

Abstract
Reactive oxygen species (ROS) have been implicated in the activation of protein kinases, DNA damage responses, and cell apoptosis. The details of how ROS regulate these intracellular biochemical and genetic processes remain to be fully understood. By establishing transformed bronchial epithelial cells through chronic low-dose arsenic treatment, we showed that the capacity of ROS generation induced by arsenic is substantially reduced in the transformed cells relative to the nontransformed cells. Such a reduction in ROS generation endows cells with premalignant features, including rapid growth, resistance to arsenic toxicity, and increased colony formation of the transformed cells. To validate these observations, the capability of ROS generation was restored in the transformed cells by treatment with inhibitors or siRNAs to silence the function of superoxide dismutase (SOD) or catalase and cell growth was determined following these treatments. Enhancement in ROS generation suppressed cell growth and colony formation of the transformed cells significantly. Despite the fact that the transformed cells showed a decreased expression of NF-κB signaling proteins IKKβ and IKKγ, the proteolytic processing p105 and p100 and NF-κB DNA binding activity were elevated in these cells. Increasing ROS generation by silencing SOD and catalase reduced the DNA binding activity of NF-κB in the transformed cells. Taken together, the transformed cells induced by arsenic exhibited a decrease in ROS generation, which is responsible for the enhanced cell growth and colony formation of the transformed cells, most likely through a sustained alternative activation of the NF-κB transcription factor.
Introduction
Environmental or occupational exposure to arsenic has been a worldwide health problem for several decades (1). In certain geographic locations, abundant arsenic can be found in the Earth’s crust, rock, soil, water, and air (2). Occupational arsenic exposure mainly occurred in the industries of mining, manufacturing, wood preservation, and agriculture (3). Emerging evidence clearly indicates that chronic exposure to arsenic or arsenic-containing compounds causes cancers of skin, bladder, liver, and kidney (4). The toxicity or carcinogenicity of arsenic varies depending on the oxidation state and chemical form of arsenic or arsenic compound. It is believed that the inorganic form of arsenic, especially inorganic trivalent arsenic (As3+), is more carcinogenic and reactive with thiol-containing molecules. The primary routes of human exposure to arsenic are inhalation, ingestion, and dermal contact. Despite extensive efforts in determining how As3+ causes malignant transformation of the normal cells in a number of experimental systems, the carcinogenic mechanism of As3+ remains poorly understood. Several important intracellular signaling pathways can be activated or regulated by As3+ exposure, such as NF-κB (5), AP-1 (6), p53 (7), Notch (8), GADD45a (9), Akt (10), PKC (11), and PML (12), which can contribute to cell growth, transformation, oncogene expression, cell apoptosis, and vasculation.
The effect of arsenic is largely achieved through both direct binding of arsenic to proteins and induction of reactive oxygen species (ROS) during the oxidation-reduction reaction of the trivalent arsenic and pentavalent arsenic compounds. Due to its similarities in atom structure and chemical characteristics with phosphorus, As5+ is able to substitute for phosphate and interfere with the posttranslational modification and function of intracellular proteins. On the other hand, As3+ is capable of binding to the thiol group of the proteins important for cellular metabolism and signaling. The most important role of As3+ in cellular biochemical and biological responses is its ability in ROS induction. Evidence supporting this notion is from studies showing induction of ROS in cells derived from either normal tissues or tumors (13). Although mitochondria have been implicated as a potential source of ROS in response to As3+ (14), accumulating data suggest that NADPH oxidase may be the primary source for the generation of superoxide anion (O2•−; refs. 15, 16). As3+ not only induces expression of the NADPH oxidase components including p47, p67, p91, and several scaffolding protein for the assembly of this complex (15) but also stimulates the enzyme activity of the NADPH oxidase by inducing phosphorylation and translocation of p47 (17). It has been generally viewed that ROS are the key mediators for As3+-induced carcinogenesis through oxidative stress and genetic mutation. Paradoxically, generation of ROS is also the one of the central mechanisms to the desired tumor cell apoptosis induced by As3+ and some chemotherapeutic reagents. In the present study, we provide evidence suggesting that the capacity of ROS generation is severely compromised in the As3+-induced transformed cells. Reduction in ROS generation endows the transformed cells resistance to As3+ cytotoxicity, rapid proliferation, anchorage-independent growth, and a sustained activation of the NF-κB transcription factor.
Materials and Methods
Reagents and cell culture
Arsenic trichloride (AsCl3) was purchased from Sigma. All antibodies used in Western blotting were purchased from Santa Cruz Biotechnology. The human bronchial epithelial cell line BEAS-2B was obtained from the American Type Culture Collection. Cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin and streptomycin.
Colony formation assay
To measure anchorage-independent growth in soft agar, 5 × 103 cells were suspended in 0.3% agar in DMEM supplemented with 1% penicillin/streptomycin, 1% l-glutamine, and 10% FBS and overlaid on 0.5% agar in the same medium in six-well plates. After 4 weeks, colonies were stained with 0.005% crystal violet, counted microscopically, and photographed with a dissection microscope.
Measurement of intracellular ROS
Cells were washed once with warm PBS and incubated with 10 μmol/L 5-(and-6)-chloromethyl-2′,7′-dichlorodihy-drofluorescein diacetate ethyl ester (CM-H2DCFDA; Molecular Probes) or 5 μmol/L dihydroethidium (DHE; Molecular Probes), respectively, in warm PBS for 30 minutes. Then, cells were washed with warm PBS and incubated in media for 30 minutes. After incubation, cells were trypsinized, washed twice with cold PBS, and analyzed by fluorescence-activated cell sorting (FACSCalibur, BD Biosciences). In some experiments, the DHE- or CM-H2DCFDA–stained cells were also subjected to immunofluorescence analysis as described previously (18).
Cell proliferation and Western blotting
Cells (1 × 105) were seeded in six-well plate, harvested after the indicated time, and counted with a Coulter Counter (Z2 Coulter Counter, Beckman-Coulter). For Western blotting analysis, cells were washed once with cold PBS and lysed with radioimmunoprecipitation assay buffer (1× PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mmol/L phenylmethylsulfonyl fluoride) supplemented with protease and phosphatase inhibitor mixtures (Sigma). Total protein (40 μg) was separated on 8% to 12% SDS-PAGE and transferred to nitrocellulose-enhanced chemiluminescence membranes (GE Healthcare). The blots were probed with various antibodies as indicated in the figures.
Electrophoretic mobility shift assay and immunofluorescence staining
Nuclear protein was extracted using NE-PER nuclear and cytoplasmic extraction reagents (Pierce). Electrophoretic mobility shift assay (EMSA) was performed using the Light-Shift Chemiluminescent EMSA kit (Pierce). In brief, the NF-κB consensus binding sequence 5′-AGTTGAGGGGACTTTCCCAGGC-3′ was end labeled with biotin as probes, and the binding reaction was performed in a final volume of 20 μL with 2 μg of nuclear extract and 2 fmol of labeled probes. For competition assay, 200× unlabeled NF-κB consensus binding oligonucleotide was added in binding reaction. For supershift assays, 2 μL of antibodies (Santa Cruz Biotechnology) were incubated at room temperature with nuclear extracts for 30 minutes before binding reaction. Then, the mixture was incubated for additional 20 minutes at room temperature, and the reactions were separated on 6% nondenaturing polyacrylamide gels. The activation status of NF-κB was also evaluated by immunofluorescence staining of the cells with antibodies against NF-κB p50 or p65 followed by analyzing the images captured by fluorescence microscopy.
Gene silencing and transfection
To knock down the specific genes of interest, artificial miRNA sequences were designed using BLOCT-iT RNAi Designer (Invitrogen). The oligonucleotides were synthesized, annealed, and inserted into the pcDNA6.2-GW/miR vector (BLOCK-iT Pol II miR RNAi Expression Vector, Invitrogen) according to the protocol provided by the manufacturer. The recombinant vector and the control plasmid pcDNA6.2-GW/miR-neg were trans-fected into the As3+-transformed cells using Lipofectamine 2000 (Invitrogen), respectively. Stably transfected cell lines with specific gene silencing of SOD2, catalase, or NF-κB subunit p50 were selected in the medium containing 10 μg/mL blasticidine. The artificial miRNA sequences inserted into the BLOCK-iT Pol II miR RNAi Expression Vector are as follows: SOD2, 5′-TGCTGAAAGGAACCAAAGTCACGTTTGTTTTGGCCACTGACTGACAAACGTGATTGGTTCCTTT-3′; catalase, 5′-TGCTGAACAGTGGAGAACCGAACTGCGTTTTGGCCACTGACTGACGCAGTTCGTCTCCACTGTT-3′; and NF-κB p50, 5′-TGCTGAATATTTGAAGGTATGGGCCAGTTTTGGCCACTGACTGACTGGCCCATCTTCAAATATT-3′.
Statistical analysis
Tests for statistical significance were done using a paired, two-tailed Student’s t test. Values represents mean ± SD (n = 3; *, P < 0.05). All data (mean ± SD) are representative of at least three independent experiments.
Results
As3+ induces cell transformation
The transformative capability of As3+ has long been established in several types of the mammalian cells (19, 20). To determine whether As3+ is capable of inducing transformation of the human bronchial epithelial cell line BEAS-2B, we cultured the cells in a medium containing 0, 0.25, 0.5, 1, and 2 μmol/L of As3+ for 24 weeks. In agreement with previous studies (20), a morphologic change of the cells from epithelioid to fibroblast-like with multiple foci occurred in all of the cells with continuous As3+ treatment for 24 weeks (Fig. 1A). Such a change was not observed in cells cultured in the As3+-free medium for 24 weeks. To further validate whether such a morphologic change of the long-term As3+-treated cells reflected transformation of the cells, we next evaluated the anchorage-independent growth of these cells using a colony formation assay in soft agar. As depicted in Fig. 1B, the long-term arsenic-treated cells (right), but not the control cells (left), form multiple colonies in soft agar. Thus, these data clearly suggested that treatment of the cells with continuous low concentration of As3+ is able to induce cell transformation.
Figure 1.
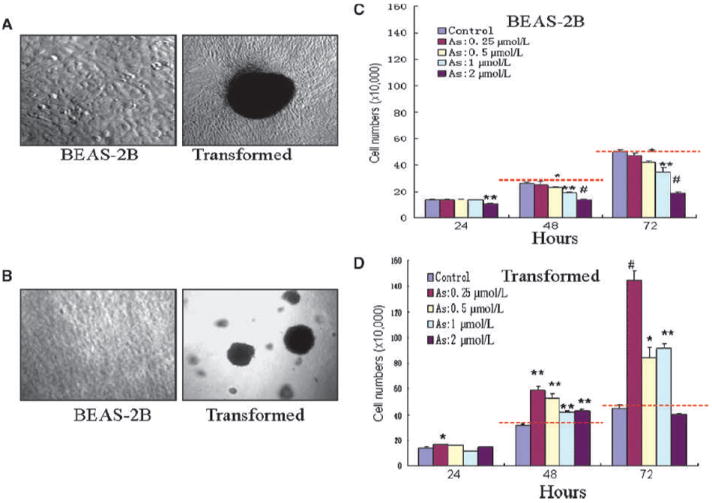
Continuous low concentration of As3+ exposure induces cell transformation. A, BEAS-2B cells maintained in As3+-free medium (left) or in a medium containing 0.25 to 2 μmol/L As3+ for 24 wk. Note the morphologic changes and multiple foci formation of the As3+-treated cells. B, As3+-transformed cells have features of anchorage-independent growth as determined by the formation of large colonies in soft agar. C, As3+ inhibits cell growth of the nontransformed cells (BEAS-2B) in a time- and dose-dependent manner. D, As3+ enhances cell growth of the transformed cells. For the purpose of comparison, red dash lines were drawn to indicate that the number of cells treated with As3+ was smaller than the control (C) or larger than the control. Columns, mean (n = 3); bars, SD. *, P < 0.05; **, P < 0.01; #, P < 0.001.
To investigate whether the As3+-transformed cells exhibit an altered cell proliferation rate under the basal and As3+ treatment condition, both nontransformed (control) and the As3+-transformed cells were treated with As3+ for 24, 48, or 72 hours followed by cell proliferation analysis. For cells without long-term As3+ treatment, a dose-dependent inhibition of cell growth was observed for cells treated with As3+ for 48 and 72 hours (Fig. 1C). At a condition of a 24-hour As3+ treatment, a marginal but statistically significant inhibition of cell growth can be seen only in cells treated with the highest dosage of As3+, 2 μmol/L. As3+ seems to be unable to inhibit growth of the As3+-transformed cells at every dosage and time point tested (Fig. 1D). In contrast, an enhanced cell proliferation was observed for these cells treated with As3+ for 48 or 72 hours (compare the cells treated with different concentration of As3+ with the control cells). Taken together, above data clearly suggested that BEAS-2B cells with a long-term low-dose As3+ treatment possess transformative features, including fast proliferation, anchorage-independent growth, and resistance to As3+ cytotoxicity.
Reduced capability of ROS generation in the As3+-transformed cells
ROS have been considered as key mediators for As3+-induced cell transformation and carcinogenesis (13). To determine whether the capacity of ROS generation was altered for the As3+-transformed cells, we next measured ROS generation in the nontransformed (control) cells and the As3+-transformed cells treated with various doses of As3+ for 24 or 72 hours through flow cytometry. O2•− and hydrogen peroxide (H2O2) were determined by DHE and CM-H2DCFDA staining, respectively. Both O2•− and H2O2 were induced in the nontransformed cells by As3+ roughly in a dose-dependent manner for 72 hours (Fig. 2A and B). In the As3+-transformed cells, unexpectedly, As3+ failed to induce ROS generation (Fig. 2C and D). In fact, As3+ treatment even compromised the generation of ROS relative to the basal level in the As3+-transformed cells, suggesting that certain ROS scavenging system was activated in these transformed cells. Reduction in ROS induction by As3+ in the transformed cells was also confirmed by immunofluorescence staining of the cells with DHE and CM-H2DCFDA, respectively, followed by analyzing images captured by fluorescence microscope (Fig. 2A and B, insets).
Figure 2.
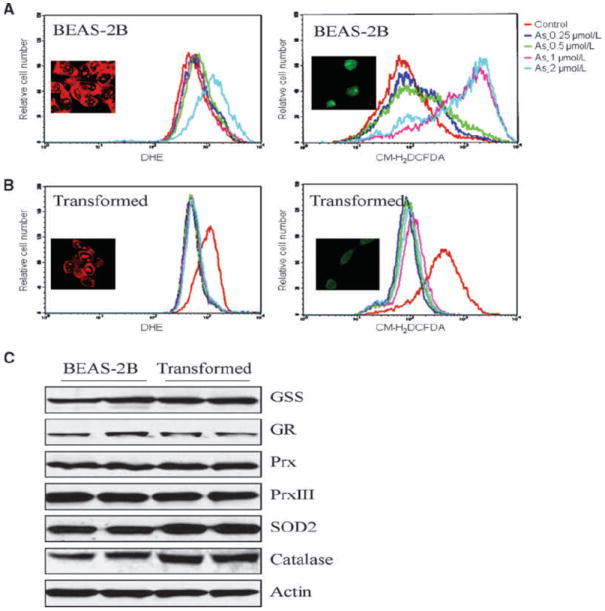
As3+ induces ROS generation in BEAS-2B cells but not in the transformed cells. A, generation of O2•− (left) and H2O2 (right) was determined in the nontransformed cells (BEAS-2B) by staining the cells with DHE and CM-H2DCFDA, respectively, followed by flow cytometry. B, generation of O2•− (left) and H2O2 (right) was determined in the transformed cells by staining the cells with DHE and CM-H2DCFDA, respectively, followed by flow cytometry. Insets in A and B are images obtained by immunofluorescence staining and fluorescence microscopic analyses for the generation of O2•− and H2O2, respectively. C, transformed cells show increased expression of SOD2 and catalase. GR, glutathione reductase.
Increased expression of superoxide dismutase 2 and catalase in the As3+-transformed cells
To delineate the mechanism of decreased generation of ROS in the As3+-transformed cells, the expression levels of several proteins important for the intracellular redox regulation were determined. To counteract ROS-induced oxidative stress, cells have evolved two antioxidation systems: thiol buffering (e.g., glutathione and thioredoxin) and enzymatic systems [e.g., superoxide dismutase (SOD), catalase, and peroxidase; ref. 21]. A comparable expression of glutathione reductase, glutathione synthetase (GSS), peroxiredoxin (Prx), and PrxIII was noted between the nontransformed and As3+-transformed cells (Fig. 2C), suggesting that these proteins are not responsible for the reduced ROS induction in the As3+-transformed cells. The As3+-transformed cells, however, showed a notable increase in the expression of catalase and SOD2 relative to the nontransformed cells. Because both SOD2 and catalase are the major cellular antioxidant enzymes responsible for the removal of O2•− and H2O2 in the cells, respectively, increased expression of SOD2 and catalase might be attributed to the decreased induction of ROS by As3+ in the transformed cells.
Increasing ROS generation suppresses growth of the As3+-transformed cells
Because above data suggest to us that transformed cells acquired features of fast growth and reduced induction of ROS generation, we reasoned that increase in ROS generation should be able to suppress the growth of the As3+-transformed cells. To test this hypothesis, we first applied specific inhibitors to block the activities of SOD and catalase in the transformed cells. As indicated in Fig. 3A and B, treatment of cells with 2-methoxyoestradiol (2-ME), a potent inhibitor of SOD, or 3-amino-1,2,4-triazole (3-AT), a catalase-specific inhibitor, caused a substantial increase of O2•− (Fig. 3A) and H2O2 (Fig. 3B). The ROS generation was further potentiated when cells were treated with both 2-ME and 3-AT.
Figure 3.
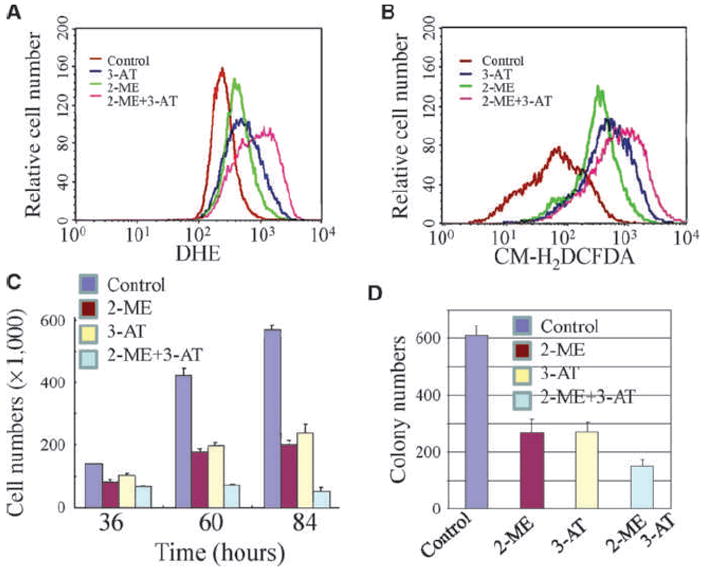
Inhibition of SOD and catalase increases ROS generation and reduces cell growth of the transformed cells. A and B, the transformed cells were treated with the SOD inhibitor 2-ME, the catalase inhibitor 3-AT, or both for 48 h. The levels of O2•− and H2O2 were determined then in these cells by DHE and CM-H2DCFDA staining, respectively, followed by flow cytometry. C, increasing ROS generation by SOD or catalase inhibition inhibits cell growth of the transformed cells. D, increasing ROS generation by SOD or catalase inhibition inhibits anchorage-independent growth of the transformed cells.
To determine whether enhanced ROS generation by inhibition of SOD and catalase can affect the growth of the As3+-transformed cells, we next measure the cell proliferation rate following the treatment of cells with SOD and/or catalase inhibitors. Treatment of cells with SOD or catalase inhibitor or combination of these two inhibitors suppressed proliferation of the transformed cells at 36, 60, and 84 hours significantly (Fig. 3C). Furthermore, boosting ROS generation by SOD or catalase inhibition also decreased anchorage-independent growth of the As3+-transformed cells (Fig. 3D).
Gene silencing of SOD2 and catalase reduced proliferation and colony formation of the As3+-transformed cells
The chemical inhibitors have suffered from off-target effects in different cellular background under many circumstances. To validate the observed effect of the SOD and catalase inhibitors in the transformed cells, we applied a mRNA silencing technique to knock down the expression of SOD2 and catalase, respectively, by artificial miRNA-based gene silencing technique in the transformed cells. Transfection of cells with the control vector exhibited no effect on the expression of SOD2 and catalase (Fig. 4A and B). An appreciable inhibition in the expression of SOD2 and catalase was observed in cells transfected with SOD2- or catalase-specific artificial miRNA (Fig. 4A and B). After 36 hours of transfection, a marginal inhibition of cell growth was noted in cells with SOD2 silencing or combination of SOD2 and catalase silencing. A pronounced inhibition of cell proliferation by either SOD2 or catalase silencing, or both, was observed at 60 and 84 hours after transfection (Fig. 4C). Silencing both SOD2 and catalase was also capable of inhibiting the anchorage-independent growth of the transformed cells (Fig. 4D).
Figure 4.
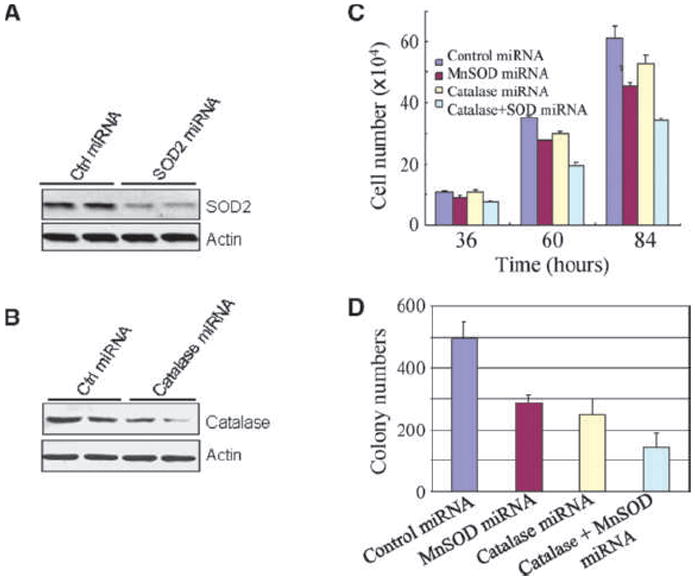
Gene silencing of SOD2 or catalase by artificial SOD2- or catalase-specific miRNA represses growth of the transformed cells. A, artificial SOD2 miRNA reduces SOD2 expression as determined by Western blotting. B, artificial catalase miRNA reduces catalase expression as determined by Western blotting. C, cell proliferation of the transformed cells was determined following transfection of the cells with SOD2 miRNA, catalase miRNA, or the combination for the indicated times. D, colony formation of the transformed cells was determined for cells transfected with the indicated miRNA by soft agar assay.
Enhanced NF-κB activation in the As3+-transformed cells
NF-κB has been viewed as a growth-promoting transcription factors by upregulating the transcription of the genes important for cell cycle and antiapoptotic responses. It should be plausible, thus, to determine whether an upregulation of NF-κB activation or activity contributes to the enhanced proliferation of the As3+-transformed cells. The expression levels of the upstream proteins for the activation of NF-κB as well as the major NF-κB family proteins were compared between the nontransformed and transformed cells by Western blotting. The transformed cells showed an expression of IKKα, IκBα, pIκBα, and RelB similar with that in the nontransformed cells, whereas the expression of IKKβ, IKKγ, and p65 was decreased (Fig. 5A). Intriguingly, the expression of NF-κB1 (p105) was significantly enhanced in the transformed cells relative to the nontransformed cells (Fig. 5B). Consequently, the level of p50 protein, a product of p105 processing, was increased in the transformed cells. Although the transformed cells showed a decrease of p100, the level of p52 was increased, suggesting a possible enhancement in the processing of p100 in the As3+-transformed cells (Fig. 5B).
Figure 5.
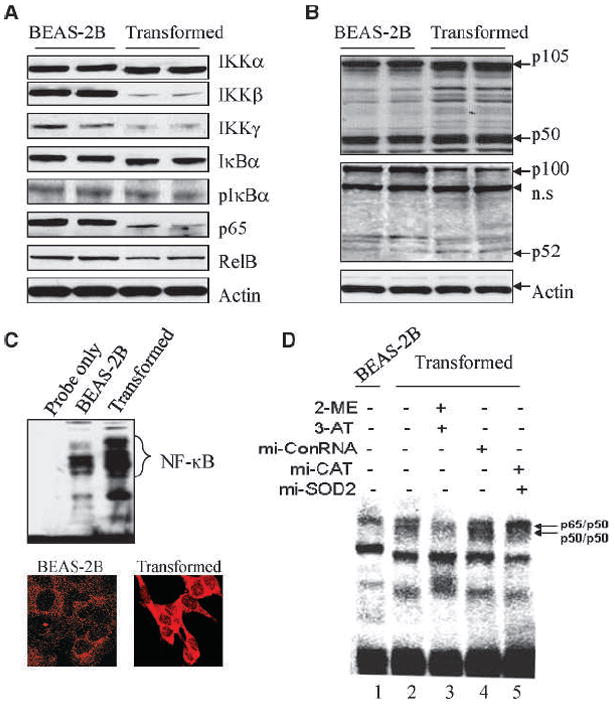
Altered expression of the NF-κB family proteins in the transformed cells. A, the protein levels of IKK subunits IκBα, pIκBα, p65, and RelB were determined by Western blotting. Actin was used as a loading control. B, expression and proteolytic processing of p105 and p100 were determined by Western blotting. C, top, NF-κB DNA binding activity was determined by EMSA; bottom, expression and nuclear translocation of NF-κB p50 protein in the BEAS-2B (left) and transformed (right) cells as determined by immunofluorescence staining using p50 antibody. D, increasing ROS generation of the transformed cells by SOD or catalase inhibition reduces NF-κB DNA binding activity. The transformed cells were treated with both SOD and catalase inhibitors (lane 3), control miRNA (lane 4), or both SOD and catalase miRNAs for 48 h. Nuclear proteins were prepared and incubated with the labeled DNA probe containing NF-κB binding site.
In correlation with the elevated expression and processing of NF-κB p105 and p100, the NF-κB DNA binding activity was increased in the transformed cells (Fig. 5C, top). In agreement with the NF-κB DNA binding activity, immunofluorescence staining using NF-κB p50 antibody indicated an increased nuclear translocation as well as overall expression of the p50 protein in the transformed cells (Fig. 5C, bottom). To determine whether reduced ROS generation is responsible for the increase in NF-κB activation, the ROS generation was resumed in the transformed cells by SOD or catalase inhibition through either specific inhibitors or artificial miRNA silencing. A decrease in NF-κB DNA binding activity was revealed in the transformed cells treated with SOD2 and catalase inhibitors (Fig. 5D, compare lane 3 with lane 2) or SOD2 and catalase siRNAs (Fig. 5D, compare lane 5 with lane 4).
Gene silencing of NF-κB p105 reduced proliferation and colony formation of the As3+-transformed cells
To understand whether enhanced NF-κB activation that resulted from a decreased generation of ROS in the transformed cells is responsible for the fast growth of the transformed cells, the rate of cell growth was determined by gene silencing of NF-κB p105 through artificial miRNA. The silencing effect was confirmed by Western blotting that showed substantial reduction of both p105 and p50 proteins (Fig. 6A). A significant decrease in cell growth was observed at 60 and 84 hours after silencing of p105 (Fig. 6B). Silencing of p105 also reduced the anchorage-independent growth of the As3+-transformed cells as determined by the colony formation in soft agar (Fig. 6C and D).
Figure 6.
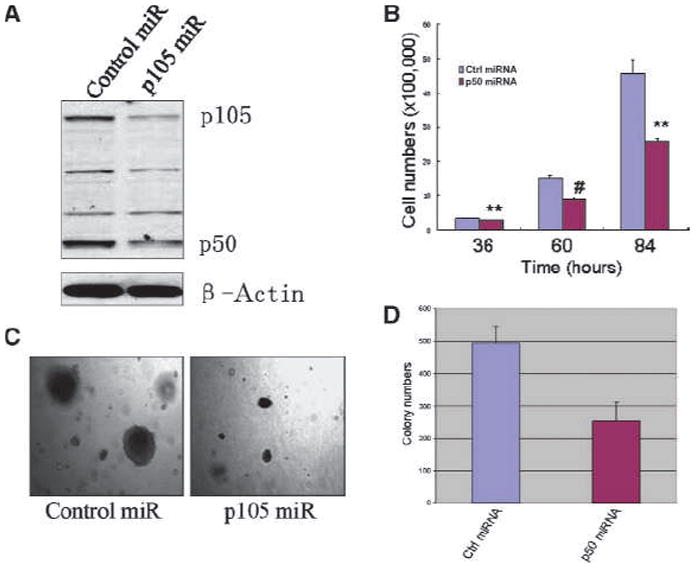
Silencing NF-κB p105 reduces cell growth and colony formation of the transformed cells. A, the silencing effect of p105 miRNA in the transformed cells was confirmed by Western blotting. B, cell proliferation was determined for the transformed cells transfected with a control miRNA or p105 (p50) miRNA. C, silencing p105 reduces anchorage-independent growth of the transformed cells. Right, note the reduced number and size of the transformed cells transfected with p105 miRNA. D, quantification of C.
Discussion
We provided evidence in the present report suggesting that treatment of the BEAS-2B cells with continuous low concentration of As3+ induces cell transformation and that the capacity of ROS generation in the transformed cells was severely compromised. Such a reduction in ROS generation seems to be responsible for fast proliferation, anchorage-independent growth, and resistance to As3+ toxicity of the transformed cells. A sustained alternative activation of NF-κB might contribute to the fast and anchorage-independent growth of these cells due to reduced ROS generation. The fast growth of the transformed cells could be reversed by either resuming the ROS production through SOD2/catalase inhibition or gene silencing of NF-κB p105/50.
ROS have long been viewed as major contributors to oxidative injury and DNA damage of tissues or cells (22). A sustained oxidative injury and unrepaired damage on DNA will be carcinogenic due to accumulation of genetic mutations that either activate oncogenes or inactivate tumor suppressors. To date, most studies addressed the procarcinogenic effect of ROS in normal or noncancerous cells. However, a considerable number of reports implicated importance of ROS in retaining tumor cell growth or facilitating chemotherapy- or radiotherapy-induced tumor cell death (23). Emerging evidence suggests that some types of cancer cells adapted a resistant program in responding to further oxidative stress by enhancing endogenous antioxidant capacity that lowers ROS and endows cell survival (24-26). This notion was supported by an earlier study using rat lung epithelial L2 cell line, which showed that exposure of the cells to low but continuous levels of exogenous oxidants resulted in an enhancement of overall cellular antioxidant capacity (27). The observation that continuous low concentration of As3+ exposure induces cells transformation associated with an increased expression of SOD2 and catalase agrees with those earlier reports. In nontransformed cells, As3+ is able to induce ROS generation in a time- and dose-dependent manner (Fig. 2). It is very likely, thus, that exposure of the cells to a continuous low concentration of As3+, which can be viewed as a condition of consistent oxidative stress, might create a selective pressure to enrich the population of cells that are able to counteract further induction of ROS. Numerous studies revealed that ROS are pivotal in inducing cell apoptosis and oxidative inactivation of NF-κB, a major antiapoptotic transcription factor (28, 29). Accordingly, cells with a reduced capacity of ROS generation will be more proliferative in comparison with cells with a higher capability of ROS induction.
The observed reduction in ROS generation of the As3+-transformed cells is further supported by several recent studies showing decreased ROS levels in normal and cancer stem cells (30-32). In hematopoietic stem cells, it was believed that activation of forkhead box Os transcription factor upregulates the expression of SOD and catalase, leading to a reduced ROS level and maintenance of long-term self-renewal (33). Reduced level of ROS was also considered as a key mechanism for the radiation resistance of the cancer stem cells within human breast tumor (30). The level of ROS is much lower in both human and murine breast cancer stem cells than the corresponding noncancerous cells. Elevated expression of some ROS-scavenging molecules, including glutamate-cysteine ligase and GSS, might be one of the mechanisms for lowering the level of ROS in these cancer stem cells (30). Given the fact that ROS are potent inducers of cell apoptosis and antagonists of NF-κB through direct oxidation of IKK kinase subunits and NF-κB p50 and p65 proteins, it is not surprising to note that some cancer cells, especially the cancer stem cells, evolved an antioxidative program to lower ROS generation and, consequently, maintain both stemness and cancer-initiating capabilities. It is also plausible to speculate that reduced level of ROS attributes to the resistance of chemotherapy and radiation of the cancer cells.
In summary, the present study provides evidence showing that As3+ is able to induce cell transformation and that ROS generation in the transformed cells was much compromised relative to the nontransformed cells. Such evidence agrees with the current concept of redox adaptation of cancer cells (22). Although it remains to be determined how transformed cells or cancer stem cells acquire the feature of lower ROS generation or induction, the findings in this report can provide new insights into the translational cancer research and therapy. It is expectable, therefore, that the use of agents to restore or induce ROS accumulation in combination with conventional chemotherapy or radiotherapy will improve the therapeutic outcomes of tumors.
Acknowledgments
Grant Support
NIH grants 5RO1CA119028-05, RO1CA116697, RO1ES015518, and RO1ES15375 (X. Shi) and National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention grant 09270036 (F. Chen).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Platanias LC. Biological responses to arsenic compounds. J Biol Chem. 2009;284:18583–7. doi: 10.1074/jbc.R900003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- 3.Wickre JB, Folt CL, Sturup S, Karagas MR. Environmental exposure and fingernail analysis of arsenic and mercury in children and adults in a Nicaraguan gold mining community. Arch Environ Health. 2004;59:400–9. doi: 10.3200/AEOH.59.8.400-409. [DOI] [PubMed] [Google Scholar]
- 4.Basu A, Mahata J, Gupta S, Giri AK. Genetic toxicology of a paradoxical human carcinogen, arsenic: a review. Mutat Res. 2001;488:171–94. doi: 10.1016/s1383-5742(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen F, Shi X. Intracellular signal transduction of cells in response to carcinogenic metals. Crit Rev Oncol Hematol. 2002;42:105–21. doi: 10.1016/s1040-8428(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 6.Dong Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ Health Perspect. 2002;110(Suppl 5):757–9. doi: 10.1289/ehp.02110s5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandoval M, Morales M, Tapia R, et al. p53 response to arsenic exposure in epithelial cells: protein kinase B/Akt involvement. Toxicol Sci. 2007;99:126–40. doi: 10.1093/toxsci/kfm153. [DOI] [PubMed] [Google Scholar]
- 8.Reznikova TV, Phillips MA, Rice RH. Arsenite suppresses Notch1 signaling in human keratinocytes. J Invest Dermatol. 2009;129:155–61. doi: 10.1038/jid.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Q, Bhatia D, Zhang Y, et al. Incorporation of an internal ribosome entry site-dependent mechanism in arsenic-induced GADD45α expression. Cancer Res. 2007;67:6146–54. doi: 10.1158/0008-5472.CAN-07-0867. [DOI] [PubMed] [Google Scholar]
- 10.Zhang QY, Mao JH, Liu P, et al. A systems biology understanding of the synergistic effects of arsenic sulfide and imatinib in BCR/ABL-associated leukemia. Proc Natl Acad Sci U S A. 2009;106:3378–83. doi: 10.1073/pnas.0813142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soucy NV, Klei LR, Mayka DD, Barchowsky A. Signaling pathways for arsenic-stimulated vascular endothelial growth factor-A expression in primary vascular smooth muscle cells. Chem Res Toxicol. 2004;17:555–63. doi: 10.1021/tx034193q. [DOI] [PubMed] [Google Scholar]
- 12.Mann KK, Miller WH., Jr Death by arsenic: implications of PML sumoylation. Cancer Cell. 2004;5:307–9. doi: 10.1016/s1535-6108(04)00089-3. [DOI] [PubMed] [Google Scholar]
- 13.Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- 14.Partridge MA, Huang SX, Hernandez-Rosa E, Davidson MM, Hei TK. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007;67:5239–47. doi: 10.1158/0008-5472.CAN-07-0074. [DOI] [PubMed] [Google Scholar]
- 15.Chou WC, Jie C, Kenedy AA, et al. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc Natl Acad Sci U S A. 2004;101:4578–83. doi: 10.1073/pnas.0306687101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Straub AC, Clark KA, Ross MA, et al. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J Clin Invest. 2008;118:3980–9. doi: 10.1172/JCI35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemarie A, Bourdonnay E, Morzadec C, Fardel O, Vernhet L. Inorganic arsenic activates reduced NADPH oxidase in human primary macrophages through a Rho kinase/p38 kinase pathway. J Immunol. 2008;180:6010–7. doi: 10.4049/jimmunol.180.9.6010. [DOI] [PubMed] [Google Scholar]
- 18.Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor κB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res. 2003;63:7689–93. [PubMed] [Google Scholar]
- 19.DiPaolo JA, Casto BC. Quantitative studies of in vitro morphological transformation of Syrian hamster cells by inorganic metal salts. Cancer Res. 1979;39:1008–13. [PubMed] [Google Scholar]
- 20.Biedermann KA, Landolph JR. Induction of anchorage independence in human diploid foreskin fibroblasts by carcinogenic metal salts. Cancer Res. 1987;47:3815–23. [PubMed] [Google Scholar]
- 21.Das KC, White CW. Redox systems of the cell: possible links and implications. Proc Natl Acad Sci U S A. 2002;99:9617–8. doi: 10.1073/pnas.162369199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 23.Lawenda BD, Kelly KM, Ladas EJ, et al. Should supplemental anti-oxidant administration be avoided during chemotherapy and radiation therapy? J Natl Cancer Inst. 2008;100:773–83. doi: 10.1093/jnci/djn148. [DOI] [PubMed] [Google Scholar]
- 24.Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance-serendipity or a causal relationship? Curr Pharm Des. 2004;10:1969–77. doi: 10.2174/1381612043384411. [DOI] [PubMed] [Google Scholar]
- 25.Ivshina AV, George J, Senko O, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- 26.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi J, Liu RM, Forman HJ. Adaptation to oxidative stress: quinone-mediated protection of signaling in rat lung epithelial L2 cells. Biochem Pharmacol. 1997;53:987–93. doi: 10.1016/s0006-2952(96)00867-2. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(–) channel. Proc Natl Acad Sci U S A. 2004;101:6770–3. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Chen F. Reactive oxygen species (ROS), troublemakers between nuclear factor-κB (NF-κB) and c-Jun NH(2)-terminal kinase (JNK) Cancer Res. 2004;64:1902–5. doi: 10.1158/0008-5472.can-03-3361. [DOI] [PubMed] [Google Scholar]
- 30.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 32.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]