (P = 0.012; Figure 6b). Next, we determined the relative amount of γ-synuclein (SNCG) mRNA by subjecting RNA preparations from 15 couples of Hq mouse retinas and 18 age-matched control retinas to RT-qPCR analysis. SNCG is highly and specifically expressed in RGCs from adult mice, and its mRNA abundance has been used by us and others as a RGC marker.15,23 The steady-state level of SNCG mRNA was significantly diminished in Hq retinas from untreated eyes, up to 44% of the control value (P < 0.0001; Figure 6c). In contrast, a consistent and statistically significant increase of 15% in its relative abundance was measured in treated eyes versus contralateral untreated eyes (P = 0.0002; Figure 6c). Thus, the partial preservation of SNCG mRNA abundance confirms that AAV2/2-NGB treatment diminished the rate of RGC loss in Hq mice.
Figure 6.
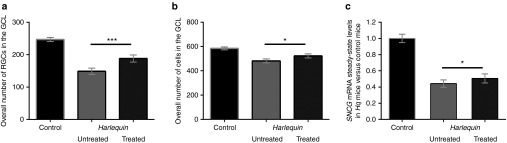
Retinal ganglion cell evaluation in retinas from Harlequin eyes treated with AAV2/2-Neuroglobin vector. (a) Retinal ganglion cell (RGC) numbers were estimated in Hq-treated and Hq-untreated eyes as well as in age-matched controls by immunodetection for BRN3A. BRN3A-positive cells in the GCL were counted in 2–4 independent sections per eye for 26 control eyes and 15 Hq pairs of eyes in which only one was subjected to AAV2/2-NGB injection. Histograms illustrate data (mean values ± SEM) corresponding to: the overall RGC number (BRN3A-positive cells in the GCL). (b) The total number of cells in the GCL was estimated by counting the number of 4′, 6-diamidino-2-phenylindole (DAPI)-stained nuclei in the GCL in 2–4 independent sections per eye for 26 control eyes and 15 Hq pairs of eyes in which only one was subjected to AAV2/2-NGB injection. (c) RT-qPCR assays were performed with total RNAs extracted from 18 control retinas and 15 pairs of retinas isolated from Hq mice in which one eye was subjected to intravitreal injection of AAV2/2-NGB. Histogram shows the steady-state levels of SNCG mRNA relative to the value assessed in RNAs from control retinas. Primers used are shown in Supplementary Table S2.
NGB overexpression protected nerve fiber integrity in Hq mice
To confirm that NGB overexpression was beneficial for RGC integrity, we evaluated both the density of their axons as well as RCCI activity in optic nerves (ONs). First, ON cross-sections were subjected to immunohistochemistry for the heavy chain subunit of neurofilaments (NF200) to detect RGC axons. Figure 7a illustrates the reduction of immunopositive dots in ONs from untreated Hq eyes relative to ONs isolated from age-matched control mice. The axonal profiles detected in ON cross-sections from treated Hq eyes confirmed that they displayed a noticeable increase in NF200-immunopositive signals relative to untreated eyes (Figure 7a). To substantiate these observations, axons (NF200 positive spots) in the optic nerves from six Hq mice in which one eye was subjected to AAV2/2-NGB treatment were counted and compared to optic nerves from Hq-untreated eyes and from age-matched controls (Figure 7b). In Hq-untreated eyes, the reduction of axons in the optic nerves was significant since it was only 46.2% relative to controls; while in optic nerves from AAV2/2-NGB-treated eyes, the overall axon number representing 76.5% of the control value. This indicates that AAV2/2-NGB administration to Hq mouse eyes attenuates RGC axonal damage. In an attempt to establish a functional link between RGC number preservation and respiratory chain integrity, we assessed respiratory chain complex activities by spectrophotometry.11,12,15 The sequential determination of rotenone-sensitive CI activity and the oligomycin-sensitive complex V (CV) activity on the same sample represent a robust and reliable enzymatic activity assay for detecting isolated and partial impairment of respiratory chain activity.12 Figure 7c,d illustrates CV and CI activity measurements for three mouse groups: (i) 38 ONs from control mice aged 6–7 months, (ii) 29 ONs from Hq eyes subjected to AAV2/2-NGB intravitreal injection, and (iii) 29 ONs from the Hq contralateral untreated eyes. In our previous study on Hq mice, we assessed respiratory chain function in ONs, and we demonstrated a severe and specific CI defect with preserved CV activity.15 In this study, the CV activity was increased by about 30% in ONs from Hq relative to controls independently of AAV2/2-NGB treatment (Figure 7c). This difference was statistically significant (P = 0.0008 and P = 0.0002 for untreated and treated eyes, respectively), while the difference of CV activities in ONs from untreated and treated eyes was not significant (P = 0.98). Nonetheless, it was obvious from the current assessments that ONs from 7–8-month-old Hq-untreated mice manifested a diminution in CI activity when compared to isogenic age-matched controls: 7.9 ± 0.5 versus 14.0 ± 0.9; 56% of the control value (P < 0.0001; Figure 7d). The ocular administration of AAV2/2-NGB protected efficiently CI function; indeed, the specific activity of CI (11.2 ± 0.6) attained in ONs from treated eyes 80% of the value measured in control mice; further, ONs from NGB-treated eyes exhibited 42% higher CI activity than ONs from their contralateral untreated eyes (P < 0.0001). Thus, high levels of NGB in RGCs from Hq mice rescued compromised CI activity presumably accounting for the RGC robustness evidenced (Figure 6b).
Figure 7.

Morphological and functional evaluation of optic nerves from Harlequin mice after ocular AAV2/2-Neuroglobin treatment. (a) Independent proximal optic nerve transversal sections (near the globe) from 7-month-old control and Hq mice were immunolabeled with antibodies against the heavy chain (200 kDa) subunit of neurofilaments (NF200, green). The scale bars are equivalent to 50 µm. (b) Bar graph of axon number in controls and Hq optic nerves; six animals per group were evaluated after immunolabeling for NF200, and two independent transversal sections were entirely counted for positive spots using the ImageJ software. Results are illustrated relative to the axon number in optic nerves from aged-matched controls. The axon number per optic nerve in Hq-untreated mice was significantly different relative to controls or Hq subjected to AAV2/2-NGB intravitreal injection (P = 0.0022). (c,d) Specific complex V (CV) and complex I (CI) enzymatic activities were assessed in single optic nerves isolated from mice aged about 7–8 months: 38 controls and 29 Hq mice in which one eye was subjected to AAV2/2-NGB intravitreal injection and the contralateral one remained untreated. The successive measurements of CI and CV activities were expressed as nanomoles of oxidized NADH/min/mg protein. Histograms illustrate CV activity (b) and CI (c) as mean ± SEM of each assay per optic nerve measured in triplicate.
NGB overexpression improves Hq mouse vision
To make the most trustworthy evidence on the protective effect of NGB against RGC degeneration besides indications gathered on cell loss prevention and CI activity protection, it is also required to assess whether the treatment leads to some preservation of visual function. We first evaluated eye fundus using a confocal scanning laser ophthalmoscope (cSLO), a reliable method for in vivo imaging the fiber bundles (RGC axon packages) composing the nerve fiber layer.24 Striations of nerve fiber layer radiating from the optic disc were clearly visible in each eye from mouse before AAV2/2-NGB administration as illustrated in Figure 8a for two 6-week-old mice. Subsequently, each area of the eye fundus (nasal, temporal, inferior, and superior) was visualized monthly until euthanasia to follow over time nerve fiber disappearance and seek for any change related to NGB overexpression. Six months after vector administration, a substantial loss of nerve fiber bundles was evidenced in the two untreated eyes shown (Figure 8a); half of the entire retinal surface appeared as being devoid of fibers (inferior and temporal area for Hq # 1 and 2 respectively). In contrast, images collected for the eyes that received AAV2/2-NGB revealed well-preserved axon tracks in all the areas visualized. We extensively evaluated 45 mice using cSLO; more than 80% of the untreated eyes did exhibit RGC axon degeneration in mice from the age of 3–4 months, while half of the eyes subjected to AAV2/2-NGB treatment exhibited high densities of fiber bundles in all the areas examined until euthanasia (up to 7 months after vector administration). Thus, NGB sustained expression, driven by the AAV2 vector, efficiently protected Hq mice against RGC axonal degeneration. Ultimately, to address the overriding question of whether AAV2/2-NGB administration confers improvements in vision to Hq mice, we assessed visual function of 2-month-old animals and at different times after gene therapy by studying their optomotor responses to rotating sinusoidal gratings (OptoMotry CerebralMechanics, http://www.cerebralmechanics.com). Visuomotor behavior was meticulously measured in isogenic age-matched controls and Hq mice to gather precise thresholds (highest spatial frequency each mouse could track) under our experimental conditions. Optomotor responses recorded from Hq mice aged 4–8 weeks (before any ocular intervention) were similar to those of wild-type mice of the same age (14 animals evaluated in each group). Figure 8b illustrates right and left eyes sensitivities for control and Hq mice: 0.44 ± 0.01 versus 0.39 ± 0.02 (P = 0.01) and 0.41 ± 0.02 versus 0.39 ± 0.02 (P = 0.21) respectively. These data indicate that visual function of Hq mice was not compromised during the first weeks of life and is in agreement with cSLO examinations in which fiber disappearance was only noticeable in 3–4-month-old Hq mice (Figure 8a). Next, we proceed to the evaluation of Hq mice which received in their left eyes AAV2/2-NGB; they were subjected to the test three times at 3 months postinjection and just before euthanasia (up to 7 months postinjection). Figure 8c illustrates clockwise and counterclockwise optokinetic tracking thresholds from Hq mice and age-matched control mice. As we have previously shown, very reduced head-tracking behavior was observed in the Hq mice aged of ~8 months;15 indeed, visuomotor behavior (n = 25) was significantly poorer than control mice (n = 25): 0.16 ± 0.023 versus 0.44 ± 0.016 cycles per degree for the clockwise direction of motion (P < 0.0001). Impressively, an optokinetic tracking threshold of 0.33 ± 0.021 cycles per degree was recorded in Hq-treated eyes, representing 78% of the value obtained in age-matched controls (0.42 ± 0.018). The decrease in visual performance in the untreated eyes was evidenced at the age of 4–5 months and worsened with age; conversely, the responses recorded from the treated eyes remained stable up to 7 months after vector administration (Figure 8c). Optokinetic tracking threshold in Hq-treated eyes was twofold superior to the one measured in untreated eyes (P < 0.0001). Therefore, NGB overexpression benefit encompassed the long-lasting protection of RGCs and their axons along with the maintenance of their functional integrity.
Figure 8.
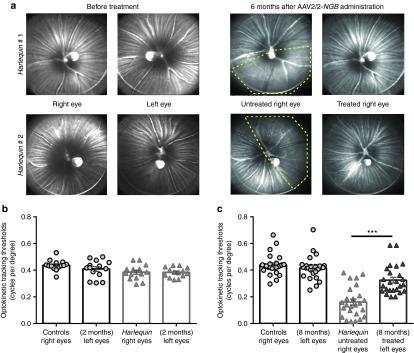
Preservation of nerve fibers in AAV2/2-neuroglobin-treated eyes protects Harlequin mouse vision. (a) Confocal scanning laser ophthalmoscope fundus imaging of two Hq mice: images were collected in 4-week-old animals before AAV2/2-NGB administration, which was performed in their left eyes (left panel). Animals were evaluated monthly thereafter; images shown in the right panel were taken 6 months postinjection. Yellow discontinued lines show retinal regions with obvious nerve fiber loss in untreated eyes; for mouse # 1, the inferior region was the most affected, while for mouse # 2, the temporal area displays few RGC axons. Eyes treated with AAV2/2-NGB exhibited a marked preservation of optic fibers in all the retinal areas examined. Out of the 45 Hq mice subjected to AAV2/2 intravitreal injection in their left eyes and extensively evaluated with SLO, more than 50% exhibited a prevention of RGC axonal damage. (b,c) The Optomotry set-up allowed the determination of the optokinetic tracking threshold measurements (cycles per degree) for left and right eyes, independently scored (clockwise and counterclockwise responses respectively) under photopic conditions. Right and left eye sensitivities) for Hq (n = 14) and control (n = 14) mice aged 6–8 weeks are illustrated in (b) as means ± SEM of measures performed 2–3 times 4–6 days apart. (c) Histogram shows right and left eye sensitivities (Optokinetic response thresholds) for 7–8-month-old control mice (n = 25) and Hq mice subjected to AAV2/2-NGB injection in their left eyes (n = 25). Hq mice were evaluated at 3 and 6–7 months postinjection (the test was performed three times each time, 2–5 days apart), values presented are the means ± SEM of measures performed 6–7 months after AAV2/2-NGB administration and before euthanasia.
Discussion
Mitochondrial disorders represent a common cause of chronic morbidity and are more prevalent than previously thought; indeed as a group, mitochondrial disorders affect at least 1 in 5,000 individuals.25 They are the result of the organelle's essential role in energy production, reactive oxygen species (ROS) biology, apoptosis, and a number of crucial intermediate metabolic pathways.26 Up to date, molecular defects observed in both mitochondrial DNA–encoded and nuclear DNA–encoded genes of mitochondrial proteins are associated with a wide spectrum of clinical problems including myopathy, encephalomyopathy, gastrointestinal syndromes, dystonia, diabetes, cancer, blindness, deafness, and cardiomyopathy.26,27 Additionally, mitochondrial impairment is a key player in the pathogenesis process of glaucoma,28 Alzheimer, and Parkinson diseases.29 Despite the huge advances in the understanding of molecular and biochemical bases underlying mitochondrial dysfunction, the ability to counteract mitochondrial pathologies remains very limited.30,31 Two aspects of mitochondrial research are critical to develop efficient therapeutics: (i) unravel the pathways which impact mitochondrial biogenesis and function32; (ii) characterize animal models that faithfully mimic human diseases to understand the physiological significance of these pathways and their tissue-specific regulation.33
Within this context, Hq mouse strain represents a powerful tool for unraveling mitochondrial function and developing safe and preventive therapies aimed at preserving mitochondrial robustness. These mice exhibit the main features of human neurodegenerative diseases due to RCCI deficiency, such as the degeneration of the retina, optic nerve, cerebellum, thalamic, striatal, and cortical regions.10,12,34 This complex phenotype is caused by the depletion of the nuclear gene AIF encoding the mitochondrial AIF, which level drops to less than 5% of the one seen in wild-type mice.10,11 AIF protein functions as a FAD-dependent NADH oxidoreductase; bovine heart studies suggested the presence of AIF in RCCI when isolated in native conditions,35 strengthening the hypothesis of AIF involvement in RCCI function or/and biogenesis. Interestingly, recent studies reported that two distinct AIF mutations were responsible for early prenatal ventriculomegaly followed by infantile encephalomyopathy and Cowchock syndrome (progressive X-linked recessive disorder with axonal neuropathy, deafness, and cognitive impairment).36,37,38
In this study, we demonstrated that: (i) the steady-state levels of the NGB mRNA and protein were diminished in Hq mouse retinas; (ii) NGB overexpression, via AAV2/2-NGB intravitreal injection, postponed RGC death, and ON degeneration leading to sustained vision preservation in these mice. Hence, the increment of NGB expression in Hq retinas significantly prevented RCCI failure and this appeared as sufficient to protect durably RGCs, as we have previously shown when Hq eyes were subjected to AAV2/2-AIF1 gene therapy.15 The evidence linking NGB and mitochondrial function has increased in the last years; several authors found NGB in close association with mitochondria using immunohistological39,40 or electron microscopy studies.41 Recently, in cultured neuronal cells, NGB was physically localized within the organelle, and NGB overexpression led to its enrichment inside the mitochondria.42 Additionally, some potential NGB-partners are mitochondrial proteins such as ATP synthase-β, cytochrome c1, and the electron-transferring flavoprotein α subunit (Etfα).43
Our aim was to shed light on the biochemical pathways underlying NGB function, for two reasons: (i) brain areas exhibiting high NGB levels in normal conditions show vulnerability to neurodegenerative diseases; accordingly, NGB protects from hypoxia/oxidative stress by eliminating ROS or preventing apoptosis3,8,44; (ii) in the retina and optic nerves,45 NGB accumulates at a concentration 100-fold higher than in the brain,46 and its overexpression was neuroprotective against ischemia by counteracting oxidative stress.39 We showed that NGB is a highly abundant protein in rodent retinas, especially in the GCL, inner nuclear layer, inner plexiform layer, and inner segments of photoreceptors (Figures 1 and 2),47 which are the main sites of oxygen consumption in retina.48 Further, we demonstrated that NGB localizes to the mitochondria in rat9 and mouse retinas (Figure 1) and that gene expression knockdown caused deleterious effects on retinal structure and function due to the direct involvement of NGB on respiratory chain function.9 Obviously, NGB becomes a candidate for maintaining or enhancing RGC mitochondrial function and somehow preserving vision in an array of ophthalmic conditions. Thus, we proceed to the reestablishment of NGB levels in Hq retinas to determine whether that could be enough for preventing RGC loss which becomes evident in animals aged 3–4 months. We designed a gene therapy using a recombinant AAV2/2 vector which harbors the ORF and the full-length 5′ and 3′UTR of NGB gene; since protein concentration depends on the translational ability of the mRNA, which resides in the UTRs located at its 5′ and 3′ ends.16 This is especially true for mRNAs transcribed in the nucleus and coding mitochondrial proteins that are translated in polysomes associated with the mitochondrial outer membrane.49 After a single administration in the vitreous body of Hq mice, NGB expression driven by the vector was sustained for up to 7 months without any harmful effect on retinal architecture or function. The noticeable increase of NGB protein levels in the GCL relative to the lower levels in untreated eyes was effective in: (i) improving RGC survival, (ii) preserving nerve fiber integrity, (iii) reducing glial cell activation, (iv) rescuing RCCI dysfunction, and (v) maintaining visual function. Thus, gene therapy for counteracting AIF depletion in Hq mouse retinas mediated by AAV2/2-NGB is almost as effective as AAV2/2-AIF1 treatment. The exact molecular step in which AIF and its NADH oxidoreductase activity is involved for preserving RCCI activity is not yet elucidated.13 The NAD(P)H-dependent NGB-reductase activity in tissue homogenates of human brain and liver14 and in Escherichia coli has been described; the NGB-reducing system was identified as the NADH flavorubredoxin oxidoreductase (FlRd-red) which shares sequence similarities with human AIF.50 NGB-reductase activity of recombinant human AIF was negligible in vitro,51 hence, the proof remains to be established in vivo, and our results suggest that AIF and NGB share some functional properties involved in the regulation of mitochondrial activity. Moreover, an interaction between AIF and NGB was evidenced in murine neuronal cells under hypoxic conditions using affinity purification mass-spectrometry.52
Obviously, the most prominent result obtained by subjecting Hq mouse eyes to AAV2/2-NGB treatment was the nearly complete and long-lasting protection against vision loss (Figure 8c). This is probably linked to the effects of NGB on the preservation of RGCs and their axons as well as RCCI activity in ONs (Figures 6 and 7). As mentioned before, NGB could share some properties with AIF; thus, its increased protein levels in AIF-depleted RGCs (Figure 4c) could counter RCCI defect by substituting itself for AIF. Although, we can envisage another scenario in which NGB, because of its more general intrinsic characteristics to reduces oxidative stress and neurotoxic effects, increases mitochondrial biogenesis and/or homeostasis.53 In this respect, our experiments on NGB knockdown in rat retinas clearly demonstrated a more general NGB role in mitochondrial energy generation; especially for respiratory chain complexes I and III function,9 which could be due to NGB ability to efficiently transfer electrons with kinetics usually attributed to cytochrome or oxidoreductase proteins.54
The successful protection against optic atrophy achieved by AAV2/2-NGB treatment is promising and opens new avenues in which NGB can be used as a therapeutic approach for counteracting neuron injury due to mitochondrial impairment.
Materials and methods
Animals. The Hq mice originate from the C57BL/6J strain: B6CBACaAw-J/A-Pdc8Hq/J harboring a spontaneous mutation in the AIF gene also known as Pdc8 (http://jaxmice.jax.org/strain/000501.html). All hemizygous (Hq/Y) males used in this study were F1 mice bred from founders having a mixed genetic background. Hemizygous (Hq/Y) males were the recipient of evaluations and gene therapy; they were compared exclusively to the littermate males from the colony. The mice were housed from one to four per cage in a temperature-controlled environment, 12-hour light/dark cycle, and free access to food and water. Studies were conducted in accordance with the statements on the care and use of animals in research of the guidelines issued by the French Ministry of Agriculture and the Veterinarian Department of Paris (Permit number DF/DF_2010_PA1000298), the French Ministry of Research (Approval number 5575) and the ethics committees of the University Paris 6 and the INSERM, Institut National de la Santé et de la Recherche Médicale (Authorization number 75–1710).
Adeno-associated viral vector and intravitreal injections. The Mus musculus NGB mRNA sequence (NM_022414.2) of 1630 base pairs (bp) was synthesized by Genscript (Piscataway, NJ). It encompasses the full-length 5′UTR (279 bp), the entire ORF encoding a 151 amino acid-long protein and two restriction sites for cloning into the pAAV-IRES-hrGFP vector (http://www.genomics.agilent.com/). The human growth hormone 1 polyadenylation signal was replaced by the full-length 3′UTR of NGB (895 bp). NGB transcription is under the control of the cytomegalovirus promoter and the β-globin intron for ensuring high levels of expression. The ORF is in frame with the 3× FLAG sequence at the C-terminus. The pAAV-IRES-hrGFP vector has a dicistronic expression cassette in which the humanized recombinant green fluorescent protein (hrGFP) is expressed as a second ORF translated from the encephalomyocarditis virus internal ribosome entry site (IRES). The final vector, named AAV2/2-NGB, contains AAV2 inverted terminal repeats (ITRs), which direct viral replication and packaging. The expression cassettes flanked by the two ITRs, were encapsidated into AAV2 shells. Vectors were produced by the “Centre de Production de Vecteurs and the INSERM UMR1089, Nantes” (http://www.atlantic-gene-therapies.fr/). For intravitreal injections, Hq mice were subjected to anesthesia with isoflurane (40 mg/kg body weight). The tip of a 33-gauge needle, mounted on a 10 µl Hamilton syringe (Hamilton Bonaduz AG, Bonaduz, Switzerland) was advanced through the sclera and 2 µl of vector suspension (2 × 109 VG) was injected intravitreally, avoiding retinal structure disruption, bleeding, or lens injury. Sixty Hq mice were subjected to AAV2/2-NGB intravitreal injection during the course of this study; five mice died from natural causes ~3 months postinjection. Forty-five mice were euthanized between 6 and 7 months after vector administration; optic nerves and retinas from these mice were all evaluated. Finally, six mice were euthanized 8 weeks after vector administration and used for NGB protein amount determinations in retinal protein extractions.
Fundus imaging by cSLO. A digital cSLO (Heidelberg Engineering, Heidelberg, Germany) was used to examine the nerve fiber layer in each cardinal area of mouse eyes before treatment and different times after vector administration as previously described.24 Briefly, all examinations were carried out in manually restrained conscious animals that were held in front of the cSLO objective after pupil dilation; the overall duration of each examination was 1 minute per eye. Stacks of 30 images were acquired at different planes of focus to capture the whole surface of the retina.
Optomotor response. Optokinetic tracking threshold was measured, under photopic conditions, by observing the optomotor responses of mice to rotating sinusoidal gratings (OptoMotry, CerebralMechanics, Alberta, Canada).55 Mice reflexively respond to rotating vertical gratings by moving their head in the direction of grating rotation. The protocol yields independent measures of right and left eye acuities based on the unequal sensitivities of the two eyes to pattern rotation: right (untreated) and left (treated) eyes are most sensitive to counter-clockwise and clockwise rotations, respectively.55,56 Once the mouse became accustomed to the pedestal, the test was initiated by presenting the mouse with a sinusoidal striped pattern that rotates either clockwise or counterclockwise and varying widths. Spatial frequency of the grating was randomly increased by the software until the animal no longer responded. The process of changing the spatial frequency of the test grating was repeated a few times until the highest spatial frequency the mouse could track is identified which defines the optokinetic tracking threshold under our experimental conditions. Experiments were performed three times independently before vector administration, 3 and 6 months postinjection, by two observers masked to the animal's treatment and previously recorded thresholds.
Retinal and optic nerve histology. Retinas and ONs were carefully collected and fixed in 4% PFA at 4 °C, cryoprotected by overnight incubation in phosphate-buffered saline (PBS) containing 30% sucrose at 4 °C. Retinas were embedded in optimum cutting temperature (OCT) compound (Neg 50; Richard-Allan Scientific, Illkirch Cedex, France), frozen in liquid nitrogen. ONs were embedded in a solution of PBS + 7.5% Type A gelatin from porcine skin (Sigma-Aldrich, St Louis, MO) and 10% sucrose and frozen in a 2-methyl-butane solution at −45 °C. Sections of retinas and ONs were cut (10 µm thickness) on a cryostat (Microm HM560, Thermo Scientific, Illkirch Cedex, France) at −20 °C and mounted on SuperFrostPlus slides.
For immunochemistry, sections of retinas and ONs were permeabilized with 0.1% Triton X-100 in PBS for 15 minutes at room temperature and treated with 3% bovine serum albumin, 0.1% Triton, and 0.05% Tween 20 in PBS for 1 hour. They were then incubated with primary antibody overnight at 4 °C. The next day, sections were washed three times in PBS and incubated with appropriate secondary antibodies and 2 μg/ml of 4′, 6-diamidino-2-phenylindole for 2 hours at room temperature with 3% bovine serum albumin, 0.1% Triton, and 0.05% Tween 20 in PBS. Finally, they were washed three times with PBS, rinsed with sterile water, and mounted on glass slides. Primary and secondary antibodies used are shown in Supplementary information (Supplementary Table S1).
Microscopic observations. Fluorescence labeling was monitored in the Cellular Imaging facility of the institute with: (i) a confocal laser scanning microscope (Olympus FV1000, Rungis, France), image acquisition was conducted with Olympus Fluoview software version 3.1. (ii) Retinal sections were also scanned with the Hamamatsu Nanozoomer Digital Pathology 2.0 HT, its Fluorescence Unit option (L11600-05) and the NanoZoomer's 3-CCD TDI camera (Hamamatsu Photonics). BRN3A-positive cells, as the estimation of overall RGCs, were assessed for each animal by manually counting 2–4 entire retinal sections as described earlier.9,15 Axon numbers were estimated by counting the fluorescent spots obtained after NF200 labeling for: (i) six optic nerves from Hq eyes subjected to AAV2/2-NGB injection, (ii) six optic nerves from the Hq contralateral untreated eyes, and (iii) six optic nerves from aged-matched controls. Images for each optic nerve were traced at ×20 magnification, and the cross-sectional area was automatically calculated with ImageJ software (National Institutes of Health, Bethesda, MD). All photographs were converted into an 8-bit gray scale image. By adjusting the contrast, brightness, and threshold values, representative images of the pictures were obtained. Next, ImageJ software assessed the total number of “particles” (i.e., axon number) in each image. Two independent optic nerves per animal were subjected to the analysis.
Transmission electron microscopy. Twelve Hq and nine control mice aged from 6 weeks to 15 months were used for ultrastructural studies. Mice were anesthetized by the intraperitoneal administration of ketamine (100 mg/kg) and xylazine (8 mg/kg) and transcardially perfused with 0.9% NaCl for 30 seconds and then with Karnovsky fixative (paraformaldehyde 2%, glutaraldehyde 2.5% in 0.1 mol/l phosphate buffer, pH 7.4) for 12 minutes. ONs were removed and postfixed in the same fixative for 1 hour at 4 °C and stored in PBS overnight at 4 °C. The samples were then rinsed briefly in water, fixed in 2% aqueous OsO4 for 45 minutes at 4 °C, and finally rinsed in water. Samples were dehydrated in a series of graded ethanol solutions (70, 95, and 100%, 3 × 10 minutes for each), then in a mixture 1/1 (v/v) of ethanol and propylene oxide (10 minutes), and finally, in pure propylene oxide (3 × 10 minutes). Next, the specimens were embedded in Epon (Electron Microscopy Sciences, Hatfield, PA) and propylene oxide at 1:1 for 2 hours and pure Epon for 2 × 12 hours at room temperature. Ultrathin (80 nm) sections were prepared using a Leica ultracut S microtome fitted with a diamond knife (Diatome histoknife Jumbo or Diatome Ultrathin). Both transverse and longitudinal sections from distal segments of optic nerves were obtained. The sections were contrast stained with 2% uranyl acetate and lead citrate and photographed in a Jeol S100 transmission electron microscope (Croisy-sur-Seine, France) fitted with an Orius SC200 digital camera (Gatan-Roper Scientific, Evry, France) for image capture.
RNA extraction and RT-qPCR assay. Total RNA from rat retinas were extracted using RNeasy Plus Mini kit (Qiagen, Courtaboeuf Cedex, France). To ensure the absence of DNA, a treatment with RNase-free DNase (Qiagen) and a subsequent cleanup with the RNeasy MinElute cleanup kit (Qiagen) were performed. Absence of DNA was confirmed by subjecting 10 ng of each RNA preparation to qPCR with specific primers for the NGB transgene and the mitochondrial ATP6 gene. One microgram of total RNA was reverse transcribed with oligo-dT using Superscript II Reverse Transcriptase (Life Technologies, Villebon sur Yvette, France). Quantitative PCR reactions were performed using ABI 7500 Fast (Life Technologies), and the specific primers listed on Supplementary Table S2. The equivalent of 10 and 2 ng of cDNAs were used per gene as template for qPCR reactions with Power Sybr green PCR Master Mix (Applied Biosystems, Life Technologies). Each biological sample was subjected to the assay in triplicates per gene; Ct values were obtained with the ABI 7500 software (v.2.0.6). Messenger RNA steady-state levels of the mitochondrial ATP6 gene was the most stable in the 48 independent samples evaluated regardless the eye treatment (Supplementary Figure S2). Therefore, to determine the relative mRNA amount of each studied gene, we used the comparative ΔΔCt method and ATP6 as normalizing gene.
Mitochondria extraction and western blotting analysis. Thirty-six retinas were isolated from 12-week-old mice (B6CBACa strain) and washed in PBS at 4 °C; they were then homogenized in extraction buffer (0.32 mol/l sucrose, 30 mmol/l Tris-HCl; pH 7.6, 5 mmol/l MgAc, 100 mmol/l KCl, 0.1% fatty acid-free bovine serum albumin, 5 mmol/l β-mercaptoethanol, and 1 mmol/l phenylmethylsulfonyl fluoride (PMSF)), and mitochondria were purified as previously described for rat retinas.9 Thirty micrograms of mitochondrial proteins were treated with 150 µg/ml of proteinase K in the presence or absence of 1% Triton X-100 (v/v) at 4 °C for 30 minutes. The reaction was stopped by addition of 1 mmol/l PMSF (Sigma-Aldrich). Samples were collected by centrifugation at 10,000g for 15 minutes at 4 °C. Three independent mitochondria purifications were subjected to the analyses.
For whole proteins extracts, single retinas were homogenized in 50 μl of 20 mmol/l 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 60 mmol/l mannitol (pH 7.2) using a 200 μl micro-hand-driven glass–glass potter at 4 °C. Large cellular debris was spun down by a low speed centrifugation (1000g for 5 minutes at 4 °C), and supernatants were subjected to protein quantification (Bradford reagent from Sigma-Aldrich) before proceeding to western blotting. After denaturation at 94 °C for 15 minutes, samples were resolved in 12 or 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and next transferred to a polyvinylidene difluoride (PVDF) membrane. Membranes were probed with antibodies against NGB, AIF, β-actin, and ATP synthase subunit β (Supplementary Table S1). Immunoreactive bands were visualized with appropriate secondary antibodies coupled to horseradish peroxidase (0.1 mg/ml) (Supplementary Table S1) followed by detection with Pierce ECL plus western blotting substrate (Pierce, Thermo Scientific). Ten, twenty, and thirty micrograms of protein extracts from: 12 controls, 6 Hq mice, which received AAV2/-NGB intravitreally and 8 Hq mice without any treatment were subjected to western blotting (in triplicates) using antibodies successively against NGB and β-actin. Theoretical molecular mass of each protein was estimated by comparing the electrophoretic properties of each specific signal in the immunoblots with the “PageRuler Plus Prestained Protein Ladder” (Pierce Protein Biology products, ThermoScientific). Signals obtained from different immunoblots were scanned and quantified with the Quantity One Analysis Software (Bio-Rad, Marnes-la-Coquette, France).
Tissue homogenate preparation and respiratory chain enzyme assays. Optic nerves were rapidly collected and kept frozen (−80 °C). Respiratory chain CI and CV enzymatic activities were measured using a Cary50 UV-Vis spectrophotometer (Agilent Technologies, Les Ulis, France), as described for ONs from mice,15 and each assay was made in triplicate. CI and CV values were converted to specific activities expressed as nanomoles of oxidized reduced form of nicotinamide adenine dinucleotide (NADH)/min/mg protein after protein quantification by the Bradford method. All chemicals were of the highest grade from Sigma-Aldrich.
Statistical analyses. Values are expressed as means ± standard error of the mean. Statistical analyses were performed with the GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA) assuming a confidence interval of 95%. Generally, the observations within each group do not fit in a normal distribution, thus nonparametric methods have been applied for evaluating the significance. Data collected from control and Hq were compared using the unpaired nonparametric significance test of Mann–Whitney (*≤0.05, **≤0.01, and ***≤0.005). Data collected from Hq-treated eyes and Hq-untreated contralateral eyes were compared using the paired nonparametric significance test of Wilcoxon (*≤0.05, **≤0.01, and ***≤0.005).
SUPPLEMENTARY MATERIAL Figure S1. Transmission electron microscopy. Figure S2. Representation of the ATP6 mRNA abundance evaluated by RT-qPCR. Table S1. List of primary and secondary antibodies used for histochemistry. Table S2. List of specific primers used in the RT-qPCR assays.
Acknowledgments
We are grateful to Pierre Rustin and Michael C. Marden for useful discussions and comments on the data. We are grateful to David Godefroy and Stéphane Fouquet (Cellular Imaging Facility of the Institut de la Vision). The main sources of support were: Fovea Pharmaceuticals (Sanofi, Ophthalmology Division), Agence Nationale pour la Recherche/Emergence-Bio, INSERM, CNRS, and the foundation “Voir et Entendre”. T.D. works for Sanofi Fovea—Ophthalmology which supported the study; an international application for a patent has been filed on the data (PCT/FR2013/052323). The invention relates to the use of neuroglobin agonist for preventing or treating mitochondrial respiratory chain complex I and/or respiratory chain complex III deficiency disease. There are no other patents, products in development, or marketed products to declare.
Supplementary Material
Transmission electron microscopy.
Representation of the ATP6 mRNA abundance evaluated by RT-qPCR.
List of primary and secondary antibodies used for histochemistry.
List of specific primers used in the RT-qPCR assays.
References
- Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- Ostojic J, Grozdanic S, Syed NA, Hargrove MS, Trent JT, 3rd, Kuehn MH, et al. Neuroglobin and cytoglobin distribution in the anterior eye segment: a comparative immunohistochemical study. J Histochem Cytochem. 2008;56:863–872. doi: 10.1369/jhc.2008.951392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RC, Guo SZ, Lee SK, Gozal D. Neuroglobin protects neurons against oxidative stress in global ischemia. J Cereb Blood Flow Metab. 2010;30:1874–1882. doi: 10.1038/jcbfm.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hümmler N, Schneider C, Giessl A, Bauer R, Walkinshaw G, Gassmann M, et al. Acute hypoxia modifies regulation of neuroglobin in the neonatal mouse brain. Exp Neurol. 2012;236:112–121. doi: 10.1016/j.expneurol.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Rajendram R, Rao NA. Neuroglobin in normal retina and retina from eyes with advanced glaucoma. Br J Ophthalmol. 2007;91:663–666. doi: 10.1136/bjo.2006.093930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski M, Wang R, Fallin MD, Bassett SS, Avramopoulos D. Neuroglobin and Alzheimer's dementia: genetic association and gene expression changes. Neurobiol Aging. 2010;31:1835–1842. doi: 10.1016/j.neurobiolaging.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Mao XO, Banwait S, Jin K, Greenberg DA. Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc Natl Acad Sci USA. 2007;104:19114–19119. doi: 10.1073/pnas.0706167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu Z, Guo S, Lee SR, Xing C, Zhang C, et al. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J Neurosci Res. 2009;87:164–170. doi: 10.1002/jnr.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechauve C, Augustin S, Cwerman-Thibault H, Bouaita A, Forster V, Célier C, et al. Neuroglobin involvement in respiratory chain function and retinal ganglion cell integrity. Biochim Biophys Acta. 2012;1823:2261–2273. doi: 10.1016/j.bbamcr.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit P, Goncalves S, Dassa EP, Brière JJ, Rustin P. The variability of the harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS One. 2008;3:e3208. doi: 10.1371/journal.pone.0003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid Redox Signal. 2011;14:2545–2579. doi: 10.1089/ars.2010.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trandafir F, Hoogewijs D, Altieri F, Rivetti di Val Cervo P, Ramser K, Van Doorslaer S, et al. Neuroglobin and cytoglobin as potential enzyme or substrate. Gene. 2007;398:103–113. doi: 10.1016/j.gene.2007.02.038. [DOI] [PubMed] [Google Scholar]
- Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Bénit P, Simonutti M, et al. Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain. 2012;135 Pt 1:35–52. doi: 10.1093/brain/awr290. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Pal JK. Role of 5'- and 3'-untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101:251–262. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Jr, Perkins GA, Giddabasappa A, Chaney S, Xiao W, White AD, et al. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol Vis. 2007;13:887–919. [PMC free article] [PubMed] [Google Scholar]
- Haverkamp S, Wässle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. [PubMed] [Google Scholar]
- Saggu SK, Chotaliya HP, Blumbergs PC, Casson RJ. Wallerian-like axonal degeneration in the optic nerve after excitotoxic retinal insult: an ultrastructural study. BMC Neurosci. 2010;11:97. doi: 10.1186/1471-2202-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas-Navarro M, Mayor-Torroglosa S, Jiménez-López M, Avilés-Trigueros M, Holmes TM, Lund RD, et al. A computerized analysis of the entire retinal ganglion cell population and its spatial distribution in adult rats. Vision Res. 2009;49:115–126. doi: 10.1016/j.visres.2008.09.029. [DOI] [PubMed] [Google Scholar]
- Surgucheva I, Weisman AD, Goldberg JL, Shnyra A, Surguchov A. Gamma-synuclein as a marker of retinal ganglion cells. Mol Vis. 2008;14:1540–1548. [PMC free article] [PubMed] [Google Scholar]
- Paques M, Simonutti M, Roux MJ, Picaud S, Levavasseur E, Bellman C, et al. High resolution fundus imaging by confocal scanning laser ophthalmoscopy in the mouse. Vision Res. 2006;46:1336–1345. doi: 10.1016/j.visres.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- Cwerman-Thibault H, Sahel JA, Corral-Debrinski M. Mitochondrial medicine: to a new era of gene therapy for mitochondrial DNA mutations. J Inherit Metab Dis. 2011;34:327–344. doi: 10.1007/s10545-010-9131-5. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–840. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
- Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013;13:12–15. doi: 10.1016/j.coph.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DS. Review of clinical trials for mitochondrial disorders: 1997-2012. Neurotherapeutics. 2013;10:307–319. doi: 10.1007/s13311-013-0176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Dogan SA, Trifunovic A. Modelling mitochondrial dysfunction in mice. Physiol Res. 2011;60 suppl. 1:S61–S70. doi: 10.33549/physiolres.932179. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V, Csaba Z, Olivier P, Lelouvier B, Schwendimann L, Dournaud P, et al. Apoptosis-inducing factor deficiency induces early mitochondrial degeneration in brain followed by progressive multifocal neuropathology. J Neuropathol Exp Neurol. 2007;66:838–847. doi: 10.1097/NEN.0b013e318148b822. [DOI] [PubMed] [Google Scholar]
- Palmisano G, Sardanelli AM, Signorile A, Papa S, Larsen MR. The phosphorylation pattern of bovine heart complex I subunits. Proteomics. 2007;7:1575–1583. doi: 10.1002/pmic.200600801. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Ben-Neriah Z, Dor-Wolman T, Shaag A, Saada A, Zenvirt S, et al. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol Genet Metab. 2011;104:517–520. doi: 10.1016/j.ymgme.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Rinaldi C, Grunseich C, Sevrioukova IF, Schindler A, Horkayne-Szakaly I, Lamperti C, et al. Cowchock syndrome is associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2012;91:1095–1102. doi: 10.1016/j.ajhg.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AS, Saraswathy S, Rehak M, Ueki M, Rao NA. Neuroglobin protection in retinal ischemia. Invest Ophthalmol Vis Sci. 2011;53:704–711. doi: 10.1167/iovs.11-7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann A, Schmidt M, Reuss S, Wolfrum U, Hankeln T, Burmester T. Divergent distribution in vascular and avascular mammalian retinae links neuroglobin to cellular respiration. J Biol Chem. 2005;280:20660–20665. doi: 10.1074/jbc.M501338200. [DOI] [PubMed] [Google Scholar]
- Hundahl CA, Allen GC, Hannibal J, Kjaer K, Rehfeld JF, Dewilde S, et al. Anatomical characterization of cytoglobin and neuroglobin mRNA and protein expression in the mouse brain. Brain Res. 2010;1331:58–73. doi: 10.1016/j.brainres.2010.03.056. [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu J, Liu N, Wang Y, Li X, Pallast S, et al. Mitochondrial distribution of neuroglobin and its response to oxygen-glucose deprivation in primary-cultured mouse cortical neurons. Neuroscience. 2012;218:235–242. doi: 10.1016/j.neuroscience.2012.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Liu N, Wang Y, Li X, Wang X. Identification of neuroglobin-interacting proteins using yeast two-hybrid screening. Neuroscience. 2012;200:99–105. doi: 10.1016/j.neuroscience.2011.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wu Y, Ren C, Lu Y, Gao Y, Zheng X, et al. The activity of recombinant human neuroglobin as an antioxidant and free radical scavenger. Proteins. 2011;79:115–125. doi: 10.1002/prot.22863. [DOI] [PubMed] [Google Scholar]
- Lechauve C, Augustin S, Roussel D, Sahel JA, Corral-Debrinski M. Neuroglobin involvement in visual pathways through the optic nerve. Biochim Biophys Acta. 2013;1834:1772–1778. doi: 10.1016/j.bbapap.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Giessl A, Laufs T, Hankeln T, Wolfrum U, Burmester T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J Biol Chem. 2003;278:1932–1935. doi: 10.1074/jbc.M209909200. [DOI] [PubMed] [Google Scholar]
- Lechauve C, Rezaei H, Celier C, Kiger L, Corral-Debrinski M, Noinville S, et al. Neuroglobin and prion cellular localization: investigation of a potential interaction. J Mol Biol. 2009;388:968–977. doi: 10.1016/j.jmb.2009.03.047. [DOI] [PubMed] [Google Scholar]
- Cringle SJ, Yu DY, Yu PK, Su EN. Intraretinal oxygen consumption in the rat in vivo. Invest Ophthalmol Vis Sci. 2002;43:1922–1927. [PubMed] [Google Scholar]
- Sylvestre J, Margeot A, Jacq C, Dujardin G, Corral-Debrinski M. The role of the 3' untranslated region in mRNA sorting to the vicinity of mitochondria is conserved from yeast to human cells. Mol Biol Cell. 2003;14:3848–3856. doi: 10.1091/mbc.E03-02-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrè A, Moschetti T, Vallone B, Brunori M. Neuroglobin: enzymatic reduction and oxygen affinity. Biochem Biophys Res Commun. 2008;367:893–898. doi: 10.1016/j.bbrc.2008.01.021. [DOI] [PubMed] [Google Scholar]
- Moschetti T, Giuffrè A, Ardiccioni C, Vallone B, Modjtahedi N, Kroemer G, et al. Failure of apoptosis-inducing factor to act as neuroglobin reductase. Biochem Biophys Res Commun. 2009;390:121–124. doi: 10.1016/j.bbrc.2009.09.078. [DOI] [PubMed] [Google Scholar]
- Haines BA, Davis DA, Zykovich A, Peng B, Rao R, Mooney SD, et al. Comparative protein interactomics of neuroglobin and myoglobin. J Neurochem. 2012;123:192–198. doi: 10.1111/j.1471-4159.2012.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Mao XO, Banwait S, DerMardirossian CM, Bokoch GM, Jin K, et al. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J. 2008;22:1737–1747. doi: 10.1096/fj.07-100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger L, Tilleman L, Geuens E, Hoogewijs D, Lechauve C, Moens L, et al. Electron transfer function versus oxygen delivery: a comparative study for several hexacoordinated globins across the animal kingdom. PLoS One. 2011;6:e20478. doi: 10.1371/journal.pone.0020478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Douglas RM, Alam NM, Silver BD, McGill TJ, Tschetter WW, Prusky GT. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis Neurosci. 2005;22:677–684. doi: 10.1017/S0952523805225166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transmission electron microscopy.
Representation of the ATP6 mRNA abundance evaluated by RT-qPCR.
List of primary and secondary antibodies used for histochemistry.
List of specific primers used in the RT-qPCR assays.