

Abstract
Inflammation is traditionally considered a defense response induced by infection or injury. However, inflammation can also be induced by tissue stress and malfunction in the absence of infection or overt tissue damage. Here we discuss the relationship between homeostasis, stress responses and inflammation. Stress responses have cell autonomous and cell extrinsic components, the latter contributing to tissue level adaptation to stress conditions. Inflammation can be thought of as the extreme end of a spectrum that ranges from homeostasis to stress response to bona fide inflammatory response. Inflammation can be triggered by two types of stimuli: extreme deviations of homeostasis or challenges that cause a disruption of homeostasis. This perspective may help to explain qualitative differences and functional outcomes of diverse inflammatory responses.
Introduction: Homeostasis, Homeostatic Range, and Stress Responses
Homeostasis is a fundamental property of biological systems. It preserves their stability by maintaining key regulated variables within an acceptable range (Buchman, 2002). It operates at the level of the entire organism, within tissue compartments, and inside individual cells. Homeostasis is best characterized at the level of the whole organism (systemic homeostasis). Here, regulated variables are well defined and include blood levels of glucose, Na+, Ca2+, and O2, blood pH and osmolarity, and core body temperature (Table 1). These variables are maintained within an acceptable dynamic range by the endocrine and autonomic nervous systems.
TABLE 1.
Representative Regulated Variables and Sensors in Systemic Homeostasis
| Regulated Variable | Sensor |
|---|---|
| Blood Pressure / Blood Volume / Na+ Concentration | Aortic Body (Aorta), Carotid Body (Carotid Artery), Atrial Volume Receptors (Heart), Juxtaglomerular Apparatus (Kidney) |
| Ca2+ / Mg2+ / PO43− Concentrations | Chief Cells (Parathyroid Gland) |
| Glucose | Islet of Langerhans (Pancreas) |
| Osmolarity | Circumventricular Organs (Hypothalamus) |
| pO2, pCO2, and pH | Aortic Body (Aorta), Carotid Body (Carotid Artery), Ventrolateral Medulla (Medulla) |
| Temperature | Thermosensory neurons (Skin), Preoptic Area (Hypothalamus) |
Tissue homeostasis has yet to be defined in terms of its regulated variables, but examples of these variables include cell number and cell composition per tissue compartment, tissue architecture (cell positioning, cell-cell interactions, and extracellular matrix abundance and composition), integrity of structural components (e.g., cell junctions and basement membrane), concentrations of O2, nutrients, and metabolic end products (e.g., CO2 and urea), as well as volume, pH, temperature, and osmolarity of interstitial fluids (Table 2).
TABLE 2.
Examples of Regulated Variables of Tissue Homeostasis
| REGULATED VARIABLE |
|---|
| Cell number per compartment |
| Cell composition (ratios of different cell types) |
| Cell-cell interactions |
| Cell positioning |
| ECM composition and abundance |
| Levels of oxygen |
| Levels of nutrients |
| Levels of metabolic waste products |
| Volume of interstitial fluid |
| pH of interstitial fluid |
| Electrolyte composition of the interstitial fluid |
| Osmolarity of interstitial fluid |
Cellular homeostasis maintains a number of regulated variables including cell volume, osmolarity, electrolyte concentration (e.g., Na+, K+, and Cl− concentrations), pH, membrane potential, and concentrations of intracellular ions, proteins, nutrients, cholesterol, oxygen, and reactive oxygen species (ROS) (Table 3).
TABLE 3.
Representative Stressors and Sensors in Cellular Homeostasis
| STRESSOR | SENSOR |
|---|---|
| Amino Acid Deprivation | ATF4, mTOR (Kroemer et al., 2010; Rutkowski and Kaufman, 2003) |
| ATP Depletion | AMPK (Sengupta et al., 2010) |
| ER Stress/UPR | ATF6, IRE-1α / XBP-1, PERK / eIF-2α (Cao and Kaufman, 2012) |
| Genotoxic Stress | p53 (Reinhardt and Schumacher, 2012) |
| Glucose Deprivation | CHREBP (Postic et al., 2007) |
| Heat Shock | HSF-1 (Anckar and Sistonen, 2007) |
| Hypoxia | HIF-1α (Weidemann and Johnson, 2008) |
| Osmotic Stress | NFAT5 (Aramburu et al., 2006) |
| Oxidative Stress | NRF2 (Nguyen et al., 2009) |
| Viral Infection | Receptors signaling through IRF3 (Holm et al., 2013) |
| Xenobiotic Stress | AHR, CAR, PXR (Pascussi et al., 2008) |
| General Environmental Stress (including Infection) | Receptors signaling through NF-κB and MAPKs (ERK, JNK, p38) (Takeuchi and Akira, 2010; Wagner and Nebreda, 2009) |
In each of these cases and at all levels (systemic, tissue, and cellular), the regulated variables have a characteristic dynamic range that is maintained by homeostatic control systems. When regulated variables change beyond the dynamic range (as a result of external perturbations and insults), the system engages in a stress response that aims to restore homeostasis (Goldstein and Kopin, 2007).
In order to maintain homeostasis, specialized sensors constantly monitor the values of regulated variables. In systemic homeostasis these sensors include endocrine cells and sensory neurons (Table 1). In cellular homeostasis the sensors are signaling proteins that detect alterations in various core processes, such as protein folding, levels of ROS, and nutrient availability (Table 3). Given the large number of regulated variables in cellular homeostasis, additional stress response pathways may be uncovered in the future. In principle it can be predicted that sufficient disruption in homeostasis of each regulated variable should elicit a corresponding stress response.
In tissue homeostasis the sensors for most regulated variables have not been studied and for the most part are unknown. It is not known, for example, how compartment size is sensed, how cell number and composition are measured, and how changes in the extracellular matrix (ECM) are monitored, even though it is clear that all these parameters are tightly controlled (Table 2). The only known sensors of tissue homeostasis are specialized in detecting extreme challenges such as infection and tissue injury. These sensors include tissue resident immune cells (particularly macrophages and mast cells) as well as somatosensory neurons (e.g., C-fiber nociceptors). Indeed, emerging evidence suggests that macrophages and C-fiber nociceptors may monitor and control tissue homeostasis not only at the extremes of infection and injury, but also during more common and less dramatic alterations of normal tissue states (Ahern, 2013; Basbaum et al., 2009; Mosser and Edwards, 2008; Nguyen et al., 2011; Pollard, 2009; Tracey, 2009).
Although it is well appreciated that both inflammatory and stress responses are protective reactions that defend homeostasis, the relationship between them is ambiguous. They can be viewed as distinct but overlapping components of a spectrum of system states (Homeostatic State – Stress Response State – Inflammatory State), each of which can be defined in terms of the maintenance of regulated variables. The system (organism, tissues, cells) is in a homeostatic state when the values of regulated variables are within an acceptable dynamic range. When the homeostatic capacity is insufficient to maintain these values, (e.g., due to external perturbations), a stress response is engaged. If the stress response is insufficient to defend homeostasis, an inflammatory response is induced. Both stress response and inflammation are engaged to eliminate the stressor (i.e. the source of perturbation), to promote adaptations to the stressor, and ultimately to return the system to the homeostatic state (Figure 1).
FIGURE 1. An Inflammatory Spectrum.
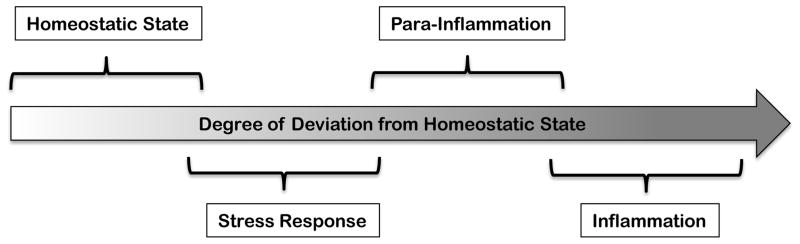
An inflammatory response is an extreme end of the spectrum that ranges from homeostatic state, to stress response, para-inflammation, and finally, inflammation.
The stress and inflammatory responses can also be distinguished by the conditions that induce them. There are principally two types of stimuli eliciting these responses: the first is an extreme deviation of regulated variables from normal values, and the second is a challenge that can cause deviation of regulated variables but is not itself a regulated variable. Challenges in the latter category include infections, allergens, toxins, and noxious xenobiotics – all of which disrupt homeostasis and need to be sensed to elicit a protective response that aims to ultimately restore homeostasis. To distinguish between the two scenarios, we will refer to the first as a stress response and the second as a defense response (Figure 2). From this perspective the responses induced by hypoxia and heat shock, for example, are stress responses, whereas responses induced by pathogens, toxins and tissue damage (i.e., the immune response, detoxification response, and tissue repair response) are defense responses. Stress responses are elicited by sensors (such as HIF-1α and HSF-1) that monitor regulated variables (oxygen and protein folding state, respectively). Defense responses are elicited by sensors that are specialized in detecting insults that can disrupt homeostasis. Thus, pattern recognition receptors and xenobiotic sensors directly detect infection and noxious chemicals, respectively. Some insults, however, cannot be detected directly, because they are too diverse and unpredictable. These include most toxins and poisons as well as majority of allergens. Sensing mechanisms in these cases are not defined but may include detection of unique noxious activities, such as disruption of tight junctions, proteolysis of ECM components, and inflammasome activation.
FIGURE 2. Stress and Defense Response Components of Inflammation.
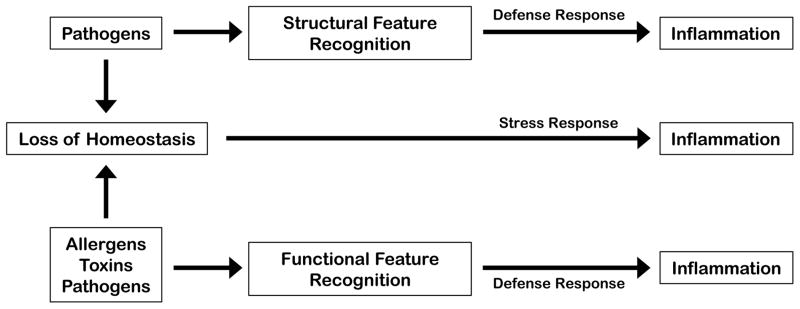
Inflammation can be induced by sensing extreme deviations from tissue homeostasis or by sensing the challenges that can cause extreme deviations of tissue homeostasis. The former are detected by sensors of regulated variables of cellular and tissue homeostasis. The latter can be induced upon direct recognition of structural features of agents that can disrupt tissue homeostasis (e.g., PAMP recognition by PRRs) or by detecting their conserved functional features, such as protease activity or chemical reactivity.
Functional Categories of Stress Response Genes
The sensing mechanisms, signaling pathways, and gene expression programs controlling cellular homeostasis are well characterized. Functionally, stress response gene products can be divided into two categories depending on whether they are involved in the elimination of a stressor or adaptation to a stress condition (Table 4). For example, gene products involved in degrading unfolded proteins, scavenging ROS, and repairing DNA damage are effectors of the stress response involved in elimination of a stressor. Stress conditions also often require specific cellular adaptations so as to minimize their negative impact: thus the unfolded protein response (UPR) not only triggers elimination of misfolded proteins but also suppresses new protein synthesis and promotes expression of chaperones to help prevent protein misfolding (the adaptation response). The genes involved in adaptation to stress are especially important when stress conditions are chronic, as is seen in long-term infection (Tabas and Glass, 2013), or when stress conditions are a normal part of specialized cellular function. For example, cells with a very high secretory demand, such as plasma cells and various exocrine and endocrine cells, experience a chronic ER stress response (Garrett et al., 2010), while kidney tubular epithelial cells experience continuous osmotic stress (Neuhofer and Beck, 2005). Consequently, these cell types develop specialized features that promote adaptation to chronic stress conditions by constitutive expression of stress-adaptation genes. In some cases the distinction between elimination and adaptation responses is less straightforward: osmotic stress triggers changes in expression of aquaporins and electrolyte transporters, and these gene products both eliminate the stressor and promote adaptation to the given stress.
TABLE 4.
Elimination and Adaptation Responses in Cellular Homeostasis
| STRESSOR | ELIMINATION RESPONSE | ADAPTATION RESPONSE |
|---|---|---|
| Amino Acid Deprivation | Induction of amino acid transporters | Autophagy |
| ER Stress/UPR | Degradation of unfolded proteins | Stabilization of misfolded proteins, elimination of non- essential translation, expansion of ER compartment |
| Genotoxic Stress | Induction of DNA repair mechanisms | Cell cycle arrest |
| Glucose Deprivation | Induction of glucose transporters | Use of alternative fuel sources (e.g., amino acids, lipids) |
| Heat Shock | Degradation of unfolded proteins | Stabilization of misfolded proteins |
| Hypoxia | Angiogenesis | Shift to anaerobic glycolysis |
| Osmotic Stress | Induction of aquaporins and electrolyte transporters | Induction of aquaporins and electrolyte transporters |
| Oxidative Stress | ROS scavenging | ROS scavenging |
Cell Intrinsic and Cell Extrinsic Stress and Defense Responses
Most of what is known about cellular stress and defense responses has to do with cell autonomous (cell intrinsic) mechanisms of stressor elimination and stress adaptation. However, there is an equally important non-cell autonomous (cell extrinsic) response containing two functional gene categories. The first category of gene products regulates the extracellular compartment (e.g., ECM remodeling, cell positioning, cell-ECM and cell-cell contacts). Genes in the second category are likely involved in paracrine communication to neighboring cells, though for the most part, these genes have not been functionally defined in the context of a stress response. Most stress conditions affect populations of cells in a given compartment, rather than just individual cells. When one cell in a population is exposed to a given challenge (e.g., hypoxia or nutrient deprivation), it is likely that other cells in the same compartment will be exposed to it as well. Because of that, cells under stress produce signals that alert other cells in the vicinity to the presence of the stressor. These signals may function to induce preemptive responses in cells that have not yet been exposed to the challenge, making them adapt more efficiently. A well known example of this phenomenon is provided by the antiviral defense response: virally infected cells produce type 1 interferons (IFNs) which act on neighboring cells to induce an antiviral state (Honda and Taniguchi, 2006; Levy et al., 2011).
Cell extrinsic stress and defense responses also afford tissue level adaptations to a challenge – adaptations that cannot be achieved at the level of individual cells. For example, hypoxia not only induces cell autonomous adaptations, such as induction of glucose transporters and a switch to anaerobic glycolysis, but it also promotes tissue level adaptations, such as angiogenesis, induced by cell extrinsic signals like VEGF and other angiogenins (Shweiki et al., 1992). In this case both cell intrinsic and extrinsic components of the hypoxia response are induced by the same sensor, HIF-1α. The cell extrinsic stress response has many overlapping components with the tissue level stress response, though the two are not equivalent when the regulated variables affected under cellular versus tissue level stress conditions are distinct. However, the cell extrinsic response does induce tissue level adaptation to stress, as illustrated most clearly by the induction of angiogenesis upon hypoxia.
Another notable example of a cell extrinsic stress response is the so called radiation-induced bystander effect, in which irradiated cells induce an irradiated phenotype in nearby, untreated cells (Mothersill and Seymour, 2004). However, the signals that communicate this information are still unknown. The best available characterization of cell extrinsic stress responses has been provided by studies in C. elegans, where there is evidence for systemic adaptations to cellular heat shock, oxidative stress, and genotoxic stress that are communicated either through sensory neurons or soluble factors (Durieux et al., 2011; Ermolaeva et al., 2013; Prahlad et al., 2008).
It is likely that every type of cellular stress and defense response has a cell extrinsic component even though most have not been characterized. Indeed, global gene expression analysis indicates that various forms of stress induce large numbers of genes whose products are either known or predicted to have extracellular functions, including many secreted proteins (Table 5). Though the functions of most of these genes in the context of cellular homeostasis are unknown, they likely operate in either the extracellular compartment or a paracrine manner to induce tissue level adaptations to stress.
TABLE 5.
Predicted Gene Products of the Cell Extrinsic Stress Response in HeLa Cells
| STRESSOR | GENE CATEGORY | SECRETED/EXTRACELLULAR GENE PRODUCT |
|---|---|---|
| ER Stress/UPR | Chemokine | CXCL3 |
| Cytokine | AIMP1 IL17RB ISG15 LIFR LTBP1 |
|
| ECM Structural Component | ADAMTSL3 COL1A2 LAMC1 NUCB2 STC2 |
|
| ECM Signaling Component | DKK1 FRZB GPC3 LRCH3 |
|
| Effector Enzyme | ADAM23 ARSJ KLK11 LIPG MMP16 |
|
| Growth Factor / Hormone | BMP8A EPO GDF15 IGFBP7 INHBA |
|
| Glucose Deprivation | Chemokine | CCL20 CXCL2 CXCL3 IL8 |
| Cytokine | IL-1A IL6 IL11 IL18 |
|
| ECM Structural Component | COL5A1 CRELD2 LAMB3 MEGF6 NUCB2 |
|
| ECM Signaling Component | DKK1 GPC5 WNT10B |
|
| Effector Enzyme | ANG LNPEP LOX LOXL2 RNASE4 |
|
| Growth Factor / Hormone | EGFR GDF15 HBEGF IGFBP3 INHBA |
|
| Heat Shock | Chemokine | CCL5 CXCL9 |
| Cytokine | AIMP1 IL6ST IL11 IL33 LIFR |
|
| ECM Structural Component | ADAMTSL1 COL21A1 CRISP3 LAMA3 STC2 |
|
| ECM Signaling Component | GPC3 GPC4 LYPD6 RSPO3 WIF1 |
|
| Effector Enzyme | ADAMTS5 GZMA LOX MMP19 PLBD1 |
|
| Growth Factor / Hormone | FLT3LG HBEGF IGF1 INHBA EPO |
|
| Hypoxia | Chemokine | CCL2 CCL28 CXCL12 CXCL14 CXCL17 |
| Cytokine | IFNA1 IFNB1 IL1A IL17A IL33 |
|
| ECM Structural Component | COL2A1 CRISP3 LAMA2 MUC1 STC1 |
|
| ECM Signaling Component | DKK2 GPC6 RSPO1 SFRP1 WNT6 |
|
| Effector Enzyme | ADAM12 ANG ARSF LOX MMP11 |
|
| Growth Factor / Hormone | ANGPT4 ANGPTL1 GDF15 IGFBP1 INHBA |
|
| Oxidative Stress | Chemokine | CCL15 CCL16 |
| Cytokine | CD109 FAM3C ISG15 |
|
| ECM Structural Component | COL9A2 CRISPLD1 HSPG2 MMRN2 THSD4 |
|
| ECM Signaling Component | NXPH2 WNT5B |
|
| Effector Enzyme | ADAMTS5 KLK5 MMP16 PLBD1 |
|
| Growth Factor / Hormone | IGF2 INHBA INSL6 VIP |
Analysis performed on previously published gene expression data (Mense et al., 2006; Murray et al., 2004; Page et al., 2006; Saito et al., 2009). Briefly, gene expression data were obtained from the NCBI GEO database and sorted for gene products exhibiting greater than or equal to two-fold induction. The relevant genes were then analyzed using NIAID DAVID Bioinformatics Resources 6.7 software. Genes were sorted by extracellular and secreted functional categories, and representative genes are displayed in the table.
Tissue level stress response and para-Inflammation
While most cells can detect disruptions of tissue homeostasis, this function is performed most efficiently by specialized sensory cells, such as tissue resident macrophages, mast cells, and sensory neurons (Figure 3). Thus, pathogen sensing is primarily delegated to macrophages (Gordon, 2007), but in addition, macrophages may also play a role in sensing more generic stressors, such as hypoxia and metabolic stress (i.e. deficit or excess in key metabolites) (Allavena et al., 2008). Macrophages are well known to produce cytokines when they sense pathogens or VEGF when they sense hypoxia, but they likely also sense other types of stressors and produce corresponding signals to orchestrate tissue level defenses and adaptations. Similarly, sensory neurons are specialized in sensing various stressors including cold and hot temperature, reactive chemicals, low pH, etc., through the use of molecular sensors such as TRP and ASIC channels (Venkatachalam and Montell, 2007). C-fiber nociceptors locally produce CGRP and Substance P upon activation by these stressors and these neuropeptides coordinate local stress adaptations known as neuro-inflammation (Julius and Basbaum, 2001).
FIGURE 3. Cell Autonomous and Non-Cell Autonomous Responses.

Cellular stress and defense responses have two components – cell autonomous (i.e., intrinsic) and non-cell autonomous (i.e., extrinsic). The former controls intracellular adaptations, while the latter modifies the extracellular environment or communicates to neighboring parenchymal cells. Stress signals also act directly on specialized cells such as macrophages and sensory neurons. These ‘professionals’ are able to detect stress at a lower threshold than parenchymal cells, and they produce cytokines and growth factors to promote restoration of function. Stressed parenchymal cells also communicate with the specialized sensory cells through largely unknown paracrine factors.
Similar to cellular stress and defense responses, which can be cell autonomous but may also involve cell extrinsic components, tissue level stress and defense responses can be tissue autonomous and/or may engage tissue extrinsic help. These tissue extrinsic responses involve recruitment of specialized cells either from other locations within the tissue (e.g., tissue resident macrophages) or from the circulation (e.g., blood monocytes). Indeed, chemokines are one common component of the extracellular response to different stressors (Hitchon et al., 2002; Kanda et al., 2006; Qian et al., 2011). Tissue extrinsic responses are particularly important when tissue autonomous responses are insufficient to resolve the insult. They also share many features with the inflammatory response. Therefore, inflammation can be viewed as a more extreme version of the tissue level stress response. Because the tissue level stress response is not equivalent to the classical acute inflammatory response in that it does not necessarily involve exudate formation and neutrophil recruitment, we refer to it here as para-inflammation (Medzhitov, 2008). There are many examples of conditions that fall under the definition of para-inflammation in that they are clearly not normal, and yet, do not include all the hallmarks of inflammation. These include aging-associated inflammation (Goto, 2008), including age-related macular degeneration (Parmeggiani et al., 2012), as well as inflammation caused by metabolic stress, including obesity (Gregor and Hotamisligil, 2011; Odegaard and Chawla, 2013). The tissue level stress response and para-inflammation are engaged when the level of stress is greater than what can be managed by cellular homeostatic mechanisms (as described above). However, if the level of stress is greater still, for example due to infection or tissue injury, these responses also become insufficient and a bone fide inflammatory response ensues (Figure 1).
Inflammation
The inflammatory response is engaged when tissue autonomous defenses are insufficient or overwhelmed. The acute inflammatory response relies upon specialized cell types (neutrophils, monocytes, eosinophils and basophils) that are typically recruited from the circulation. The sensory cells involved in initiating the inflammatory response are the same as the ones involved in tissue level stress and defense responses: tissue resident macrophages, mast cells (in some tissues) and sensory neurons (mainly nociceptors). These sensory cells detect noxious insults (pathogens, toxins, irritants, etc.) either directly (e.g., through pattern recognition receptors), or indirectly through their effect on tissue homeostasis (e.g., by sensing unscheduled cell death, ECM degradation products, tissue damage, etc.) (Medzhitov, 2010). Thus, according to the definition introduced above, inflammation has both a stress response component and a defense response component (Figure 2).
First, inflammation can be induced as a result of extreme deviations in regulated variables of cellular and tissue homeostasis (i.e., the stress response component of inflammation). Thus, inflammation is at the extreme end of the spectrum of adaptive responses aimed to protect tissue homeostasis. Second, inflammation can be induced by agents that cause disruption of tissue homeostasis, including pathogens, toxins and xenobiotics (i.e., the defense response component of inflammation). These agents can be sensed using two strategies: a direct mechanism which relies upon recognition of structural features, or an indirect mechanism, which relies upon recognition of functional features.
Whenever possible, direct recognition of the disruptive agent is used to elicit the inflammatory response, even before it has a chance to cause any harm. Pattern recognition receptors (PRRs) of the innate immune system are the most prominent example of this sensing mechanism. Sensing bacterial lipopolysaccharide (LPS), for example, will elicit an inflammatory response even though LPS by itself does not cause disruption of homeostasis. The rationale for such response is that the presence of LPS in normally sterile tissues signifies the presence of pathogens that can disrupt homeostasis and cause tissue damage. Thus, the strategy of direct recognition can detect an agent before it can cause any harm; its recognition can be dissociated from the negative consequences of its presence. As such, LPS can cause an inflammatory response regardless of whether it is present in the context of a pathogen or not.
The second strategy of recognition is used for agents that can cause disruption of homeostasis or tissue damage but cannot be sensed directly. This category includes many allergens, toxins, and poisons. These agents are sensed through recognition of functional features, such as characteristic enzymatic activity, membrane pore formation, or chemical conjugation (adduct formation). Thus, some noxious stimuli can be sensed by TRP channels on nociceptor neurons. For example, TRPA1 can detect many noxious chemicals, including formalin, acrolein (tear gas), isothiocyanates (mustard, horseradish, and wasabi oils), allicin (garlic), and cinnamaldehyde (cinnamon), through sensing of shared functional chemical features such as highly reactive electrophile groups (Bautista et al., 2013; Venkatachalam and Montell, 2007). The NLRP3 inflammasomes, on the other hand, monitor membrane integrity and can sense pore-forming toxins, crystals, and many other noxious stimuli (Rathinam et al., 2012). Similarly, many allergens are sensed by detection of their enzymatic activities or other functional features (Palm et al., 2012). In all these cases, functional feature recognition is coupled with the induction of a defensive inflammatory response that promotes adaptation and elimination of the noxious stimulus. In addition, this strategy of sensing is also used by nociceptors to trigger non-inflammatory protective responses to low levels of noxious stimuli before they can cause much damage.
Conclusions and perspectives
Inflammation has traditionally been considered to be a defense response induced by infection or injury. It is increasingly appreciated, however, that chronic inflammation can accompany many pathological states without infection or injury, such as in adipose tissue of obese humans and animals (Hotamisligil, 2010). It has been shown, for example, that when the fat storage capacity of adipocytes is exceeded, an ER stress response is initiated resulting in production of inflammatory cytokines – a condition referred to as metabolic inflammation (Hotamisligil, 2006). Recent findings along these lines increasingly suggest a close connection between inflammatory and stress responses, though the nature of that relation remains somewhat ambiguous. Here we suggest two perspectives to address this issue.
Inflammation can be viewed as the end of the spectrum of mechanisms that maintain and defend homeostasis. This spectrum is formed by homeostatic mechanisms that operate under normal conditions: stress and defense responses that are engaged when homeostatic capacity is insufficient; para-inflammation – a tissue level stress response that has some but not all of the characteristics of inflammation; and finally inflammation proper, which is induced when other mechanisms are insufficient or incapable to maintain homeostasis.
We distinguish two types of stimuli that can induce inflammation. The first are extreme deviations of regulated variables, which cannot be handled by homeostatic mechanisms. This type of inflammation is an extension of a stress response. The second are stimuli (including pathogens, toxins, and allergens) that are not regulated variables themselves but can affect regulated variables thereby disrupting homeostasis. These agents can be sensed either directly through structural feature recognition, as is the case with pattern recognition, or indirectly through their functional features, such as enzymatic activities and disruption of membrane integrity. The inflammatory responses induced by the two types of stimuli are presumably qualitatively distinct. Inflammation induced by deviation of regulated variables is likely to contain homeostasis-restoring components, inflammation induced by infection is coupled with induction of antimicrobial immune responses, and inflammation induced by tissue damage involves a tissue reparative component. Future mechanistic studies should define these distinct components and their contribution to defense of homeostasis and inflammatory pathologies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CITATIONS
- Ahern GP. Transient receptor potential channels and energy homeostasis. Trends in endocrinology and metabolism: TEM. 2013;24:554–560. doi: 10.1016/j.tem.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Critical reviews in oncology/hematology. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Anckar J, Sistonen L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Advances in experimental medicine and biology. 2007;594:78–88. doi: 10.1007/978-0-387-39975-1_8. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Drews-Elger K, Estrada-Gelonch A, Minguillón J, Morancho B, Santiago V, López-Rodríguez C. Regulation of the hypertonic stress response and other cellular functions by the Rel-like transcription factor NFAT5. Biochemical pharmacology. 2006;72:1597–1604. doi: 10.1016/j.bcp.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: A gatekeeper for inflammation. Annual review of physiology. 2013;75:181–200. doi: 10.1146/annurev-physiol-030212-183811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman TG. The community of the self. Nature. 2002;420:246–251. doi: 10.1038/nature01260. [DOI] [PubMed] [Google Scholar]
- Cao SS, Kaufman RJ. Unfolded protein response. Current biology : CB. 2012;22:R622–626. doi: 10.1016/j.cub.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Costanzo LS. Physiology. Philadelphia, PA: Saunders/Elsevier; 2010. p. 1.p. xi.p. 493. online resource. [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolaeva MA, Segref A, Dakhovnik A, Ou HL, Schneider JI, Utermöhlen O, Hoppe T, Schumacher B. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature. 2013;501:416–420. doi: 10.1038/nature12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Kopin IJ. Evolution of concepts of stress. Stress (Amsterdam, Netherlands) 2007;10:109–120. doi: 10.1080/10253890701288935. [DOI] [PubMed] [Google Scholar]
- Gordon S. The macrophage: past, present and future. European Journal of Immunology. 2007;37(Suppl 1):S9–17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- Goto M. Inflammaging (inflammation + aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Bioscience trends. 2008;2:218–230. [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis and rheumatism. 2002;46:2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- Holm CK, Paludan SR, Fitzgerald KA. DNA recognition in immunity and disease. Current Opinion in Immunology. 2013;25:13–18. doi: 10.1016/j.coi.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nature Reviews Immunology. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K-i, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Marié IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Current opinion in virology. 2011;1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Mense SM, Sengupta A, Zhou M, Lan C, Bentsman G, Volsky DJ, Zhang L. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiological genomics. 2006;25:435–449. doi: 10.1152/physiolgenomics.00315.2005. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothersill C, Seymour CB. Radiation-induced bystander effects--implications for cancer. Nature reviews Cancer. 2004;4:158–164. doi: 10.1038/nrc1277. [DOI] [PubMed] [Google Scholar]
- Murray JI, Whitfield ML, Trinklein ND, Myers RM, Brown PO, Botstein D. Diverse and specific gene expression responses to stresses in cultured human cells. Molecular biology of the cell. 2004;15:2361–2374. doi: 10.1091/mbc.E03-11-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhofer W, Beck FX. Cell survival in the hostile environment of the renal medulla. Annual review of physiology. 2005;67:531–555. doi: 10.1146/annurev.physiol.67.031103.154456. [DOI] [PubMed] [Google Scholar]
- Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of biological chemistry. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A. The immune system as a sensor of the metabolic state. Immunity. 2013;38:644–654. doi: 10.1016/j.immuni.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page TJ, Sikder D, Yang L, Pluta L, Wolfinger RD, Kodadek T, Thomas RS. Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Molecular bioSystems. 2006;2:627–639. doi: 10.1039/b606129j. [DOI] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484:465–472. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmeggiani F, Romano MR, Costagliola C, Semeraro F, Incorvaia C, D’Angelo S, Perri P, De Palma P, De Nadai K, Sebastiani A. Mechanism of inflammation in age-related macular degeneration. Mediators of inflammation. 2012;2012:546786. doi: 10.1155/2012/546786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annual Review of Pharmacology and Toxicology. 2008;48:1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Trophic macrophages in development and disease. Nature Reviews Immunology. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annual review of nutrition. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nature Immunology. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends in genetics : TIG. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Developmental cell. 2003;4:442–444. doi: 10.1016/s1534-5807(03)00100-x. [DOI] [PubMed] [Google Scholar]
- Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K, Tsuruo T, Tomida A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer research. 2009;69:4225–4234. doi: 10.1158/0008-5472.CAN-08-2689. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science (New York, NY) 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. Reflex control of immunity. Nature Reviews Immunology. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annual Review of Biochemistry. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature reviews Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death and Differentiation. 2008;15:621–627. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]