

Abstract
The success of adoptively transferred tumor-directed T cells requires them to survive and expand in vivo. Most tumors, however, employ immune evasion mechanisms, including the production of inhibitory cytokines that limit in vivo T-cell persistence and effector function. To protect tumor-directed T cells from such negative influences, we generated a chimeric cytokine receptor in which the interleukin (IL) 4 receptor exodomain was fused to the IL7 receptor endodomain. We thereby inverted the effects of tumor-derived IL4 so that the proliferation and activation of tumor directed cytotoxic T cells was enhanced rather than inhibited in the tumor microenvironment, resulting in superior antitumor activity. These transgenic T cells were only activated in the tumor environment since triggering required exposure to both tumor antigen (signal 1) and tumor-derived IL4 (signal 2). This selectivity supports future clinical adaptation.
Introduction
Adoptively transferred tumor-directed T cells can effectively traffic to distant tumor sites, penetrate even bulky disease, and kill disseminated tumor cells in a range of malignancies, including Hodgkin lymphoma, nasopharyngeal carcinoma, neuroblastoma, and melanoma.1,2,3,4,5,6 While infusion of such effector T cells may benefit some patients with malignant disease, most tumors employ an array of immune evasion mechanisms that allow them to escape destruction by the infused cells. These mechanisms include the downregulation of costimulatory molecules and the upregulation of coinhibitory receptors such as PD1 and cytotoxic T-lymphocyte antigen 4 (CTLA4) or the production of soluble inhibitory/Th2-polarizing cytokines such as transforming growth factor (TGF) β, interleukin (IL) 10, IL13, and IL4, all of which serve to limit in vivo T-cell persistence and effector function.7,8,9
Investigators have neutralized tumor-derived inhibitory signals by using checkpoint blockade antibodies directed to inhibitory receptors on T cells such as CTLA4, PD1, and its ligand (PDL1), an approach that has been shown to enhance immune responses to tumors and improve clinical outcomes.10,11,12,13 An alternative approach is to genetically engineer the T cells to be resistant to tumor inhibition. For example, Bollard and colleagues demonstrated that the inhibitory effects of TGFβ on T cells could be negated by forced expression of a dominant-negative TGFβ receptor type II (dnTGFβ-RII) in tumor-directed T cells, prolonging their persistence and enhancing tumor elimination in mice bearing TGFβ-expressing tumors.14,15 We are currently assessing the safety and efficacy of such dnTGFβ-RII–modified tumor-specific T cells in patients with relapsed/refractory Hodgkin or non-Hodgkin lymphoma.
We have now extended our T-cell engineering approach to move beyond neutralization of inhibitory cytokines to the active reversal of their effects, so that an immunosuppressive signal becomes immunostimulatory. The advantages of this approach are twofold: first, this modification should augment the function and survival of the modified cells in the otherwise suppressive milieu of the tumor. Second, it will allow the T cells to persist and sustain function predominantly at the tumor site, since only there will the engineered T cells encounter both signal one (antigen) and signal two (immunosuppressive/stimulatory cytokine). In other words, the approach should be both generally safe and locally effective.
To test the feasibility of this approach, we chose to focus on the inhibitory Th2 cytokine IL4, which has been found at elevated levels in many different tumors including Hodgkin's lymphoma, breast, prostate, and pancreatic cancer, where it has been reported to favor tumor growth by inhibiting tumor-directed Th1-polarized effector T-cell responses.16,17,18,19,20 Under physiological conditions, IL4 receptor engagement activates a signal cascade that downregulates proinflammatory and upregulates anti-inflammatory (Th2-polarizing) cytokines. To reverse these inhibitory effects, we fused the IL4 receptor exodomain (cytokine-binding portion) to the signaling endodomain of the IL7 receptor, a Th1 cytokine receptor, and used a retroviral construct to express the chimeric receptor (IL4/7 ChR) in tumor-directed T cells.
We show that upon IL4 engagement, the IL4/7 ChR signals via the IL7 endodomain, supporting the maintenance of a Th1 phenotype in effector cells and augmenting their proliferation and cytotoxic function, thereby enhancing both their persistence and in vivo antitumor activity.
Results
Transforming an immunosuppressive T-cell signal into an immunostimulant
Tumor-directed T cells may be inhibited in vivo by high levels of tumor-associated IL4. Following engagement with its cognate receptor on T cells, IL4 induces Stat6 phosphorylation, activating a signal cascade that downregulates proinflammatory (Th1-polarizing) and upregulates anti-inflammatory (Th2-polarizing) cytokines (Figure 1a). To reverse these inhibitory effects, we constructed a retroviral vector encoding a fusion between the cytokine-binding portion of the IL4 receptor exodomain and the signaling endodomain of the IL7 receptor (a Th1 cytokine receptor) (IL4/7 ChR) (Figure 1c). Upon IL4 engagement, this novel chimeric cytokine receptor should signal via the IL7 receptor endodomain resulting in phosphorylation of Stat5 (pStat5) and the transmission of a Th1 signal in transgenic T cells (Figure 1b). To confirm expression of the IL4/7 ChR, we transduced Epstein-Barr virus (EBV)–specific T cells and assessed their transduction efficiency by flow cytometric analysis, measuring expression of both the IL4 receptor and an incorporated mOrange marker sequence. Figure 1d shows a representative example, with 40.8% of cells expressing both markers. The transduction efficiency ranged from 30.4 to 56% (n = 6 donors; data not shown).
Figure 1.
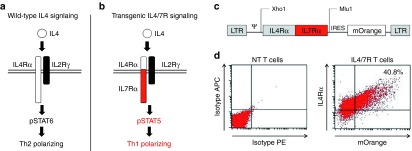
Transforming the immunosuppressive IL4 signal into an immunostimulant. Schematic of IL4 signaling through (a) wild-type and (b) chimeric cytokine receptors. (c) Retroviral vector map of chimeric cytokine receptor (IL4/7 ChR). (d) The transgenic expression of IL4/7 ChR on EBV-specific T cells as detected by mOrange and the IL4 receptor exodomain using an anti-IL4 receptor antibody.
Transgenic IL4/7 ChR signaling
To assess the function of IL4/7 ChR, we first measured transgene signaling by determining pStat5 levels after exposure of control nontransduced (NT) and IL4/7 ChR transgenic EBV-specific T cells to IL4 (1,000 U/ml). As a positive control, we exposed the same cells to IL2 (50 U/ml), since both native and modified T cells constitutively express the receptor for this cytokine. As anticipated, exposure to IL2 induced pStat5 in both NT and transgenic IL4/7 ChR EBV-specific T cells treated. By contrast, IL4 exposure induced pStat5 only in IL4/7 ChR transgenic cells (Figure 2a).
Figure 2.

Transgenic IL4/7 ChR signaling. (a) IL2 induces Stat5 phosphorylation in both control (NT) and IL4/7 ChR EBV-directed T cells, whereas IL4 induces Stat5 phosphorylation only in IL4/7 ChR EBV-directed T cells. Black and red lines represent the profile of phosphorylated Stat5 in NT and transgenic T cells, respectively. RNA microarray expression profile of >20,000 target genes in control NT and IL4/7 ChR T cells exposed to IL4. As an additional control condition, EBV-directed T cells were modified to express the IL7 receptor48 and subsequently exposed to either IL4 or IL7. Panel (b) shows a heat map of differentially expressed gene transcripts. (c) Finally, the cytokine production profile of NT and IL4/7 ChR cells exposed to IL4 was evaluated by luminex assay using supernatant harvested 24 hours after antigenic stimulation. The prototypic Th2 cytokine IL13 is shown in the left panel, whereas prototypic Th1/proinflammatory cytokines IFNγ, GM-CSF, and IL8 are shown in the right panel.
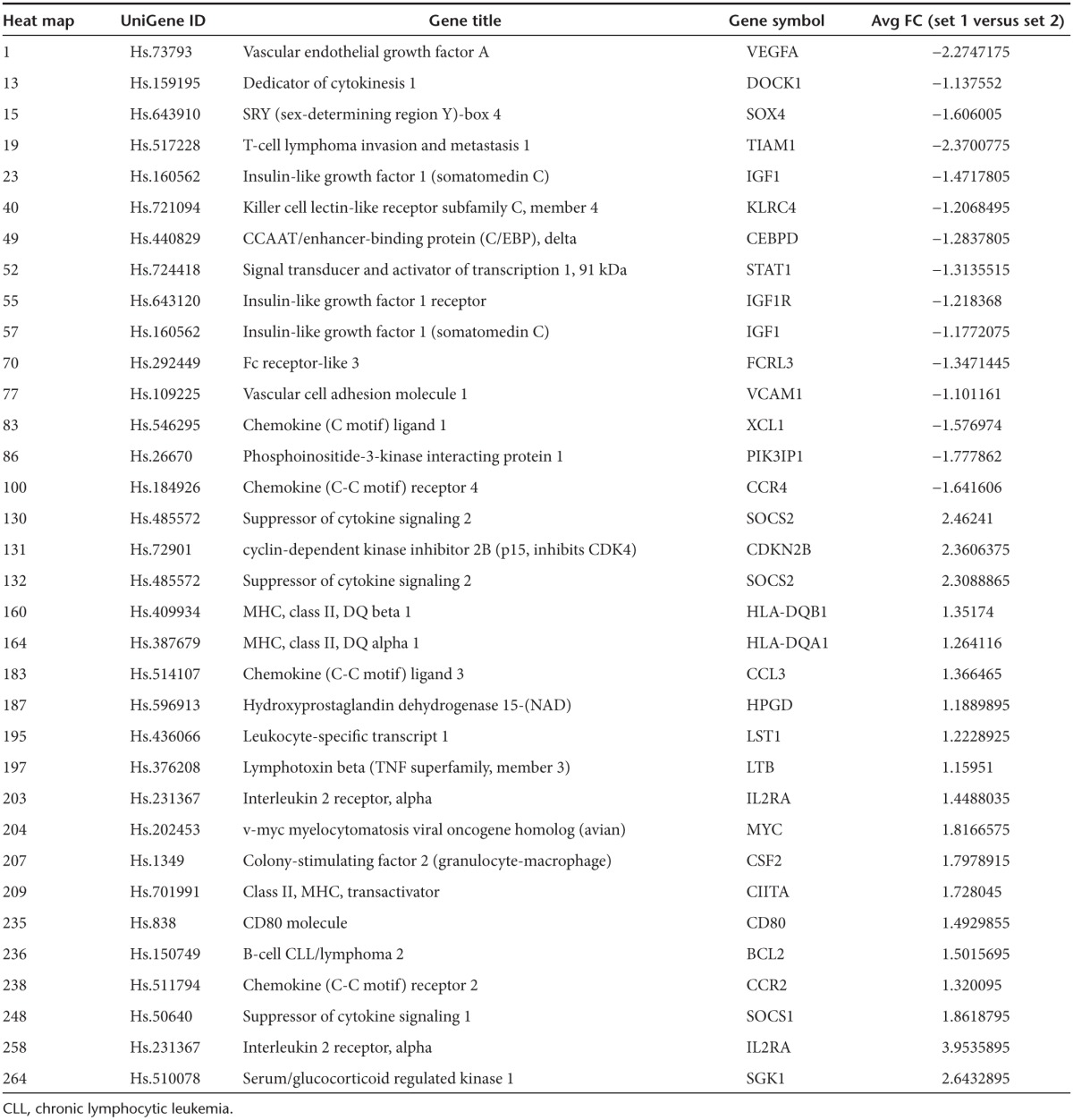
We next evaluated whether engagement of IL4 with IL4/7 ChR T cells induced the same downstream signaling events as IL7, by comparing the RNA expression profile of >20,000 target genes in control NT and IL4/7 ChR T cells exposed to IL4. Addition of IL4 to NT T cells activated the native IL4 receptor pathway and increased expression of CEBP and CCR4 (Figure 2b and Table 1), both transcriptional targets of pStat6, and elevated levels of VEGFA, VCAM1, EGF1, and Stat1—all of which have been described to increase following IL4 exposure.21,22,23 We also observed a rise in transcripts for SOX4, a transcription factor that enhances Th2 differentiation as well as XCL1, which has been shown to have a negative effect on Th1 responses as well as IL2 and IFNγ production. Exposure of IL4/7 ChR-modified T cells to IL4, however, had reciprocal effects: with decreased expression of CEBP, CCR4, VEGFA, VCAM1, EGF1, and Stat1 but increased expression of Stat5a/b target genes, including MYC, IL2 receptor α, Bcl2, and SOCS 1 and 2, which are characteristic of the IL7 receptor signaling pathway.24,25 We also detected downstream targets of the PI3 kinase signaling cascade such as serum- and glucocorticoid-inducible kinase Sgk1, which increases GLUT1 expression in the plasma membrane—a well known effect of IL7 signaling.26 Finally, exposure of IL4/7 ChR-modified T cells to IL4 upregulated CD80, HLA class II, and CCR2 expression, observations consistent with the activation of IL7-associated pathways (Figure 2b).
Table 1. Thirty four differentially expressed genes (comparison between set 1 (IL4/7 ChR- and IL7R transgenic cells + IL4) and set 2 (NT and IL7R transgenic cells + IL4)).
We next assessed the cytokine profile of IL4/7 ChR transgenic T cells exposed to IL4 since this is a prototypic Th2 cytokine, while IL7 induces a Th1-polarized cytokine profile. We exposed NT and IL4/7 ChR T cells to antigen and IL4 and subsequently assessed the cytokine profile using a luminex array. Figure 2c shows that NT T cells produce the Th2-associated cytokine IL13 in response to IL4. In contrast, exposure of IL4/7 ChR-modified cells to IL4 induces prototypic Th1 cytokines including IFNγ, granulocyte-macrophage colony-stimulating factor, and IL8, while levels of Th2 cytokines such as IL13 were substantially lower than those produced by IL4-exposed NT T cells. Thus, exposure of IL4/7 ChR-modified antigen-specific T cells to IL4 induces downstream signaling events and cytokine production consistent with activation of the IL7 receptor, thereby maintaining a Th1-polarized T-cell population even in the presence of IL4.
Selective expansion of transgenic T cells in the presence of IL4 cytokine
To discover whether the IL4/7 ChR could support the expansion of transgenic T cells in conditions that mimic the tumor milieu, we cultured both control and transgenic EBV-specific T cells in the presence of IL4 (1,000 U/ml) and assessed their proliferation in a 3-day 3H incorporation assay. As anticipated, control NT T cells proliferated minimally in response to IL4 (12,205 ± 1,021 counts per minute (CPM)). IL4/7 ChR transgenic cells, however, showed high levels of 3H incorporation in the presence of IL4 (107,436 ± 5,371 CPM) that were comparable to those observed in the presence of IL2 (97,325 ± 3,227 CPM) (Figure 3a, representative donor of n = 4 tested). To assess the effects of chronic IL4 exposure, we supplemented control and transgenic T cells with IL4 (1,000 U/ml) twice a week for 3 weeks, along with weekly antigen stimulation. Expansion was measured by cell counting using trypan blue exclusion to distinguish live and dead cells. Although NT T cells failed to expand in the presence of IL4 (day 0: 1 × 106 cells; day 21: 0.47 ± 0.79 × 106 versus 24.6 ± 7.64 × 106 total cells, IL4 versus IL2, respectively) (P = 0.00045), IL4/7 ChR transgenic T cells cultured under the same conditions expanded to the same degree as cells cultured in IL2 (18.5 ± 2.21 × 106 versus 21.17 ± 2.88 × 106 total cells, respectively—day 21) (P = 0.11; n = 4) (Figure 3b). Moreover, IL4-driven T-cell expansion produced positive selection for IL4/7 ChR (mOrange-expressing) T cells, as detailed for a representative donor in Figure 3c and summarized for five donors in Figure 3d (P = 0.001). Overall, culture in IL4 for 1 week enriched transduced (mOrange+) cells from a mean of 39 ± 11% on day 0 to 75 ± 8% on day 7. Importantly, transgenic expression of the IL4/7 ChR and progressive selection by culture with IL4 did not impair the inherent cytolytic ability or specificity of the cells nor did it impact their capacity to respond to other common γ chain cytokines. As shown in Supplementary Figure S1A, transgenic cells that had been expanded and selected with antigen and IL4 for >3 weeks were able to kill autologous but not allogeneic EBV-transformed B cells at levels similar to that of their NT counterparts, and they continued to expand when exposed to antigen and either IL2 (50 U/ml) or IL15 (10 ng/ml) (Supplementary Figure S1B).
Figure 3.

Selective expansion of transgenic T cells in the presence of IL4 cytokine. (a) Proliferation of NT and transgenic EBV-specific T cells in the presence of IL2 or IL4 as measured in a 3-day 3H incorporation assay. (b) Expansion of NT and IL4/7 ChR EBV-directed T cells after repeated weekly stimulation with autologous EBV-LCL and IL2 or IL4. NT and IL4/7 ChR EBV-directed T cells expanded at equivalent levels in response to antigen and IL2 (black solid line). In contrast, IL4/7 ChR transgenic but not NT EBV-specific T cells expanded when stimulated with EBV-LCL and IL4 (red solid line) (n = 4). The percentage of IL4/7 ChR+ cells remained unchanged when IL4/7 ChR EBV-directed T cells were stimulated with EBV-LCL+IL2, but the transgenic population significantly increased when IL4 was substituted. Panel (c) shows a representative donor, whereas panel (d) shows summary data for 5 donors tested.
Safety profile of IL4/7 ChR transgenic T cells
Since we observed both expansion and selection of transgenic T cells cultured in the presence of IL4, we next evaluated whether prolonged cytokine exposure could produce a T-cell population capable of antigen- or cytokine-independent growth. We cultured transgenic T cells with antigen (EBV-transformed B cells) and IL4 cytokine for 3 weeks, and thereby selected mOrange/IL4/7 ChR+ T cells (from 56% at day 0 to 96.1% at day 21; Figure 4a). We further studied these enriched cells. As shown in Figure 4b, the presence of either antigen alone or cytokine alone was insufficient to promote the growth of these selected transgenic T cells, which instead remained dependent on the presence of both signals (n = 5).
Figure 4.

Safety profile of IL4/7 ChR transgenic T cells. With weekly antigenic stimulation in the presence of IL4, the transgenic IL4/7 ChR+ cells had a selective advantage resulting in their outgrowth. Panel (a) shows a representative donor where the transgenic population increased from 56 to 96.1% over a period of 21 days. (b) However, the growth of these selected IL4/7 ChR EBV-specific T cells remained both antigen and cytokine dependent, as removal of either EBV-LCL or IL4 resulted in culture failure (n = 5).
In vivo activity of 4/7R transgenic T cells in the presence of IL4-producing tumors
To compare the antitumor effects of IL4/7 ChR and NT T cells, we engrafted severe combined immunodeficiency (SCID) mice subcutaneously with IL4-producing FFLuc-positive autologous EBV-transformed B cell tumors (5 × 106 cells/mouse). After engraftment (measured by increasing luminescence on two or more consecutive analyses—14–20 days after tumor inoculation), mice were divided into four groups that received either NT EBV-directed T cells (2 × 107/animal) without exogenous cytokine, NT EBV-directed T cells with exogenous IL2 (2,000 U/animal) administered three times per week, IL4/7 ChR transgenic EBV-directed T cells (2 × 107/animal), or no treatment (n = 8–28 animals/group), and tumor growth was measured by bioluminescence imaging and calipers. Figure 5a summarizes tumor growth on day 14 posttreatment. In animals that did not receive EBV-directed T cells, the tumor signal progressively increased (mean: 101.1 ± 31.9-fold change in luminescence) with an average tumor volume of 1,639 ± 128 mm3 (Figure 5b), and by day 25 after tumor injection, all animals were euthanized due to tumor burden (Figure 5c). In mice receiving unmodified EBV-directed T cells, there was an initial antitumor response with an early decrease in signal immediately (~5 days) posttreatment (data not shown). Without exogenous cytokine support, however, the benefit was transient and the tumor progressed from day 10 onwards (Figure 5a), reaching an average tumor volume of 935 ± 240 mm3 (n = 18) (Figure 5b) and resulting in euthanasia of all animals by day 36 (Figure 5c). Mice receiving NT EBV-specific T cells and IL2 showed a significant decrease in tumor signal for >2 weeks (Figure 5a and b), but tumor eventually progressed, resulting in euthanasia by day 52 (Figure 5c). Strikingly, animals receiving IL4/7 ChR transgenic cells showed benefit from the treatment, even in the absence of exogenous cytokine support. Thus, we observed a decrease in the tumor signal, originating from the tumor core, as detected by bioluminescence imaging (Figure 5d), and computed tomography (CT) imaging showed a hyperdense structure that correlated with a necrotic tumor center (Figure 5e). Overlaying these bioluminescence and CT images showed that IL4/7 ChR transgenic T-cell treatment produced central tumor necrosis that radiated to the periphery (Figure 5f and Supplementary Videos S1–S3). These changes were not evident in the other treatment groups. As a consequence of this response, median survival was increased so that 32% of the mice (9 of 28 animals) were long-term survivors (Figure 5c).
Figure 5.

In vivo activity of IL4/7 ChR transgenic T cells in the presence of IL4-producing tumors. Severe combined immunodeficiency mice engrafted subcutaneously with FFluc-positive EBV-LCLs engineered to produce IL4 (5 × 106/animal) were subsequently divided into four groups. A control group did not receive any T cells (tumor alone: open square), whereas the remaining mice were treated with (i) NT T cells without additional cytokines (NT T cells: triangle with gray shading), (ii) NT T cells with exogenous IL2 provided three times weekly (NT T cells + IL2: black triangle), or (iii) IL4/7 ChR T cells without exogenous cytokines (4/7R T cells: red circle). Panel (a) shows tumor growth as monitored using an in vivo imaging system and reported as fold change in bioluminescence, and panel (b) shows tumor volume (mm3) as measured using calipers. (c) The long-term survival advantage provided by IL4/7 ChR transgenic T cells. (d) Bioluminescence images of representative mice from the tumor alone group as well as animals receiving either NT or IL4/7 ChR T cells. (e) The CT imaging performed on the same animals, whereas panel (f) shows the bioluminescence/CT overlay.
Discussion
We have demonstrated the feasibility of protecting adoptively transferred effector T cells from inhibitory, tumor-produced cytokines by inverting a tumor-derived suppressive signal to enhance antitumor activity. The chimeric cytokine receptor (IL4/7 ChR) we describe bound to tumor-produced IL4 and activated the downstream signaling cascade associated with a native IL7 receptor, so that engagement of the IL4/7 ChR with IL4, a prototypic Th2 cytokine, supported the maintenance of a Th1 (effector) phenotype and augmented cell proliferation and antitumor function of adoptively transferred cells.
Although adoptively transferred, tumor-directed T cells can produce potent antitumor effects in vivo, most tumors employ multiple strategies to avoid immune-mediated elimination. These can be broadly grouped into two categories: (i) strategies that prevent T-cell recognition such as target antigen and major histocompatibility complex downregulation and (ii) strategies that limit T-cell persistence and effector function including the recruitment of inhibitory cells (e.g., myeloid suppressor cells, regulatory T cells), the expression of inhibitory ligands/receptors (e.g., CTLA4, PD1, and PDL1), and the production of soluble inhibitory/Th2-polarizing cytokines, several of which also support tumor growth.7,9
Investigators have counteracted these immune evasion tactics by the systemic administration of agents designed to nonspecifically deplete suppressor/inhibitory cells and by making “space” for the infused effector cells (e.g., chemotherapy or antibody-mediated lymphodepletion).2 More recently, targeted agents have been successfully tested that modulate a particular aspect of the tumor microenvironment. These include checkpoint antibodies designed to selectively block coinhibitory receptors, such as CTLA4, PD1, or its ligand, PDL1, in order to amplify antitumor immune responses or the administration of TGF-β–specific neutralizing antibodies or soluble TGFβ receptor, designed to act as specific competitors in vivo.11,12,13,27–29 Unfortunately, the systemic delivery of these immune-modulating agents has led to in vivo toxicities since they affect T cells irrespective of their target antigen specificity. For example, patients who received the CTLA4-blocking antibodies ipilimumab and tremelimumab developed off-target T-cell–mediated autoimmune disorders most commonly affecting the skin (rash, pruritus, vitiligo), bowel (diarrhea, colitis), liver (hepatitis, elevated liver enzymes), and the pituitary or other endocrine glands (hypophysitis, hypothyroidism, thyroiditis, adrenal insufficiency).11 In an effort to reduce off-target toxicities, we have instead engineered tumor-directed T cells ex vivo, so that on subsequent infusion, they are protected from the negative effects of inhibitory cytokines present at the tumor site, without modulating the broader population of endogenous T cells, thereby avoiding autoimmune reactivity. We previously described expression of a dominant-negative TGFβ receptor (DNR-type II) that binds the TGFβ present in abundance in the tumor milieu but fails to transmit the inhibitory signal mediated by engagement of a wild-type receptor.14 T cells that express DNR-II have prolonged persistence and enhanced tumor elimination in mice bearing TGFβ-expressing tumors and may show similar benefits in humans with relapsed/refractory Hodgkin's disease or non-Hodgkin lymphoma.15 We now describe how it is feasible to go beyond simply neutralizing an inhibitory cytokine signal and actively reverse its effects.
We chose to invert the signal transmitted by IL4, since this prototypic Th2 cytokine has wide-ranging immunosuppressive effects in the tumor microenvironment. IL4 polarizes tumor-directed CD4+ T cells to a type 2 phenotype and reduces effector CD8+ T-cell numbers and cytolytic activity. In addition, the cytokine activates suppressive M2 and tumor-associated macrophages (TAM), which further suppress adaptive immunity, and promote tumor growth, invasion, metastasis, and stromal remodeling. Finally, IL4 has been shown to act as a growth factor for a variety of tumors including breast, prostate, and pancreatic cancer.16,17,18,19,20
To invert the inhibitory effects of IL4 on effector T cells, we chose to fuse the IL4 receptor exodomain with the endodomain of the IL7 receptor since both receptors share the common IL2 γ chain expressed by all T cells but IL7 receptor signaling promotes homeostatic proliferation and supports effector T-cell survival and memory formation.30,31 The IL4/7 ChR receptor α chain exodomain fusion produced canonical wild-type IL7 receptor signaling when engineered T cells were exposed to IL4, including upregulation of pStat5 target genes including MYC, IL2 receptor α, and Bcl2.32 Under physiological conditions, signal transduced through the native IL7 receptor is terminated by upregulation of SOCS 1 and 2, which are downstream targets of pStat5 and directly inhibit the activated Jak pathway by acting as pseudosubstrates and promoting the degradation of the signaling complex. Our results showed that this negative feedback loop remained fully functional following engagement of the IL4/7 ChR by IL4, suggesting that this modification would not promote autonomous growth of effector T cells. Consistent with this observation, IL4/7 ChR-modified T cells ceased to proliferate or persist once cytokine was withdrawn.
Although manipulations (such as lymphodepletion, infusion of cytokines, or incorporation of costimulatory endodomains)1,33,34,35 may increase the expansion and potency of infused T cells, such actions may cause toxicity due to the paucity of true tumor-specific target antigens. Thus, the in vivo expansion and activation of T cells that recognized tumor-associated antigens may in fact result in “on-target, off-tumor” effects resulting in the damage of healthy tissues expressing the targeted antigen. These severe and even fatal consequences have been observed in subjects who received chimeric antigen receptor (CAR) T cells targeting antigens such as carbonic anhydrase IX or human epidermal growth factor receptor 2, expressed both by malignant cells and cells of the liver and lung, respectively.36,37,38 However, in the latter case, it is likely that the cell dose infused (1 × 1010 cells) was also a factor that contributed to the toxicity since our group has targeted human epidermal growth factor receptor 2 clinically with cell doses ranging from 1 × 104/m2 to 1 × 108/m2 with no dose-limiting toxicity (S. Gottschalk, personal communication).
Hence, it is crucial that efforts to enhance the functionality of tumor-directed T cells in vivo and overcome tumor immune inhibition become operative only in the tumor environment and not in normal tissues. In other words, advances in the safety and efficacy of T cells will require them to become capable of recognizing patterns of gene expression that are different between normal and malignant cells, rather than relying on single antigenic markers. One means of achieving such pattern recognition is to incorporate receptors who, once engaged, function as the Boolean operators “AND,” “OR,” and “NOT.” The IL4/7 ChR we describe is one such example of an “AND” operator, since optimal activation will occur only in engineered T cells that encounter both their specific tumor antigen and the IL4 present at high concentrations in the tumor site. Other investigators have developed alternative “AND” receptors. For example, Wilkie et al.39 modified activated T cells with two engineered receptors: a first-generation CAR targeting the breast cancer-expressed human epidermal growth factor receptor 2 coupled with CD3ζ and a chimeric costimulatory receptor (CCR) in which Muc1 specificity was coupled with CD28. The investigators showed that the dual-targeted T cells received complementary signals when both receptors were engaged, leading to potent cytotoxicity and synergistically enhanced proliferation only in the presence of tumor cells expressing both targets. Kloss et al. extended this approach to a prostate cancer model and demonstrated that dual-targeted T cells expressing a first-generation CAR targeting PSCA and a CCR with specificity for PSMA coupled to CD28 and 41BB did not react to tissues expressing either PSCA or PSMA alone but produced potent T-cell activation only upon encounter with the two coexpressed antigens.40 The disadvantage of the dual-antigen complementation approach, however, is that antigen loss variants readily occur in most tumors, and the more antigens that are required to be present on a tumor for successful T-cell recognition to occur, the higher the probability that a mutational event will render the tumor nonstimulatory.33,41 While mutation of the tumor/tumor microenvironment may also lead to loss of IL4 secretion, the absence of this cytokine would have multiple detrimental effects on tumor growth and its evasion of an effective immune response, limiting the benefits of such a change. The use of dual receptor complementation and the expression of an IL4/7 ChR are not mutually exclusive and ultimately may prove synergistic for both safety/specificity and efficacy.
Finally, as with any genetic modification of hemopoietic cells that provides a positive growth signal, there is a concern that there will be long-term dangers of malignant transformation.42,43 While this possibility will need careful consideration, we think the risk of transformation is low since we are modifying fully differentiated T cells rather than hematopoietic stem cells, and our modification is to the IL7 α endodomain, which is triggered exclusively by IL4 cytokine, and not to the endodomain of the common γ chain which is shared by the receptors for multiple cytokines.30,31,44,45,46 Certainly, our modified cells remain dependent on antigenic signals and exogenous growth factors, and we found no evidence for autonomous growth. Hence, expression of an IL4/7 ChR on tumor-targeted T cells can invert the effects of a tumor-derived inhibitory cytokine and thereby enhance the persistence and antitumor activity observed.
Materials and Methods
Donor and cell lines. Peripheral blood mononuclear cells, obtained from healthy volunteers with informed consent under an IRB-approved protocol, were used to generate EBV-specific T-cell lines and EBV-transformed lymphoblastoid cell lines (EBV-LCLs). EBV-LCLs were generated with concentrated supernatants of the B95-8 cell line, as previously described,46 and were maintained in LCL media (RPMI 1640 (Hyclone Laboratories, Logan, UT), 10% FBS (Hyclone Laboratories, Logan, UT) and 2 mmol/l l-glutaMAX (Gibco by Life Technologies, Grand Island, NY)) in G-Rex flasks (Wilson Wolf Manufacturing, New Brighton, MN).47
Generation of retroviral construct and retroviral transduction. We synthesized (DNA 2.0, Menlo Park, CA) a codon-optimized sequence encoding the signal peptide and extracellular domain of the human IL4 receptor α chain fused with the transmembrane and intracellular domain of IL7 receptor, with the restriction sites Xho1 and Mlu1 incorporated up- and downstream, respectively. After enzymatic digestion with Xho1 and Mlu1, the IL4/7 ChR DNA insert was incorporated into a SFG retroviral vector containing an internal ribosomal entry site element and the fluorescent marker mOrange.41
Retroviral supernatant were produced using 293T cells, which were cotransfected with 3.7 µg of the IL4/7 ChR retroviral vector, 3.7 µg of the Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol, and 2.5 µg of the RDF plasmid containing the sequence for the RD114 envelope, using the GeneJuice transfection reagent (Novagen, Billerica, MA). Retroviral supernatant was collected at 48 and 72 hours posttransfection, filtered (using a 0.45-mm filter), and stored at −80 °C.
EBV-specific T-cell generation and transduction. EBV-specific T cells were prepared using peripheral blood mononuclear cells, autologous EBV-LCLs, and recombinant human IL2 (IL2, 50 U/ml; NIH, Bethesda, MD), as previously described.46 EBV-specific lines obtained after the third stimulation were transduced with the 4/7R ChR retroviral supernatant, as previously described.48 In brief, 2 days after stimulation with EBV-LCLs, T cells were plated in 24-well plates precoated with a recombinant fibronectin fragment (FN CH-296; Retronectin; Takara Shuzo, Otsu, Japan). After the addition of retroviral supernatant and IL2 (100 U/ml), T cells were spun at 1,000g for 30 minutes at room temperature and then transferred to a 37 °C incubator and maintained at 5% CO2. Three days later, EBV-specific T cells were collected, transduction efficiency was analyzed by flow cytometry, and the cells were maintained in culture by weekly stimulation with EBV-LCLs and IL2 (50 U/ml), IL4 (1,000 U/ml), or IL15 (5 ng/ml) (R&D Systems, Minneapolis, MN).
Characterization of T cells in vitro
Flow cytometry. For all flow cytometric analyses, samples were acquired on a Gallios Flow cytometer, and the data analyzed using Kaluza software (Beckman Coulter, Brea, CA). Antibodies were purchased from Becton-Dickinson (BD, Franklin Lakes, NJ), and the isotype controls were IgG1-PE, IgG1-PerCP, IgG1-FITC, or IgG1-APC. Phosphate-buffered saline (PBS, Sigma, St. Louis, MO) with 2% FBS and 0.1% sodium azide (Sigma) was used as a wash buffer, and PBS with 0.5% paraformaldehyde (Sigma) was used as fixative solution. For staining, cells were harvested, washed once with wash buffer, pelleted, and antibodies were added in saturating amounts (5 l). After 15-minute incubation at 4 °C in the dark, cells were washed twice, fixed, and analyzed.
Surface staining. Transgenic populations were detected by analyzing mOrange positive cells on the PE channel and expression of the IL4 receptor using an APC-conjugated IL4 receptor antibody.
Phospho flow. To detect the phosphorylation of Stat5, control NT T cells and IL4/7 ChR T cells were collected 7 days after antigen stimulation and rested in serum-free Iscove's Modified Dulbecco's Medium (GIBCO, Grand Island, NY) media for 24 hours without any cytokines. Subsequently, T cells were stained with a monoclonal antibody, which bound to Tyr-694 and conjugated with Alexa Fluor 647 (from Becton-Dickinson).
Measurement of cytokine levels. The Milliplex Human Cytokine Immunoassay (Cat# HSCYTO-60SKPMX13) (Millipore, Billerica MA), which detects IL1β, IL2, IL4, IL5, IL6, IL7, IL8, IL10, IL12p70, IL13, IFNγ, GM-CSF, and TNFα, was used to assess cytokine levels in culture supernatant collected from NT and IL4/7 ChR cells 24 hours poststimulation. The assay was performed as per manufacturers' instructions, and samples were analyzed on a Luminex 200 (XMAP Technology, Luminex Corporation, Austin, TX).
Gene expression profiling. To assess the gene expression profile of NT and transgenic T cells, we took cells from each condition 4 days after the last antigenic stimulation, exposed the cells to IL4 for 24 hours and then harvested and the cell pellet was snap frozen. Due to the lack of IL7 receptor expression on EBV-specific T cells,48 we additionally modified these cells to transgenically express this molecule48 and cultured these cells in the presence of either IL4 or IL7. RNA was extracted from these samples using an RNeasy Kit from Qiagen (Valencia, CA). RNA expression profiling was performed by Genome Exploration USA (Memphis, TN). In brief, total RNA was processed by the random primed RT-IVT-RT method using the Ambion WT Expression Kit (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Fragmented cDNA was hybridized for 17 hours at 45 °C to GeneChip PrimeView Human Gene Expression Arrays (Affymetrix, Santa Clara, CA). The PrimeView array is comprised of more than 530,000 probes (>43,000 probe sets) covering over 36,000 transcripts and variants, which, in turn, represent more than 20,000 genes mapped through UniGene or via RefSeq annotation. Expression data were analyzed following background correction, quantile normalization, and probe set signal summarization by robust multi-array average method.49 Differential expression was identified in pairwise comparisons of single arrays using an absolute fold change threshold >2. Punnett square analysis was further used to identify differential expression when comparing two pairs of arrays. Unsupervised hierarchical clustering and heat map generation were performed in GeneMaths XT using absolute or row mean centered log2-transformed RMA signal values. Probe set clustering was performed by the UPGMA method (unweighted pair group method using arithmetic averages) using Pearson correlation or Euclidean distance as the similarity metric; sample clustering was performed by UPGMA or complete linkage methods using Pearson correlation as the similarity metric.
Chromium release assay. We evaluated the cytotoxic activity of EBV-specific T cells using a standard 4-hour 51Cr release assay, as previously described45,46 with autologous and mismatched EBV-LCLs as target cells.
Proliferation assay. For proliferation assays, 1 × 105 NT and IL4/7 ChR T cells were cultured with γ-irradiated (40 Gy) autologous EBV-LCLs in 200 μl T-cell media (RPMI supplemented with 45% Click's medium (Irvine Scientific, Santa Ana, CA), 2 mmol/l GlutaMAX, and 10% FBS) in the presence of either IL4 (1,000 U/ml), IL2 (50 U/ml), or without exogenous cytokine. The cocultures were plated in triplicate in U-bottom 96-well plates (Falcon; Becton-Dickinson). On day 4, cultures were pulsed with 1 μCi 3H-thymidine per well (Amersham Biosciences, Piscataway, NJ) and harvested onto strips (Brandel, Gaithersburg, MD) 15–18 hours later using a PHD cell harvester. 3H-thymidine uptake was measured using in a β-scintillation counter (TriCarb 2910 TR; Perkin Elmer, Waltham, MA). The experiments were performed in triplicate.
In vivo antitumor activity in a xenograft model. To assess the antitumor effects of EBV-specific T cells expressing IL4/7 ChR, we used a severe combined immunodeficiency mouse model and an in vivo imaging system as previously described.47,48 EBV-LCLs were transduced with a retroviral vector encoding for IL4 cytokine and eGFP-FFLuc and sorted based on GFP expression using a MoFlo flow cytometer (Cytomation, Fort Collins, CO). ICR severe combined immunodeficiency mice (8–10 weeks old) were purchased from Taconic (Germantown, NY), engrafted subcutaneously with 5 × 106 eGFP-FFLuc-IL4/EBV-LCLs, and tumor cell growth was monitored weekly using bioluminescence imaging and calipers. After tumor engraftment (defined as an increase in tumor signal in at least two consecutive bioluminescence measurements and a tumor diameter >5 mm), mice were subdivided in different treatment groups. In vivo imaging was performed by administering d-luciferin (150 mg/kg) intraperitoneally, and animals were analyzed using the Xenogen-In Vivo Imaging System, as previously described.47,48 The intensity of the signal was measured as total photon/s/cm2/sr (p/s/cm2/sr). Mouse experiments were performed in accordance with Baylor College of Medicine's Animal Husbandry guidelines.
CT imaging of tumors in vivo. The CT images were acquired at the Small Animal Imaging Facility of Texas Children's Hospital using a Siemens Inveon MM preclinical scanner (Siemens AG, Knoxville, TN). In each scan, 360 projections were acquired covering a full rotation of the gantry, with a source-to-detector distance of 312.91 mm and a source-to-COR distance of 183.92 mm. The X-ray tube voltage and current were set at 80 kVp, 500 µA, with an exposure time of 650 ms per projection. During the procedure, the animals were anesthetized with 1.5 to 2% of isoflurane.
The CT slices were reconstructed using COBRA (Exxim Computing Corporation, Pleasanton, CA) on a volume of 640 × 640 × 992, 70 µm isotropic voxels. The natural contrast of the images provided a clear visualization of the necrotic tissue and a faint contrast at the interface of the tumor and the surrounding tissue. This was used to segment the tumor tissue manually using Inveon Research Workplace (Siemens AG, Knoxville, TN). The 3D volume renderings were generated using OsiriX (open source software, available online: http://www.osirix-viewer.com).
Statistical analysis. All in vitro data are presented graphically and summarized by mean ± SD or SEM. When a P value was <0.05, a mean difference was accepted as statistically significant. For the bioluminescence experiments, intensity signals were log-transformed and summarized using mean ± SD at baseline and multiple subsequent time points for each group of mice. Changes in intensity of signal from baseline at each time point were summarized.
SUPPLEMENTARY MATERIAL Figure S1. Specificity and proliferative capacity of NT and IL4/7 ChR T cells in vitro. Video S1. Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an untreated animal. Video S2. Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with NT T cells. Video S3. Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with 4/7 ChR T cells.
Acknowledgments
This work was supported by grants from the National Institutes of Health-National Cancer Institute (P01 CA094237 and P50 CA126752) as well as the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine. J.F.V. is supported by an Idea Development Award from the Department of Defense Prostate Cancer Research Program (no. W81XWH-11-1-0625). The authors thank Texas Children's Hospital for the use of the Small Animal Imaging Facility, and we also appreciate the support of the Flow Cytometry and Cell and Vector Production shared resources in the Dan L Duncan Cancer Center support grant P30CA125123. H.E.H. is supported by a Dan L. Duncan Chair and M.K.B. by a Fayez Sarofim Chair.
Supplementary Material
Specificity and proliferative capacity of NT and IL4/7 ChR T cells in vitro.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an untreated animal.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with NT T cells.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with 4/7 ChR T cells.
References
- Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straathof KC, Bollard CM, Popat U, Huls MH, Lopez T, Morriss MC, et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus–specific T lymphocytes. Blood. 2005;105:1898–1904. doi: 10.1182/blood-2004-07-2975. [DOI] [PubMed] [Google Scholar]
- Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood. 2007;110:2838–2845. doi: 10.1182/blood-2007-05-091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting epstein-barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808. doi: 10.1200/JCO.2013.51.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia WK, Teo M, Wang WW, Lee B, Ang SF, Tai WM, et al. Adoptive T-cell transfer and chemotherapy in the first-line treatment of metastatic and/or locally recurrent nasopharyngeal carcinoma. Mol Ther. 2014;22:132–139. doi: 10.1038/mt.2013.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol. 2009;157:9–19. doi: 10.1111/j.1365-2249.2009.03912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94:41–53. doi: 10.1189/jlb.1212631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- Lacuesta K, Buza E, Hauser H, Granville L, Pule M, Corboy G, et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J Immunother. 2006;29:250–260. doi: 10.1097/01.cji.0000192104.24583.ca. [DOI] [PubMed] [Google Scholar]
- Pangault C, Amé-Thomas P, Ruminy P, Rossille D, Caron G, Baia M, et al. Follicular lymphoma cell niche: identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia. 2010;24:2080–2089. doi: 10.1038/leu.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G, et al. IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J Immunol. 2004;172:5467–5477. doi: 10.4049/jimmunol.172.9.5467. [DOI] [PubMed] [Google Scholar]
- Roca H, Craig MJ, Ying C, Varsos ZS, Czarnieski P, Alva AS, et al. IL-4 induces proliferation in prostate cancer PC3 cells under nutrient-depletion stress through the activation of the JNK-pathway and survivin up-regulation. J Cell Biochem. 2012;113:1569–1580. doi: 10.1002/jcb.24025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Todaro M, Lombardo Y, Francipane MG, Alea MP, Cammareri P, Iovino F, et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008;15:762–772. doi: 10.1038/sj.cdd.4402305. [DOI] [PubMed] [Google Scholar]
- Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- Faffe DS, Flynt L, Bourgeois K, Panettieri RA, Jr, Shore SA. Interleukin-13 and interleukin-4 induce vascular endothelial growth factor release from airway smooth muscle cells: role of vascular endothelial growth factor genotype. Am J Respir Cell Mol Biol. 2006;34:213–218. doi: 10.1165/rcmb.2005-0147OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Li WQ, Hofmeister RR, Young HA, Hodge DR, Keller JR, et al. Distinct regions of the interleukin-7 receptor regulate different Bcl2 family members. Mol Cell Biol. 2004;24:6501–6513. doi: 10.1128/MCB.24.14.6501-6513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barata JT, Cardoso AA, Nadler LM, Boussiotis VA. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1) Blood. 2001;98:1524–1531. doi: 10.1182/blood.v98.5.1524. [DOI] [PubMed] [Google Scholar]
- Palmada M, Boehmer C, Akel A, Rajamanickam J, Jeyaraj S, Keller K, et al. SGK1 kinase upregulates GLUT1 activity and plasma membrane expression. Diabetes. 2006;55:421–427. doi: 10.2337/diabetes.55.02.06.db05-0720. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Weber JS, Maio M, Neyns B, Harmankaya K, Chin K, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24:2174–2180. doi: 10.1093/annonc/mdt161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton CP, Abraham DJ. Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001;13:505–511. doi: 10.1097/00002281-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, Schluns KS. Functions of γC cytokines in immune homeostasis: current and potential clinical applications. Clin Immunol. 2009;132:153–165. doi: 10.1016/j.clim.2009.03.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh ST. Structural insights into the common γ-chain family of cytokines and receptors from the interleukin-7 pathway. Immunol Rev. 2012;250:303–316. doi: 10.1111/j.1600-065X.2012.01160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow MA, Lee G, Gillis S, Yancopoulos GD, Alt FW. Interleukin-7 induces N-myc and c-myc expression in normal precursor B lymphocytes. Genes Dev. 1992;6:61–70. doi: 10.1101/gad.6.1.61. [DOI] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. 2014;22:623–633. doi: 10.1038/mt.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Bear AS, Morgan RA, Cornetta K, June CH, Binder-Scholl G, Dudley ME, et al. Replication-competent retroviruses in gene-modified T cells used in clinical trials: is it time to revise the testing requirements. Mol Ther. 2012;20:246–249. doi: 10.1038/mt.2011.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, et al. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. 2009;17:880–888. doi: 10.1038/mt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specificity and proliferative capacity of NT and IL4/7 ChR T cells in vitro.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an untreated animal.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with NT T cells.
Flythrough of a volume-rendering CT scan showing the tumor mass (highlighted in green) in an animal treated with 4/7 ChR T cells.