

Abstract
Current inotropic therapies used to increase cardiac contractility of the failing heart center on increasing the amount of calcium available for contraction, but their long-term use is associated with increased mortality due to fatal arrhythmias. Thus, there is a need to develop and explore novel inotropic therapies that can act via calcium-independent mechanisms. The purpose of this study was to determine whether fast α-myosin molecular motor gene transfer can confer calcium-independent positive inotropy in slow β-myosin-dominant rabbit and human failing ventricular myocytes. To this end, we generated a recombinant adenovirus (AdMYH6) to deliver the full-length human α-myosin gene to adult rabbit and human cardiac myocytes in vitro. Fast α-myosin motor expression was determined by Western blotting and immunocytochemical analysis and confocal imaging. In experiments using electrically stimulated myocytes from ischemic failing hearts, AdMYH6 increased the contractile amplitude of failing human [23.9±7.8 nm (n=10) vs. AdMYH6 amplitude 78.4±16.5 nm (n=6)] and rabbit myocytes. The intracellular calcium transient amplitude was not altered. Control experiments included the use of a green fluorescent protein or a β-myosin heavy chain adenovirus. Our data provide evidence for a novel form of calcium-independent positive inotropy in failing cardiac myocytes by fast α-myosin motor protein gene transfer.—Herron, T. J., Devaney, E., Mundada, L., Arden, E., Day, S., Guerrero-Serna, G., Turner, I., Westfall, M., Metzger, J. M. Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by α-myosin motor gene transfer.
Keywords: cardiac gene transfer, ischemic heart failure, adenoviral gene transfer
Heart failure is a clinical syndrome that represents the final common pathway for many diseases of cardiac muscle dysfunction and is a main cause of death in the Western world (1). Current pharmacological therapy for acute heart failure relies on classic inotropic agents directed at increasing intracellular calcium mobilization, including β-adrenergic agonists or phosphodiesterase inhibitors. These calcium-based therapies can provide short-term benefits of improved cardiac contractility, but their long-term use is associated with increased mortality resulting from fatal arrhythmias (2, 3). Other agents, including myofilament calcium sensitizers, may offer promise for long-term therapeutic inotropic support (4), but their use has been associated with an increased risk for atrial fibrillation (5). More recently, a number of gene-based therapies for heart failure have been proposed. These gene therapy approaches have been largely calcium-centric by focusing on increasing calcium load in sarcoplasmic reticulum by either gene delivery of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA2a) and/or knockdown of phospholamban (6, 7). Increased activity of the ATP-dependent SERCA2a pump runs the risk of increasing energy demand in the energy-starved failing heart and subjects the myocardium to increased calcium load, which has been associated with deleterious effects on heart performance during ischemia/reperfusion (8). In this light, there is a great need to investigate alternative approaches that do not require intracellular calcium-handling manipulation to increase the inotropic state of the failing heart. Here we present a novel mechanism for calcium-independent positive inotropy by implementing fast α-myosin molecular motor gene transfer to failing cardiac ventricular myocytes.
Cardiac myosin is the heart’s molecular motor that transduces chemical energy from ATP hydrolysis into mechanical energy to drive each heartbeat. In the heart, the MYH6 gene encodes the α-myosin heavy chain (α-MyHC) gene, the mammalian heart’s fast molecular motor. It has been well established in small-animal models of heart failure and other cardiac-related diseases that α-MyHC expression is lost and replaced by the slower β-MyHC motor isoform (9,10,11,12,13). Expression of β-MyHC is considered to be energetically favorable in the failing heart, but this might be offset by depressed cardiac function (14, 15). In the nonfailing human heart, there is a small, but significant, amount of the faster α-MyHC expressed. In failing human hearts, α-MyHC expression is reduced to undetectable levels, such that these diseased hearts exclusively express the β-MyHC motor isoform, a mechanically slower and less powerful molecular motor (16,17,18). It is currently unclear whether this small shift in the myosin isoform expression has direct functional effects on the contractile function of β-MyHC-dominant ventricular myocytes of rabbits and humans, for example. Previous models used to manipulate the relative expression of the cardiac myosin isoforms have included hormonal manipulation (11, 19,20,21) and transgenic approaches (14, 22, 23). These models have provided insight into functional effects of myosin isoform switching but have inherent limitations. Manipulation of thyroid hormone levels is a common means of altering the relative myosin isoform expression, but thyroid hormone also regulates the expression of other key cardiac regulatory proteins, including the SERCA2a pump. In transgenic animal models, it may be difficult to distinguish the primary effects of myosin isoform shifts from secondary compensatory mechanisms that may be in play to account for the presence of the myosin transgene (24). To circumvent these limitations, here we have for the first time used acute gene transfer of the full-length MYH6 gene to increase α-MyHC expression in β-MyHC-dominant adult cardiac myocytes in culture.
It is unclear whether α-MyHC gene transfer will enhance cardiac function in a calcium-independent manner. Recently, acute genetic replacement of α-MyHC with the β-MyHC gene in α-MyHC-dominant rodent cardiac myocytes was shown to directly depress cardiac myocyte contractility independent of changes in the intracellular calcium transient amplitude (25). The importance of myosin as a therapeutic target for heart failure is underscored by the recent development of small-molecule compounds that directly target and speed the kinetics of cardiac myosin. These compounds offer the promise of calcium-independent positive inotropy for failing myocardium (26, 27). Recently, successful surgical and pharmacological therapy for cardiomyopathy and heart failure in humans has been associated with restoration of α-MyHC expression, supporting an association between the cardiac myosin isoform expression profile and heart performance (28,29,30). Collectively, these studies led to the hypothesis tested here that fast α-MyHC gene transfer will enable calcium-independent positive inotropy in failing ventricular myocytes.
MATERIALS AND METHODS
α-MyHC cloning and vector production
The cDNA for human α-MyHC measures 5820 bp and was cloned using a combination of PCR-based techniques. The 3′ end was isolated by PCR from a human heart cDNA library (Clontech, Mountain View, CA, USA), and a C-terminal FLAG epitope tag was engineered, also with PCR techniques. To control for the presence of the FLAG epitope, separate plasmids were also generated with an N-terminal FLAG epitope or with no FLAG. The 5′ sequence was not represented in the library and was isolated by RT-PCR from a sample of normal ventricular muscle tissue. Complete sequence accuracy was confirmed by the DNA Sequencing Core at the University of Michigan. High titer recombinant adenovirus was generated using the AdMax system (Microbix, Toronto, ON, Canada).
Immunoblot detection and indirect immunofluorescence
α-MyHC protein expression was detected using the unique FLAG tag epitope that was engineered into the C-terminal rod region or the N-terminal region of the molecule (Fig. 1A). Cultured adult myocytes were collected in standard Laemmli sample buffer 24 and 48 h after gene transfer, and proteins were subsequently separated by electrophoresis on acrylamide gels (4–12% gradient) and transferred to nitrocellulose membranes for immunoblotting. The M2 FLAG mouse monoclonal antibody (1:1000; Sigma-Aldrich Corp., St. Louis, MO, USA) was used for specific detection of adenovirus-directed human α-MyHC protein expression (Fig. 1B). The MF20 monoclonal antibody (1:1000) developed by Donald Fischman was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology (Iowa City, IA, USA). MF20 antibody was used to determine the total amount of myosin. Western blots were also probed with the mouse monoclonal 5C5 actin antibody (1:2500; Sigma-Aldrich Corp.) to ensure equal protein loading (Fig. 1B). Horseradish peroxidase-labeled secondary antibody (1:2500; BD Pharmingen, San Jose, CA, USA) was used in combination with enhanced chemiluminescence for antibody detection. Protein quantification was determined by densitometry of scanned films using commercially available software (Quantiscan; Biosoft, Cambridge, UK).
Figure 1.
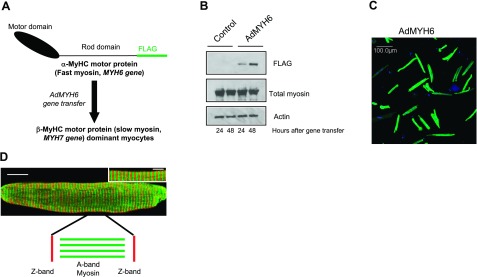
AdMYH6 gene transfer in healthy β-MyHC-dominant rabbit cardiac myocytes. A) A recombinant adenovirus (AdMYH6) was designed to deliver and express the full-length human α-MyHC fast myosin motor in adult cardiac myocytes. B) Western blotting shows that α-MyHC expression was detectable 24 h after viral gene transfer and increased 48 h after gene transfer. C) Immunocytochemical analysis and low magnification (×20) demonstrate highly efficient (∼98%) gene transfer using AdMYH6. Green fluorescence represents the presence of the FLAG epitope; nuclear staining is blue (DAPI). D) Immunocytochemical analysis and high-resolution confocal imaging demonstrate that α-MyHC protein was expressed homogenously across the length and width of cardiac myocytes (whole-cell image, scale bar=20 μm) and incorporated appropriately in the sarcomere A band (inset, scale bar=5 μm) in between the Z lines (red, α-actinin).
Indirect immunofluorescence and confocal imaging were performed as before (25) to identify the spatial localization of α-MyHC protein in the cardiac sarcomere after gene transfer. Myocytes were fixed (4% paraformaldehyde; Sigma-Aldrich Corp.) on laminin-coated coverslips after 2 d in primary culture. The anti-FLAG antibody produced in rabbit was used to detect the FLAG-tagged α-MyHC molecule by immunofluorescence and a mouse monoclonal antibody against α-actinin (clone EA-53; Sigma-Aldrich Corp.) was used to label the sarcomeric Z band. Goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA) were used to detect the rabbit FLAG antibody. Goat-anti-mouse secondary antibodies conjugated to Texas Red were used to detect α-actinin in the Z band (Fig. 1D). 4′,6-Diamidino-2-phenylindole (DAPI) (1 nM in PBS; Sigma-Aldrich Corp.) was used to stain the nuclei (Fig. 1C). Confocal imaging of myocytes was performed with sequential laser firing using an Olympus FluoView confocal laser scanning microscope (FV500; Olympus, Tokyo, Japan).
Adult cardiac myocyte isolation, culture, and in vitro gene transfer
New Zealand White rabbits (2–3 kg) were used for this study, and all animal use and care was approved by the Care and Use Committee of the University of Michigan and supported by the Unit for Laboratory Animal Medicine. Myocytes were isolated after enzymatic perfusion with collagenase type 2 (Worthington Biochemical Corp., Lakewood, NJ, USA) and hyaluronidase (Sigma-Aldrich Corp.) through the aorta and cultured as described previously for rodent myocyte isolation (25). Human myocytes were isolated at the time of cardiac transplantation surgery from the explanted heart of a 69-yr-old male patient with ischemic cardiomyopathy and heart failure. The use of human tissue was approved by the institutional review board of the University of Michigan. Human myocytes were isolated and maintained in culture as described previously (31). Acute gene transfer using recombinant adenoviruses was performed as before with multiplicity of infection (500 MOI) and high efficiency (25). In healthy rabbit cardiac myocytes, control adenoviruses included the use of AdMYH7, AdMYH6 with a 3′FLAG, AdMYH6 with a 5′FLAG, and AdMYH6 with no FLAG. In failing human cardiac myocytes, AdGFP was used to control for adenoviral transduction.
Ischemic heart failure model
We performed chronic left circumflex artery (LCX) ligation on adult rabbits as described previously (32, 33). Animals underwent a left thoracotomy procedure while maintained under isoflurane (2–4%) anesthesia after sedation with a subcutaneous injection of ketamine (10 mg/kg) and xylazine (5 mg/kg) and intravenous injection of thiopental (15 mg/kg). After the fourth intercostal space was entered and the pericardium was cut, the LCX was visualized and ligated with 7-0 Prolene suture. Pre- and postoperative analgesia was administered (0.03 mg/kg s.c. Buprenex) as recommended by veterinarians of the Unit for Laboratory Animal Medicine, and all surgical procedures were approved by the Care and Use Committee of the University of Michigan. Electrocardiogram monitoring and the appearance of myocardial pallor were used to verify successful coronary artery occlusion. Significant S-T segment elevation was routinely observed after LCX ligation. The thoracotomy was closed (4-0 Vicryl suture; Ethicon, Somerville, NJ, USA) after placement of a chest tube (16-gauge Angiocath), and muscle layers were sutured in multiple layers. A subcuticular suture (4-0 monocryl; Ethicon) was used, and the incision was closed with tissue adhesive (Indermil; Covidien, Mansfield, MA, USA). Echocardiography (HP Sonos 5500; Hewlett-Packard, Palo Alto, CA, USA) was performed before and after surgery to verify myocardial injury and to measure ejection fraction. Animals were euthanized and cardiac myocytes were enzymatically isolated when the ejection fraction was less than 50% (Fig. 6B).
Figure 2.
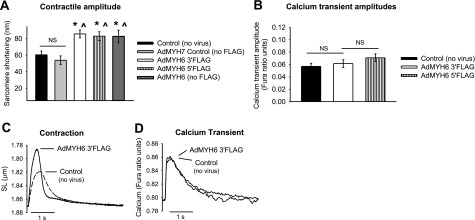
Functional effects of AdMYH6 gene transfer on healthy rabbit cardiac myocytes. AdMYH6 gene transfer potentiates sarcomere shortening, whereas the calcium transient is unaltered. All experiments were performed 48 h after gene transfer. A) Summary of AdMYH6 effects on contraction amplitude. AdMYH7 control values did not differ from nontransduced control values (53.9±5.23 nm, n=18 vs. 60.5±4.5 nm, n=74). AdMYH6 increased contraction amplitude regardless of the presence or location of the FLAG epitope. AdMYH6 3′FLAG amplitude = 85.8 ± 4.8 nm, n = 86; AdMYH6 5′FLAG amplitude = 83.1 ± 5.23 nm, n = 37; AdMYH6 no FLAG amplitude = 82.3 ± 7.7 nm, n = 19. B) Calcium transient amplitudes were the same between nontransduced control (0.057±0.005, n=38), AdMYH6 3′FLAG-treated (0.062±0.006, n=34), and AdMYH6 5′FLAG-treated (0.069±0.006, n=32) cardiac myocytes. C, D) Representative recordings of sarcomere length (SL) shortening (C) and intracellular calcium transients (D). *P < 0.05 vs. control (no virus); ∧P < 0.05 vs. AdMYH7 control; ANOVA with Tukey post hoc between-group comparisons.
Figure 3.
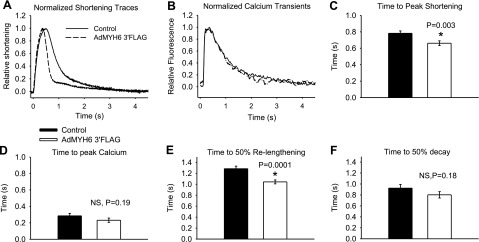
α-MyHC gene transfer speeds healthy rabbit myocyte shortening and relengthening kinetics independent of the calcium transient dynamics. A) Normalized sarcomere length-shortening traces show that AdMYH6 gene transfer sped sarcomere shortening and relengthening. B) Normalized intracellular calcium transient traces show that the kinetics of calcium release and decay were unaffected by AdMYH6 gene transfer. C) Time to peak of sarcomere shortening was faster in AdMYH6-transduced myocytes (0.66±0.03 s, n=86) than in nontransduced myocytes (0.78±0.03 s, n=74). D) Time to peak of the calcium transient, however, did not differ between the two groups (0.28±0.03 s, n=38; 0.23±0.03 s, n=34). E) Time to 50% relaxation was reduced by AdMYH6 gene transfer compared with that in nontransduced control myocytes. F) Time to 50% decay of the calcium transient was not affected. All values are means ± se. *P < 0.05 vs. control, two-tailed t test. Actual P values are shown in figure.
Figure 4.

AdMYH6 gene transfer speeds the maximal rates of sarcomere shortening and relengthening in healthy β-MyHC-dominant rabbit myocytes. A) Comparison of maximal rate of sarcomere shortening in control and AdMYH6-treated myocytes. B) Comparison of maximal rate of sarcomere relengthening in control (n=74) and AdMYH6-treated myocytes (n=86). *P < 0.05; unpaired t test.
Figure 5.
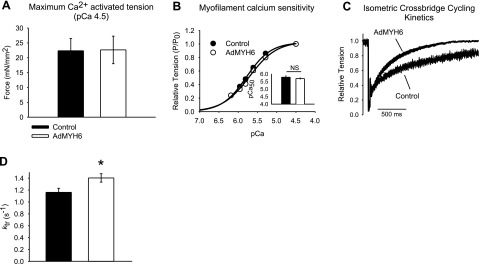
AdMYH6 effects on rabbit cardiac myocyte maximal calcium-activated (pCa 4.5) isometric contractile function. A) Maximal isometric tension generation of skinned (1% Triton X-100) cardiac myocytes was not affected by AdMYH6 gene transfer. B) Myofilament calcium sensitivity did not differ between control (•) and AdMYH6-treated (○) myocytes. P, tension development; P0, maximal tension development; P/P0, relative tension. C, D) Maximal rate of isometric cross-bridge cycling was faster in AdMYH6-treated cardiac myocytes vs. control myocytes (1.40±0.07 s−1, n=16 vs. 1.16±0.07 s−1, n=16). All values are means ± se. *P < 0.05 vs. control; two-tailed t test.
Figure 6.
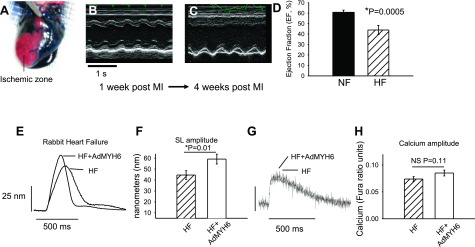
α-MyHC gene transfer increases the contractility of failing rabbit cardiac myocytes. A) An ischemic model of heart failure was implemented in adult rabbits. Successful coronary occlusion was confirmed by retrograde perfusion of Evans Blue dye. B, C) Representative m-mode echocardiography images of animals with a nonfailing heart (NF; B) and heart failure (HF; C) show that cardiac function was severely depressed after chronic LCX occlusion. D) Ventricular ejection fraction was depressed in HF vs. NF animals (43.9±2.2%, n=4 vs. 60.6±1.2%, n=4). E) Representative sarcomere length (SL) shortening measurements in HF cardiac myocytes 2 d after in vitro gene transfer with AdMYH6 (HF+AdMYH6) or with no virus treatment (HF). F) Summary data show that α-MyHC gene transfer increased SL shortening amplitude in electrically paced (1 Hz) single myocytes. HF amplitude = 44.6 ± 4.0 nm, n = 59; HF + AdMYH6 amplitude = 59.2 ± 4.5 nm, n = 50. G) Representative calcium transient recordings show no effect of AdMYH6 gene transfer on intracellular calcium cycling. H) Summary data show that the intracellular calcium transient amplitude was not significantly affected by α-MyHC gene transfer. HF amplitude = 0.074 ± 0.004, n = 53; HF + AdMYH6 amplitude = 0.085 ± 0.005 ratio units, n = 48.
Single cardiac myocyte functional analysis
Myocyte function was measured 48 h after in vitro gene transfer for all experiments. Sarcomere shortening and intracellular calcium concentrations were monitored simultaneously as before, using electrically stimulated preparations (25). In brief, coverslips of cardiac myocytes were loaded with Fura-2AM (2 μM, 10 min, room temperature) to monitor intracellular calcium transients. Fura-2 fluorescence was measured using an IonOptix spectrophotometer (stepper switch; IonOptix, Milton, MA, USA). Initially Fura was excited by 360-nm light (the Fura isosbestic point, a calcium-independent measure of Fura fluorescence) and then continuously with 380-nm light (calcium-dependent measure of Fura fluorescence). Emitted fluorescence (510 nm) was detected by a photomultiplier tube. Sarcomere shortening was collected (240 Hz) by real-time fast Fourier transform of the myocyte video signal using commercially available data acquisition software (IonOptix). Experiments were performed on a heated platform (37°C, PM Series; Warner Instruments, Hamden, CT, USA) on the stage of an inverted microscope (Nikon, Tokyo, Japan), and myocytes were visualized using an oil-immersion objective (S Fluor ×40, 1.3 NA). Field stimulation (40 V, 0.2 or 1 Hz) was accomplished using the MyoPacer (IonOptix). In all experiments, myocytes were kept in M199 medium (1.8 mM Ca2+; Sigma-Aldrich Corp.). Typical recordings of intracellular calcium transients and contraction are shown in Fig. 2. Only myocytes with a resting sarcomere length ≥1.75 μm that followed the field stimulation protocol 1:1 were used for this study. The average resting sarcomere length for healthy rabbit cells used here appears in Table 1.
TABLE 1.
Diastolic sarcomere lengths of healthy rabbit cardiac myocytes used for functional studies
| Treatment | Resting sarcomere length (μm) |
|---|---|
| Control (no virus) | 1.87 ± 0.05 (n=74) |
| AdMYH7 control (no FLAG) | 1.84 ± 0.07 (n=43) |
| AdMYH6 3′FLAG | 1.86 ± 0.05 (n=86) |
| AdMYH6 5′FLAG | 1.87 ± 0.04 (n=37) |
| AdMYH6 (no FLAG) | 1.81 ± 0.06 (n=21) |
No difference was found between any of the groups.
Myofilament and myosin function was more directly assessed using permeabilized myocyte preparations (1% Triton X-100), and calcium sensitivity and actomyosin kinetics were measured as before (25, 34). Typical force recordings (taken 48 h after gene transfer) and isometric cross-bridge cycling measurement are shown in Fig. 5.
Statistical analysis
All values are expressed as means ± se. We compared continuous variables by unpaired t tests or 1-way ANOVA with Tukey post hoc between-group comparisons. Paired t tests were used to compare in vivo ejection fractions before and after chronic LCX ligation.
RESULTS
AdMYH6 gene transfer and protein expression
A recombinant adenoviral vector (AdMYH6) was constructed for highly efficient gene transfer of the full-length human MYH6 gene to adult cardiac myocytes in vitro. MYH6 gene expression was driven by the cytomegalovirus promoter, and a FLAG epitope was cloned into the 3′ end of the gene to facilitate α-MyHC motor protein expression analysis. As outlined in Fig. 1A, AdMYH6 was used to express α-MyHC in rabbit and human adult cardiac myocytes that predominantly express β-MyHC. Adult rabbit ventricular cardiac myocytes were used here because it is well established that, similar to human hearts, they express predominantly β-MyHC with α-MyHC, making up none or a very small component of the total myosin protein (35). After gene transfer, α-MyHC protein expression was detected by Western blot using an antibody directed against the FLAG epitope (Fig. 1B). Notably, total myosin protein content remained unchanged as α-MyHC protein expression increased over 48 h after transduction, suggesting that the endogenous β-MyHC was stoichiometrically replaced by α-MyHC (Fig. 1B). By densitometry analysis of exposed film, α-MyHC accounted for ∼21% of the total myosin 24 h after adenoviral gene transfer and ∼30% of the total myosin after 48 h. The efficiency of gene transfer was near 100% (Fig. 1C), and α-MyHC protein was incorporated homogenously across the entire length and width of the cardiac myocytes, specifically in the sarcomere A band (Fig. 1D; green) between the Z bands (Fig. 1D; red). Similar α-MyHC expression results were obtained using AdMYH6 in which the FLAG epitope was cloned at the N-terminal end of the myosin motor.
AdMYH6 functional effects in healthy β-MyHC-dominant rabbit ventricular myocytes
The functional effects of AdMYH6 gene transfer on single cardiac myocyte contractile function and intracellular calcium transients were determined using electrical field stimulation (0.2 Hz). Contraction amplitude of healthy rabbit adult cardiac myocytes was significantly increased (P<0.05) by α-MyHC gene transfer in the absence of any significant change in the intracellular calcium transient amplitude (Fig. 2). In control experiments the AdMYH7 virus had no effect on myocyte contractility, similar to a previous report (25). The presence of the FLAG tag epitope fused to the α-MyHC protein did not affect the positive inotropic effects of α-MyHC gene transfer (Fig. 2A, B). Normalization of contraction (Fig. 3A) and calcium (Fig. 3B) traces shows that AdMYH6 gene transfer sped the kinetics of contraction and myocyte relaxation without concomitant effects on the kinetics of the calcium transient (Fig. 3). Kinetic data were calculated relative to the time of electrical stimulation. AdMYH6-transduced myocytes reached the peak of contraction sooner than nontransduced cells (Fig. 3C), but the time to the peak of the calcium transient was not different (Fig. 3D). In addition, the time to 50% sarcomere relengthening, an index of cellular diastolic function, was significantly abbreviated in AdMYH6 myocytes (Fig. 3E), but the time to 50% decay of the calcium transient was unaffected. Likewise, the myocyte shortening and relengthening velocities (Fig. 4) were faster in AdMYH6-transduced myocytes.
AdMYH6 effects on healthy rabbit cardiac myofilament tension development and calcium sensitivity
Next we sought to determine the subcellular mechanism responsible for the augmentation of myocyte function caused by AdMYH6 gene transfer. Myofilament function was assessed to determine whether AdMYH6 gene transfer affected total tension-generating capacity and/or myofilament calcium sensitivity of isometric tension development. To do this we used skinned myocyte preparations in which the cell membrane was permeabilized with 1% Triton X-100 as described previously (11, 25). This preparation allows for direct measurement of myofilament and myosin function. The calcium sensitivity of isometric force was determined using solutions with free calcium ranging from pCa 9.0 to pCa 4.5. Maximal calcium-activated tension (pCa 4.5) did not differ between control and AdMYH6 3′FLAG-transduced myocytes (Fig. 5A). Calcium sensitivity of tension did not differ between groups (Fig. 5B).
AdMYH6 effects on myofilament tension development kinetics
To further explore the mechanism of calcium-independent positive inotropy by AdMYH6 we directly measured the kinetic properties of the myofilaments of control and AdMYH6-transduced myocytes by measuring the kinetics of isometric tension redevelopment (ktr) after a rapid slack restretch maneuver described previously (25, 36, 37). In these experiments, conducted during maximal calcium activation (pCa 4.5), skinned cardiac myocytes were rapidly shortened and restretched to the original isometric length. After this mechanical maneuver tension initially falls to zero because of the transition of myosin heads from being strongly bound (force-bearing) to actin to being weakly bound (non-force-bearing) to actin. The kinetics of tension redevelopment after this maneuver is an index of the rate at which myosin heads can transition from non-force-bearing to force-bearing states (36). Isometric cross-bridge cycling was significantly faster in AdMYH6-treated myocytes than in control myocytes (Fig. 5C, D).
AdMYH6 effects on the contractile function of failing rabbit cardiac myocytes
The promising results obtained in healthy rabbit cardiac ventricular myocytes prompted us to next test whether fast α-MyHC motor gene transfer would potentiate contractility of failing cardiac myocytes. First this hypothesis was tested using a rabbit model of ischemic heart failure (3, 38). Ischemic heart failure was induced surgically by chronic coronary artery ligation. Successful ligation of the LCX was confirmed by retrograde perfusion through the aorta using Evans Blue dye (Sigma-Aldrich Corp.) (Fig. 6A). In this assay, perfused regions of the heart become blue and the ischemic or nonperfused area remains pink. Cardiac function was evaluated in vivo by echocardiography of animals after the thoracotomy and LCX ligation. Compared with nonfailing rabbit hearts (Fig. 6B), the left ventricle of animals with heart failure was akinetic (Fig. 6C), and the ejection fraction was significantly depressed (Fig. 6D) by 3 wk after surgery. The heart failure phenotype was accompanied by significant left ventricular remodeling after LCX ligation and myocyte contractility was significantly attenuated in acutely isolated myocytes as reported by others (39). Cardiac myocytes from failing hearts were isolated and again in vitro gene transfer was performed using AdMYH6. α-MyHC gene transfer increased the contraction amplitude of electrically paced failing myocytes (1 Hz, P<0.01) (Fig. 6E, F) without affecting the intracellular calcium transient amplitude (Fig. 6G, H). In control experiments, gene transfer of the full-length human β-MyHC using AdMYH7 did not affect the contraction of failing myocytes, similar to healthy rabbit myocytes (Fig. 2) and as reported previously (25).
AdMYH6 effects on human failing myocytes
To test whether results from animal studies could translate to human myocytes, we next used AdMYH6 to treat ventricular myocytes from the explanted failing heart of a 69-yr-old male patient with ischemic cardiomyopathy and heart failure. Tissue was collected at the time of cardiac transplantation (University of Michigan Health Systems) and transported to the laboratory on ice-cold cardioplegia. In vitro gene transfer was performed as above, and AdGFP was used as a control viral vector. Human myocytes were maintained in primary culture, and functional analysis was performed 48 h after gene transfer. Successful viral transduction using AdGFP was confirmed by epifluorescent imaging of adenovirus-treated human myocytes. Figure 7A shows robust green fluorescent protein (GFP) expression 48 h after gene transfer. Notably, the human myocytes maintain their rod-shaped morphology over this course of time in primary culture (Fig. 7A). In functional assays, human cardiac myocytes were studied 48 h after gene transfer. Sarcomere shortening and relengthening were recorded in electrically stimulated (40 V, 1 Hz) myocytes. Compared with untreated heart failure myocytes and control heart failure myocytes treated with AdGFP, AdMYH6 gene transfer enhanced the contractile amplitude of these failing human cardiac myocytes (P<0.003) (Fig. 7B, C).
Figure 7.
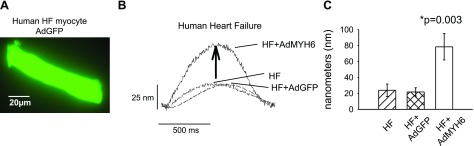
AdMYH6 gene transfer augments failing human cardiac myocyte contraction. A) Fluorescent image of a single human cardiac myocyte demonstrates successful adenovirus-mediated gene transfer of GFP. Image was taken 48 h after gene transfer. B) Representative sarcomere length shortening measurements in human failing ventricular cardiac myocytes. α-MyHC gene transfer enhanced the contractile amplitude of failing human cardiac myocytes paced at 1 Hz. C) Summary of data. HF amplitude = 23.9 ± 7.8 nm, n = 10; HF+AdGFP amplitude = 21.8 ± 5.2nm, n = 5; HF + AdMYH6 amplitude = 78.4 ± 16.5 nm, n = 6. All values are means ± se. *P < 0.05 vs. control, two-tailed t test. Actual P values are shown in figure. Human myocyte data were analyzed by 1-way ANOVA as there were 3 groups to compare: HF, HF + AdGFP, and HF + AdMYH6.
DISCUSSION
We have used here, for the first time to our knowledge, a recombinant adenovirus (AdMYH6) to deliver and express the full-length human α-MyHC gene in β-MyHC-dominant ventricular myocytes (Fig. 1). In healthy rabbit myocytes, AdMYH6 gene transfer increased the contraction amplitude and kinetics of electrically stimulated myocytes with no effect on the intracellular calcium transient amplitude (Fig. 2). At the myofilament level, AdMYH6 gene transfer did not affect total tension development or the calcium sensitivity of tension development (Fig. 5A, B). Rather, AdMYH6 gene transfer sped the rate of cross-bridge cycling (Fig. 5C, D) and thus sped the kinetics of intact myocyte shortening and relengthening (Figs. 3 and 4). α-MyHC gene transfer also increased the contraction amplitude of failing cardiac myocytes isolated from rabbits with ischemic heart failure (Fig. 6) and from an ischemic failing human heart (Fig. 7). This study represents the first report in which α-MyHC gene transfer was used as an in vitro therapeutic strategy to augment failing cardiac myocyte contractility.
Myosin isoforms and heart failure
There is much interest in the significance of MyHC isoform expression changes in human heart failure (40). α-MyHC accounts for ∼10% of the total cardiac myosin in normal healthy human hearts with β-MyHC making up the remaining 90% (15, 17). α-MyHC expression decreases to undetectable levels in the failing human cardiac ventricle (1, 16, 17) and with recovery of left ventricular function the α-MyHC expression returns to normal levels (28). It is a common suggestion that the disappearance of α-MyHC in the failing heart is nature’s compensatory mechanism aimed to increase myocardial energy efficiency (22). α-MyHC is reportedly a less energetically efficient motor protein than β-MyHC (41, 42), so loss of α-MyHC in an energy-starved failing heart could be considered an energy-sparing mechanism (42). Consistent with other reports, our data suggest that moderate levels of α-MyHC actually enhance cardiac function (22, 23, 43), perhaps at a higher energetic cost. Using α-MyHC transgenic rabbits, Robbins and colleagues reported that under severe stress, α-MyHC expression may be cardioprotective and β-MyHC is detrimental (22). In heart failure the loss of the faster, more powerful α-MyHC motor is predicted to be detrimental to cardiac performance because the rates of force development and relaxation would be slowed, twitch force would be reduced, and whole-heart power output would be lower (15, 20, 25, 37, 43).
Calcium-independent positive inotropy by α-MyHC gene transfer
Here we report calcium-independent positive inotropy by α-MyHC gene transfer in β-MyHC-dominant myocytes. This finding is consistent with a previous report showing calcium-independent negative inotropy by β-MyHC gene transfer in α-MyHC-dominant myocytes (25). Motor protein gene transfer does not affect myofilament Ca2+ sensitivity (25). This finding is consistent with in vitro motility assays in which myosin isoforms were compared (44) and in single cardiac myocytes in which myosin isoform expression was manipulated by thyroid hormone (15, 19). Rather, the mechanism of calcium-independent inotropy by myosin isoform shifts lies in the well known kinetic differences of ATP hydrolysis and cross-bridge cycling for each myosin isoform. α-MyHC has markedly higher ATPase activity, faster shortening velocity, and thus faster rates of force development than β-MyHC (41, 45,46,47). Thus, the faster kinetics of the weak to strong cross-bridge transition due to the expression of the faster α-MyHC motor (Fig. 5C, D) provides a mechanism to explain the calcium-independent augmentation of contractile performance observed here in membrane-intact cardiac myocytes. Moreover, recent mathematical model predictions of the kinetics of an isometric twitch demonstrate that cardiac muscle with greater expression of α-MyHC has greater peak force production in response to a fixed Ca2+ transient (15). At the whole organ level, modeling also indicates that small increases of α-MyHC would also accelerate the rate of ventricular pressure development (dP/dt) and facilitate ejection of blood and also diastolic filling (−dP/dt) in response to a limited-duration Ca2+ transient (15). Experimental results reported here are in good agreement with this mathematical model of calcium-independent myosin effects on twitch dynamics. Augmentation of cardiac contractility in vivo by fast α-MyHC motor gene transfer by the same calcium-independent mechanism is likely but awaits further experimentation.
How much α-MyHC is required to augment cardiac contractility?
It was originally believed that the human cardiac ventricles normally expressed 100% β-MyHC, thus precluding any isoform shifts in the failing heart (48). Therefore, myosin isoform shifts were not thought to contribute to contractile dysfunction in heart failure. More recent work has shown that α-MyHC accounts for ∼10% of the total ventricular myosin in normal adult human hearts (17) (range from 7 to 13%), and this amount disappears in the failing heart in which 100% β-MyHC is found. In addition, it was reported that α-MyHC mRNA accounts for 25–35% of the total myosin mRNA in normal human hearts and decreases to only 2% in end-stage failing hearts (16, 18). Since these seminal reports, a long-standing question has been whether these small shifts in myosin isoform expression are sufficient to cause cardiac dysfunction. A collection of studies suggest that cardiac muscle performance is sensitive to very small changes of α-MyHC expression. Increasing α-MyHC by just 12% is sufficient to increase single cardiac myocyte power output by ∼50% (43). In transgenic rabbits that express 15% α-MyHC, ventricular papillary muscles reportedly generate 20% greater peak power output than nontransgenic samples expressing 100% β-MyHC (49). Finally, mathematical modeling predicts that just 10% α-MyHC (90% β-MyHC) can enhance myocardial twitch kinetics and amplitude independent of the calcium transient (15). Here we report that increasing α-MyHC content to 30% by gene transfer significantly enhances single intact cardiac myocyte contraction. Collectively, data suggest that small shifts in α-MyHC can indeed affect cardiac function.
Taken together, our results support the application of MYH6 gene transfer as a new mechanism to increase the contractility of failing human cardiac myocytes without obligate alterations in the intracellular calcium transient. Because most therapies for the diseased heart target modifications in intracellular calcium handling, the advent of cardiac molecular motor engineering, either directly or indirectly via potential microRNA strategies (50), may offer a unique therapeutic strategy for the failing heart.
References
- McKinsey T. A., Olson E. N. Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest. 2005;115:538–546. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone S. V., Kaplinsky E. J. Calcium sensitizer agents: a new class of inotropic agents in the treatment of decompensated heart failure. Int J Cardiol. 2005;103:248–255. doi: 10.1016/j.ijcard.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Cohn J. N., Goldstein S. O., Greenberg B. H., Lorell B. H., Bourge R. C., Jaski B. E., Gottlieb S. O., McGrew F., 3rd, DeMets D. L., White B. G. A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure: Vesnarinone Trial Investigators. N Engl J Med. 1998;339:1810–1816. doi: 10.1056/NEJM199812173392503. [DOI] [PubMed] [Google Scholar]
- Antoniades C., Tousoulis D., Koumallos N., Marinou K., Stefanadis C. Levosimendan: beyond its simple inotropic effect in heart failure. Pharmacol Ther. 2007;114:184–197. doi: 10.1016/j.pharmthera.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Cleland J. G. F., Freemantle N., Coletta A. P., Clark A. L. Clinical trials update from the American Heart Association: REPAIR-AMI, ASTAMI, JELIS, MEGA, REVIVE-II, SURVIVE, and PROACTIVE. Eur J Heart Fail. 2006;8:105–110. doi: 10.1016/j.ejheart.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Del Monte F., Harding S. E., Schmidt U., Matsui T., Kang Z. B., Dec G. W., Gwathmey J. K., Rosenzweig A., Hajjar R. J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Monte F., Harding S. E., Dec G. W., Gwathmey J. K., Hajjar R. J. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross H. R., Kranias E. G., Murphy E., Steenbergen C. Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol Heart Circ Physiol. 2003;284:H683–H690. doi: 10.1152/ajpheart.00567.2002. [DOI] [PubMed] [Google Scholar]
- Fitzsimons D. P., Patel J. R., Moss R. L. Aging-dependent depression in the kinetics of force development in rat skinned myocardium. Am J Physiol Heart Circ Physiol. 1999;276:H1511–H1519. doi: 10.1152/ajpheart.1999.276.5.H1511. [DOI] [PubMed] [Google Scholar]
- Mercadier J. J., Lompre A. M., Wisnewsky C., Samuel J. L., Bercovici J., Swynghedauw B., Schwartz K. Myosin isoenzymic changes in several models of rat cardiac hypertrophy. Circ Res. 1981;49:525–532. doi: 10.1161/01.res.49.2.525. [DOI] [PubMed] [Google Scholar]
- Metzger J. M., Wahr P. A., Michele D. E., Albayya F., Westfall M. V. Effects of myosin heavy chain isoform switching on Ca2+-activated tension development in single adult cardiac myocytes. Circ Res. 1999;84:1310–1317. doi: 10.1161/01.res.84.11.1310. [DOI] [PubMed] [Google Scholar]
- Rundell V. L. M., Geenen D. L., Buttrick P. M., de Tombe P. P. Depressed cardiac tension cost in experimental diabetes is due to altered myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol. 2004;287:H408–H413. doi: 10.1152/ajpheart.00049.2004. [DOI] [PubMed] [Google Scholar]
- Rundell V. L. M., Manaves V., Martin A. F., de Tombe P. P. Impact of β-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005;288:H896–H903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- Krenz M., Robbins J. Impact of β-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- Locher M. R., Razumova M. V., Stelzer J. E., Norman H. S., Patel J. R., Moss R. L. Determination of rate constants for turnover of myosin isoforms in rat myocardium: implications for in vivo contractile kinetics. Am J Physiol Heart Circ Physiol. 2009;297:H247–H256. doi: 10.1152/ajpheart.00922.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes B. D., Minobe W., Abraham W. T., Rizeq M. N., Bohlmeyer T. J., Quaife R. A., Roden R. L., Dutcher D. L., Robertson A. D., Voelkel N. F., Badesch D. B., Groves B. M., Gilbert E. M., Bristow M. R. Changes in gene expression in the intact human heart: downregulation of α-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315–2324. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S., Minobe W., Bristow M. R., Leinwand L. A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- Nakao K., Minobe W., Roden R., Bristow M. R., Leinwand L. A. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–2370. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons D. P., Patel J. R., Moss R. L. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol (Lond) 1998;513:171–183. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron T. J., Korte F. S., McDonald K. S. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol. 2001;281:H1217–H1222.l. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- Korte F. S., Herron T. J., Rovetto M. J., McDonald K. S. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol. 2005;289:H801–H812. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- James J., Martin L., Krenz M., Quatman C., Jones F., Klevitsky R., Gulick J., Robbins J. Forced expression of α-myosin heavy chain in the rabbit ventricle results in cardioprotection under cardiomyopathic conditions. Circulation. 2005;111:2339–2346. doi: 10.1161/01.CIR.0000164233.09448.B1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff J. C., Hewett T. E., Factor S. M., Vikstrom K. L., Robbins J., Leinwand L. A. Expression of the beta (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. Am J Physiol Heart Circ Physiol. 2000;278:H412–H419. doi: 10.1152/ajpheart.2000.278.2.H412. [DOI] [PubMed] [Google Scholar]
- Michele D. E., Metzger J. M. Contractile dysfunction in hypertrophic cardiomyopathy: elucidating primary defects of mutant contractile proteins by gene transfer. Trends Cardiovasc Med. 2000;10:177–182. doi: 10.1016/s1050-1738(00)00067-0. [DOI] [PubMed] [Google Scholar]
- Herron T. J., Vandenboom R., Fomicheva E., Mundada L., Edwards T., Metzger J. M. Calcium-independent negative inotropy by β-myosin heavy chain gene transfer in cardiac myocytes. Circ Res. 2007;100:1182–1190. doi: 10.1161/01.RES.0000264102.00706.4e. [DOI] [PubMed] [Google Scholar]
- Solaro J. R. CK-1827452, a sarcomere-directed cardiac myosin activator for acute and chronic heart disease. IDrugs. 2009;12:243–251. [PubMed] [Google Scholar]
- Teerlink J. A novel approach to improve cardiac performance: cardiac myosin activators: [E-pub ahead of print] Heart Fail Rev. 2009 doi: 10.1007/s10741-009-9135-0. [E-pub ahead of print] doi:10.1007/s10741-009-9135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes B. D., Gilbert E. M., Abraham W. T., Minobe W. A., Larrabee P., Ferguson D., Wolfel E. E., Lindenfeld J., Tsvetkova T., Robertson A. D., Quaife R. A., Bristow M. R. Myocardial gene expression in dilated cardiomyopathy treated with β-blocking agents. N Engl J Med. 2002;346:1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- Butter C., Rastogi S., Minden H. H., Meyhöfer J., Burkhoff D., Sabbah H. N. Cardiac contractility modulation electrical signals improve myocardial gene expression in patients with heart failure. J Am Coll Cardiol. 2008;51:1784–1789. doi: 10.1016/j.jacc.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Rastogi S., Mishra S., Gupta R., Sabbah H. Reversal of maladaptive gene program in left ventricular myocardium of dogs with heart failure following long-term therapy with the acorn cardiac support device. Heart Fail Rev. 2005;10:157–163. doi: 10.1007/s10741-005-4643-z. [DOI] [PubMed] [Google Scholar]
- Day S. M., Westfall M. V., Fomicheva E. V., Hoyer K., Yasuda S., Cross N. C. L., D'Alecy L. G., Ingwall J. S., Metzger J. M. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med. 2006;12:181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- Mahaffey K. W., Raya T. E., Pennock G. D., Morkin E., Goldman S. Left ventricular performance and remodeling in rabbits after myocardial infarction: effects of a thyroid hormone analogue. Circulation. 1995;91:794–801. doi: 10.1161/01.cir.91.3.794. [DOI] [PubMed] [Google Scholar]
- Maurice J. P., Shah A. S., Kypson A. P., Hata J. A., White D. C., Glower D. D., Koch W. J. Molecular β-adrenergic signaling abnormalities in failing rabbit hearts after infarction. Am J Physiol Heart Circ Physiol. 1999;276:H1853–H1860. doi: 10.1152/ajpheart.1999.276.6.H1853. [DOI] [PubMed] [Google Scholar]
- Metzger J. M. Myosin binding-induced cooperative activation of the thin filament in cardiac myocytes and skeletal muscle fibers. Biophys J. 1995;68:1430–1442. doi: 10.1016/S0006-3495(95)80316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litten R. Z., Martin B. J., Buchthal R. H., Nagai R., Low R. B., Alpert N. R. Heterogeneity of myosin isozyme content of rabbit heart. Circ Res. 1985;57:406–414. doi: 10.1161/01.res.57.3.406. [DOI] [PubMed] [Google Scholar]
- Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci U S A. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S., Kentish J. C. Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and relaxation in rat and guinea pig skinned trabeculae. Circ Res. 1998;83:179–186. doi: 10.1161/01.res.83.2.179. [DOI] [PubMed] [Google Scholar]
- Maurice J. P., Shah A. S., Kypson A. P., Hata J. A., White D. C., Glower D. D., Koch W. J. Molecular β-adrenergic signaling abnormalities in failing rabbit hearts after infarction. Am J Physiol Heart Circ Physiol. 1999;276:H1853–H1860. doi: 10.1152/ajpheart.1999.276.6.H1853. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G., Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- Marian A. J. On mice, rabbits, and human heart failure. Circulation. 2005;111:2276–2279. doi: 10.1161/01.CIR.0000167559.13502.9A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubarsch C., Goulette R. P., Litten R. Z., Martin B. J., Mulieri L. A., Alpert N. R. The economy of isometric force development, myosin isoenzyme pattern and myofibrillar ATPase activity in normal and hypothyroid rat myocardium. Circ Res. 1985;56:78–86. doi: 10.1161/01.res.56.1.78. [DOI] [PubMed] [Google Scholar]
- Sugiura S., Kobayakawa N., Fujita H., Yamashita H., Momomura S. I., Chaen S., Omata M., Sugi H. Comparison of unitary displacements and forces between 2 cardiac myosin isoforms by the optical trap technique : molecular basis for cardiac adaptation. Circ Res. 1998;82:1029–1034. doi: 10.1161/01.res.82.10.1029. [DOI] [PubMed] [Google Scholar]
- Herron T. J., McDonald K. S. Small amounts of α-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res. 2002;90:1150–1152. doi: 10.1161/01.res.0000022879.57270.11. [DOI] [PubMed] [Google Scholar]
- Schoffstall B., Brunet N. M., Williams S., Miller V. F., Barnes A. T., Wang F., Compton L. A., McFadden L. A., Taylor D. W., Seavy M., Dhanarajan R., Chase P. B. Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform. J Physiol (Lond) 2006;577:935–944. doi: 10.1113/jphysiol.2006.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D. E., Work S. S., Wright R. K., Alpert N. R., Warshaw D. M. Smooth, cardiac, and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11–19. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- Hoh J. F. Y., McGrath P. A., Hale P. T. Electrophoretic analysis of multiple forms of rat cardiac myosin: Effects of hypophysectomy and thyroxine replacement. J Mol Cell Cardiol. 1978;10:1053–1060. doi: 10.1016/0022-2828(78)90401-7. [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Reggiani C. G. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- Gorza L., Mercadier J. J., Schwartz K., Thornell L. E., Sartore S., Schiaffino S. Myosin types in the human heart: an immunofluorescence study of normal and hypertrophied atrial and ventricular myocardium. Circ Res. 1984;54:694–702. doi: 10.1161/01.res.54.6.694. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Palmer B. M., James J., Wang Y., Chen Z., VanBuren P., Maughan D. W., Robbins J., LeWinter M. M. Effects of cardiac myosin isoform variation on myofilament function and crossbridge kinetics in transgenic rabbits. Circ Heart Fail. 2009;2:334–341. doi: 10.1161/CIRCHEARTFAILURE.108.802298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooij E., Sutherland L. B., Qi X., Richardson J. A., Hill J., Olson E. N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]