

Abstract
The chemical structures of DNA-protein crosslinks (DPCs) are necessary for understanding mechanisms of mutagenesis, but structural analysis of DPCs is limited because the modifications are bulky. Using a combination of LC-tandem MS and chemical modification approaches, we have characterized the structures of various DNA-ethylene-O6-alkylguanine-DNA alkyltransferase (AGT) crosslinks.
Keywords: DNA-protein crosslinks, Hydrogenation, Mutations, Mass spectrometry, DNA adducts, O6-Alkylguanine-DNA alkyltransferase
O6-Alkylguanine-DNA alkyltransferase (AGT, MGMT) is involved in the repair of the promutagenic lesion O6-methyl (Me) guanine and, to a lesser extent, O4-Me thymine. AGT constitutes an important repair system against the mutagenic, carcinogenic, and toxic effects of simple alkylating agents. The repair mechanism involves a direct transfer of the alkyl group from the O6 atom of guanine to the active site cysteine (Cys145) of the AGT protein in a stoichiometric, direct damage reversal pathway.[1,2] Apart from methyl groups, AGT can also repair larger alkyl groups at the O6 atom of guanine including ethyl (Et), 2-chloroethyl, butyl, benzyl, and pyridyloxobutyl.[1,2]
Although AGT is a repair protein, it has been shown to paradoxically augment the toxicity of 1,2-dibromoethane (DBE, also called ethylene dibromide) (Scheme 1).[3] Overexpression of human AGT (hAGT) or AGTs from other species enhances the mutagenicity and lethality of DBE in Escherichia coli and mammalian cells.[3] This unusual enhancement of DBE toxicity stems from the increased reactivity of the active site Cys145 residue that readily reacts with the bis-electrophile DBE to form a half-mustard. The half-mustard intermediate subsequently cyclizes to yield an episulfonium ion that can react to form a covalent DNA-ethylene-AGT crosslink (based on the demonstrated chemistry with glutathione (GSH)) (Scheme 1).[4] Formation of a DNA-protein crosslink (DPC) is facilitated by the DNA-binding properties of AGT.[1] In prokaryotes, DPCs are repaired by both nucleotide excision repair and homologous recombination (HR), while in eukaryotes HR appears to be the major pathway.[5]
Scheme 1.
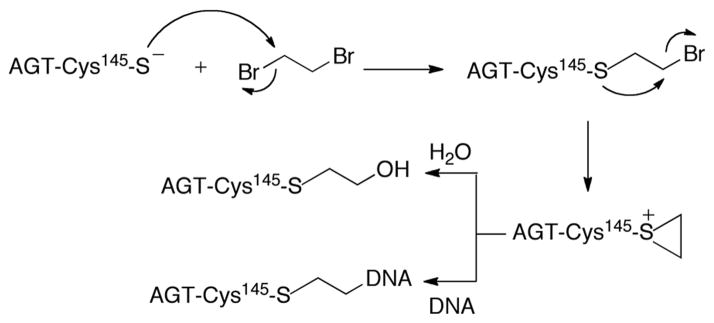
Mechanism of DBE derived crosslink formation by AGT
DBE has been widely used as a gasoline additive, as well as in pesticides and soil fumigants. DBE is carcinogenic in rats and toxic and mutagenic in microorganisms, plants, insects, and humans. Following reports of its toxicity, use of DBE has been drastically reduced. Earlier studies with DBE indicated that products formed via microsomal oxidation and by the action of GSH transferase (GST) may be responsible for its toxicity, apart from the AGT pathway. Similar to AGT, the detoxication pathway involves GST catalysis of the reaction of GSH with DBE to form a GSH-half mustard (S-(2-bromoethyl)GSH) and finally the toxic episulfonium ion intermediate. The episulfonium ion reacts rapidly with DNA to form various guanine and adenine adducts.[6] Of these, a heat labile guanyl-N7 adduct, S-[2-(N7-guanyl)ethyl]GSH, accounts for ~95% of total DNA adducts and is associated with G:C to A:T transition mutations.[6] hGST was also shown to enhance the mutagenicity of DBE in Salmonella typhimurium tester strains TA100 and TA1535.[7] Although the various bioactivation pathways of DBE are well characterized, the extent to which each pathway (activation by microsomal oxygenases, GST, or AGT) contributes to DBE-induced genotoxicity is still not clear.
The DNA adducts and mutational spectra resulting from GSH-DBE episulfonium ion have been extensively studied.[6,8] In contrast, detection and structural characterization of crosslinks resulting from the AGT-DBE episulfonium ion remain mostly unexplored. To the best of our knowledge, the N7-guanyl adduct is the only DNA-ethylene-AGT crosslink that has been detected to date.[9] Using gel-shift assays with synthetic poly A, T, C, or GC oligonucleotides, Liu et. al.[9] showed that the AGT-DBE episulfonium ion is capable of crosslinking to all the four bases, with the apparent preference (based on gel shift assays) being G>T>C>A. In addition, the major type of mutation observed in S. typhimurium (YG7108) and E. coli TRG8 cells was a G:C to A:T transition mutation.[9] Typically, labile adducts (e.g., guanyl-N7) will produce G:C to T:A transversion mutations due to depurination and the A-coding rule.[10] The fact that DBE (in the presence of AGT) produces G:C to A:T transition mutations[9] also suggests the presence of other adducts.
Structural characterization of DPCs has been largely limited to labile adducts owing to the ease with which the alkylated protein can be separated from DNA. In contrast, structural characterization of non-labile covalent DPCs, e.g. with AGT, is a challenge. The available methods for the characterization of DNA modifications involve digestion of the DNA to nucleosides followed by LC-MSn analysis and comparison with authentic standards. In the case of DNA-ethylene-AGT crosslinks, digestion of the DNA and protein to nucleosides and amino acids is inhibited due to steric hindrance for the hydrolases. Chemical methods of digestion of complexes are generally too harsh to preserve the linkages for structural characterization. MS methods of detection are optimized either for nucleic acids or protein/peptides (negative vs positive electrospray ionization (ESI)) and, therefore, in the case of DPCs the presence of one negates the other.
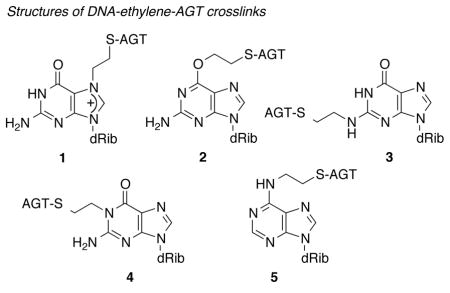
Herein we report the detection and structural characterization of various DNA-ethylene-AGT crosslinks using LC-tandem mass spectrometric and chemical approaches. Using these approaches, one labile and four non-labile covalent DPCs at guanine and adenine bases (the N2, O6, and N1 atoms of guanine and N6 of adenine, structures 2–5) have been systematically identified.
Our initial goal was to determine the formation of any non-labile covalent DPCs formed in the presence of AGT and DBE. We previously reported a LC-tandem MS (LC-MS/MS or LC-MSn)-based approach for the detection of oligonucleotide-peptide crosslinks resulting from trypsin digestion of an oligonucleotide-alkyl-AGT complex.[11] Although DPCs are resistant to nuclease and protease digestions, trypsin can efficiently digest the oligonucleotide-alkyl-AGT complex to a 12-mer peptide.[9] Accordingly, we reacted a 15-base pair GC-rich double-stranded synthetic oligonucleotide with AGT in the presence of DBE. The results clearly indicated that the AGT-DBE episulfonium ion forms non-labile crosslinks in DNA (Figures S1, S2, Supporting Information),.
The above results confirmed the presence of non-labile crosslinks but did not provide any information regarding the base specificity of the DPCs. In CID, the DNA bases mostly dissociate as neutral species. However, the possibility that the presence of the attached peptide might allow the base to dissociate as a charged species led us to search for peptide-ethylene-base species in the CID spectra. Contrary to our expectation, we did not find any fragment peak corresponding to peptide-ethylene-G or C (m/z 1450 or 1490 for C or G adducts, respectively) in the case of the GC-rich oligonucleotide (Figure S1e, Supporting Information). However, when the experiment was repeated with a double-stranded T5G2T4 oligonucleotide (designed for preferential fragmentation at G bases compared to T, thereby increasing the yields of the various fragments resulting from G), a peak with m/z 1450 and corresponding to the fragment peak of peptide-ethylene-G was detected (Figure S2, Supporting Information). We also performed a similar experiment with a double-stranded (12-base pair) (AT)6 oligonucleotide. Unlike the GC-rich oligonucleotide, in the CID spectrum we did find a peak at m/z 1474.6 that corresponds to the adenine base connected to the 12-mer peptide through the ethylene linkage (Figure S4, Supporting Information). Together these results indicate that AGT-DBE episulfonium ion can form non-labile crosslinks at G and A bases in DNA.
The LC-tandem MS approach with the oligonucleotides established the presence of non-labile DNA-ethylene-AGT crosslinks on G and A residues, but it did not provide any information regarding the structures of the DPCs. Moreover, the use of small oligonucleotides has limited capability in dealing with the effects of sequence variation. In order to elucidate the structure of the DPC, we realized that the complex would need to be degraded to the single nucleoside level. Accordingly, we utilized a reductive desulfuration approach to convert DNA-ethylene-peptide crosslinks into DNA- Et adducts.[12] Reductive desulfurization is a well-established chemical procedure for breaking molecules by attack on a thioether or other sulfur atom. Raney nickel was first employed by Bougault et al.,[12a,13a] and nickel boride (NaBH4/NiCl2) is also effective.[12b,13b,c] We previously utilized nickel boride in the initial characterization of the major DBE-GSH DNA adduct by converting it to N7-EtG by nickel boride treatment.[12b] In this case we used a Raney nickel procedure that had been applied by Hecht et al.[12a] to tRNA, because of the issues of solubility (in aqueous buffer) and the adsorption of DNA and nucleosides on the nickel. The complex was first digested with either trypsin or proteinase K to a small peptide, followed by removal of the peptide by Raney nickel reductive desulfuration. This process leaves only an Et group (derived from the DBE part of the DPC) on the DNA, which can then be readily digested to the nucleoside level with nucleases and phosphatases for characterization by LC-MS and comparison with authentic standards. To include all possible sequence variations, sheared calf thymus DNA was used (Scheme 2). A study with a set of three nucleosides containing an ethylene-GSH modification indicated yields of 89–91% for the Raney nickel reduction (Table S3, Supporting Information).
Scheme 2.
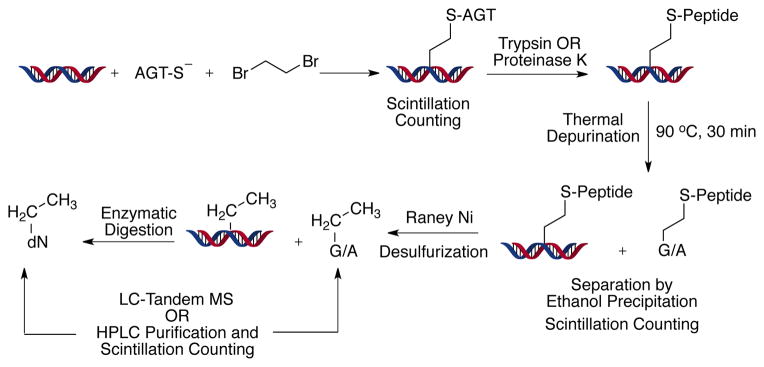
Flow chart for the detection and quantitation of various DPC and DNA adducts
DNA was incubated with DBE and AGT to generate DPCs and then subjected to various procedures, described in the experimental section (Supporting Information). LC-MS/MS analysis of the final reaction product using the m/z 280→164 transition (characteristic neutral loss of -116 for deoxyribonucleosides) for Et-dA adducts gave a peak at tR 3.6 min. CID of the m/z 164 peak gave a fragment ion at m/z 136, consistent with an authentic sample of N6-EtdA (5) (Figure S5, Supporting Information). Together these results clearly indicate formation of the DNA-ethylene-AGT crosslink at the N6 atom of dA in the DNA (although the presence of an initial crosslink at the N1 atom of dA followed by Dimroth rearrangement cannot be excluded).
In principle there are three potential sites in dG (N2, N1, O6) that could form non-labile DPCs with the AGT-DBE episulfonium ion. Authentic standards of these three ethyl-dG adducts were used in the LC method: tR N1-EtdG (4) 3.2 min, N2-EtdG (3) 3.6 min, and O6-EtdG (2) 4.0 min. LC-MS3 of the deglycosylated MH+-116 fragments showed very distinct fragmentation patterns: major ion m/z 163 for N2-EtdG and m/z 152 for N1-EtdG and O6-EtdG. LC-MS/MS analysis using the m/z 296→180 transitions gave a peak at tR 3.6 and a very small peak at tR 4.1 min. CID of the m/z 180 fragments gave a peak at tR 3.6 min with a major fragment ion at m/z 163 and another peak at tR 4.1 min, with a major fragment ion at m/z 152. Based on the tR (~3.2, 3.5, and 4.0 min for N1-, N2-, and O6-EtdG, respectively) and fragmentation of the authentic standard (Figures S6 and S7, Supporting Information), the tR 3.6 and 4.1 min peaks are N2-EtdG and O6-EtdG, respectively. These peaks were not seen in control reactions where either AGT or AGT and DBE are omitted (Figure S9, Supporting Information). From the LC-tandem MS results we conclude that DPCs form at the N2 and O6 positions of dG in the DNA. The finding that the O6-guanyl-AGT is not cleaved by AGT itself is consistent with the lack of AGT activity on an O6-dG butyl crosslink we reported earlier.[11b]
Calf thymus DNA was treated with 14C-DBE and AGT as described previously (Scheme 2). An aliquot of the precipitated DNA-(14CH2)2-AGT complex was analyzed by scintillation counting and showed that the reaction with AGT yielded significantly higher (~6 nmol) levels of DPCs (both labile and non-labile) compared to the control reactions (Figure 1a). The reaction mixtures were subsequently digested with proteinase K and ethanol-precipitated to remove the digested peptides and heated at 90° C for 30 min to hydrolyze the labile DNA-ethylene-peptide complexes. Of the total DNA-ethylene-AGT complexes formed, ~80% were found to be labile and 20% non-labile (Figure 1a and c). To determine if DPCs are also formed at N3-dA, the precipitate was subjected to reductive desulfuration with Raney nickel to generate N7-dG- and/or N3-dA Et adducts (Scheme 2). The only labile adduct detected was the N7-dG one and the N3-dA adduct was not present. Further, no evidence for an N3-adenyl-linked typtic peptide was seen with LC-MSn of the soluble fraction.
Figure 1.

Yields of the various types of DPCs formed in the presence of AGT and 14C-DBE. a) Total DPCs (labile + non-labile). b) Labile DPCs. c) Non-labile DPCs. d) Relative yields of labile DPCs, N7-G adducts measured as N7-Et guanine. e) Yields of various non-labile DPCs (N1-dG, N2-dG, and O6-dG and N6-dA) measured as their corresponding Et adducts (n = 3, ± SD).
The precipitate, containing the non-labile adducts, was subjected to Raney nickel desulfurization and DNA digestion to convert DNA-(14CH2)2-peptide complexes into Et adducts (vide supra). The various Et adducts were separated by HPLC after spiking with carrier N1-, N2-, and O6-EtdG and N6-EtdA adducts (monitoring A260) (Figure S10, Supporting Information). The radioactivity associated with the various adducts in the DNA+AGT+DBE samples was significantly higher than in the control samples of DNA+DBE. The yields of the various adducts were in the order N6-EtdA ~ N2-EtdG > O6-EtdG ~ N1-EtdG. These results are consistent with the LC-MS data; in the LC-MS3 analysis the yield of the O6-EtdG adduct was just above the detection limit and the N1-EtdG could not be detected, but with 14C-DBE both adducts were detected.
Mutational studies involving overexpression of the repair protein AGT in the presence of DBE in cell culture systems show G:C to A:T transition mutations.[9,14] The observation that only ~20% of the mutations in bacteria can be attributed to depurination,[9] although ~80% of the adducts are labile (Figure 1), suggests that the non-labile GPCs are much more mutagenic. In vitro primer extension assays with intact proteins have not been reported, to our knowledge. However, primer extension assays with DNA-peptide crosslinks have revealed that translesion polymerases can bypass 4- to 10-amino acid peptide adducts.[15] Recent work from our group with DNA-butudiene diepoxide-GSH crosslinks at the N6 position of dA (the major adduct detected in vivo) has shown that a number of polymerases can also bypass it with high fidelity.[16] Electroporation of an hAGT-butadiene diepoxide conjugate into mammalian cells caused both cytotoxicity and mutations,[17] further establishing the relevance of the AGT pathway in bioactivation. The mutations resulting from DPCs may be due to misincorporation by translesion polymerases or HR.[5,18]
Knowing the chemical structures of the DPCs is extremely important for understanding the mechanisms of mutagenesis. Using a combination of LC-tandem MS, chemical modification, and radioisotope approaches we have detected and characterized the structures of various DNA-ethylene-AGT crosslinks. Three non-labile modifications were found at the N1, N2, and O6 atoms of guanine and one at the N6 atom of adenine (structures 2–5), consistent with our previous reports with the GSH-DBE episulfonium ions.[6]
Supplementary Material
Footnotes
This work was supported in part by United States Public Service Grants R01 ES010546 and P30 ES00267.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Goutam Chowdhury, Department of Biochemistry, Vanderbilt University School of Medicine, 638 RRB, 2220 Pierce Ave., Nashville, TN 37232, USA, Fax: 1 (615) 322-4349
Dr. Sung-Hee Cho, Department of Biochemistry, Vanderbilt University School of Medicine, 638 RRB, 2220 Pierce Ave., Nashville, TN 37232, USA, Fax: 1 (615) 322-4349
Prof. Dr. Anthony E. Pegg, Email: aep1@psu.edu, Department of Cellular and Molecular Physiology, Pennsylvania State University College of Medicine, 500 University Drive, Hershey, PA 17033, USA
Dr. F. Peter Guengerich, Email: f.guengerich@vanderbilt.edu, Department of Biochemistry, Vanderbilt University School of Medicine, 638 RRB, 2220 Pierce Ave., Nashville, TN 37232, USA, Fax: 1 (615) 322-4349
References
- 1.Pegg AE. Chem Res Toxicol. 2011;24:618–639. doi: 10.1021/tx200031q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guengerich FP, Fang Q, Liu L, Hachey DL, Pegg AE. Biochemistry. 2003;42:10965–10970. doi: 10.1021/bi034937z. [DOI] [PubMed] [Google Scholar]
- 3.Liu L, Pegg AE, Williams KM, Guengerich FP. J Biol Chem. 2002;277:37920–37928. doi: 10.1074/jbc.M205548200. [DOI] [PubMed] [Google Scholar]
- 4.Peterson LA, Harris TM, Guengerich FP. J Am Chem Soc. 1988;110:3284–3291. [Google Scholar]
- 5.Reardon JT, Sancar A. Methods Enzymol. 2006;408:189–213. doi: 10.1016/S0076-6879(06)08012-8. [DOI] [PubMed] [Google Scholar]
- 6.Cmarik JL, Humphreys WG, Bruner KL, Lloyd RS, Tibbetts C, Guengerich FP. J Biol Chem. 1992;267:6672–6679. [PubMed] [Google Scholar]
- 7.Thier R, Pemble SE, Kramer H, Taylor JB, Guengerich FP, Ketterer B. Carcinogenesis. 1996;17:163–166. doi: 10.1093/carcin/17.1.163. [DOI] [PubMed] [Google Scholar]
- 8.Valadez JG, Guengerich FP. J Biol Chem. 2004;279:13435–13446. doi: 10.1074/jbc.M312358200. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Hachey DL, Valadez G, Williams KM, Guengerich FP, Loktionova NA, Kanugula S, Pegg AE. J Biol Chem. 2004;279:4250–4259. doi: 10.1074/jbc.M311105200. [DOI] [PubMed] [Google Scholar]
- 10.Sagher D, Strauss B. Biochemistry. 1983;22:4518–4526. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 11.a) Chowdhury G, Guengerich FP. Angew Chem, Int Ed. 2008;47:381–384. doi: 10.1002/anie.200703942. [DOI] [PubMed] [Google Scholar]; b) Fang Q, Noronha AM, Murphy SP, Wilds CJ, Tubbs JL, Tainer JA, Chowdhury G, Guengerich FP, Pegg AE. Biochemistry. 2008;47:10892–10903. doi: 10.1021/bi8008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Hecht SM, Kirkegaard LH, Bock RM. Proc Natl Acad Sci U S A. 1971;68:48–51. doi: 10.1073/pnas.68.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ozawa N, Guengerich FP. Proc Natl Acad Sci U S A. 1983;80:5266–5270. doi: 10.1073/pnas.80.17.5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Bougault J, Cattelain E, Chabrier P. Bull soc chim. 1938;5:1699–1712. [Google Scholar]; Truce WE, Roberts FE. J Org Chem. 1963;28:961–964. [Google Scholar]; c) Truce WE, Perry FM. J Org Chem. 1965;30:1316–1317. [Google Scholar]
- 14.Kalapila AG, Pegg AE. Mutat Res. 2010;684:35–42. doi: 10.1016/j.mrfmmm.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, Lloyd RS. J Biol Chem. 2008;283:17075–17082. doi: 10.1074/jbc.M801238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SH, Guengerich FP. Chem Res Toxicol. 2013;26:1005–1013. doi: 10.1021/tx400145e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tretyakova NY, Michaelson-Richie ED, Gherezghiher TB, Kurtz J, Ming X, Wickramaratne S, Campion M, Kanugula S, Pegg AE, Campbell C. Biochemistry. 2013;52:3171–3181. doi: 10.1021/bi400273m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Fang Q. Mutat Res. 2013;741–742:1–10. doi: 10.1016/j.mrfmmm.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reardon JT, Sancar A. Proc Natl Acad Sci U S A. 2006;103:4056–4061. doi: 10.1073/pnas.0600538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.