

SUMMARY
Defects in the Piwi/piRNA pathway lead to transposon desilencing and immediate sterility in many organisms. We found that the C. elegans Piwi mutant prg-1 became sterile after growth for many generations. This phenotype did not occur for RNA interference mutants with strong transposon silencing defects and was separable from the role of PRG-1 in transgene silencing. Brief periods of starvation extended the transgenerational lifespan of prg-1 mutants by stimulating the DAF-16/FOXO longevity transcription factor. Constitutive activation of DAF-16 via reduced daf-2 insulin/IGF-1 signaling immortalized prg-1 strains via RNA interference proteins and histone H3 lysine 4 demethylases. In late-generation prg-1 mutants, desilencing of repetitive segments of the genome occurred, and silencing of repetitive loci was restored in prg-1; daf-2 mutants. This study reveals an unexpected interface between aging and transgenerational maintenance of germ cells, where somatic longevity is coupled to a genome silencing pathway that promotes germ cell immortality in parallel to the Piwi/piRNA system.
INTRODUCTION
Somatic cells accumulate stress that limits proliferation within a single generation, whereas germ cells are effectively immortal as they proliferate from one generation to the next. Genetic studies of C. elegans have revealed that telomerase-mediated telomere maintenance is essential for germ cell immortality (Meier et al., 2006) and that several histone modification enzymes contribute to germline maintenance over generations (Andersen and Horvitz, 2007; Buckley et al., 2012; Katz et al., 2009; Xiao et al., 2011). Although deficiency for telomerase in humans is likely to contribute to proliferative aging of somatic cells (Armanios and Blackburn, 2012), other pathways that promote germ cell immortality could be specific to the germ cells, or could reveal new connections between the germline and somatic aging.
Piwi is an Argonaute protein that associates with a diverse class of small RNAs that are abundant in germ cells termed Piwi-Interacting RNAs (piRNAs) (Juliano et al., 2011). Conserved functions for Piwi include suppression of transposons and self-renewal of germline or meristematic stem cells. Deficiency for Piwi and Piwi-like genes in Drosophila, Arabidopsis and vertebrate males results in immediate sterility (Juliano et al., 2011). Further, mating of Drosophila females that lack piRNAs targeting a transposon class with males that possess the transposon yields F1 progeny with a temperature-sensitive embryonic lethal phenotype termed hybrid dysgenesis, accompanied by transposon-induced genome instability (Juliano et al., 2011; Kidwell et al., 1977). Hybrid dysgenesis may be related to the strong immediate sterility phenotype that is accompanied by large-scale desilencing of transposons in Piwi mutants.
C. elegans has two closely related Piwi homologs, PRG-1 and PRG-2, but only deficiency for PRG-1 has phenotypic consequences (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008). PRG-1 is expressed in germ cells, and prg-1 mutants were previously reported to display temperature-sensitive sterility accompanied by transposition of the Tc3 transposon, but not other DNA transposons (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008). These phenotypes could conceivably be related to hybrid dysgenesis in Drosophila (Juliano et al., 2011; Kidwell et al., 1977). In addition, PRG-1 was recently shown to initiate silencing of foreign transgenes (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012), though silencing is then maintained by a number of factors, including small interfering RNA proteins that are responsible for silencing many active transposons in C. elegans.
Here we report that outcrossed prg-1 mutants display a previously unrecognized Piwi phenotype - transgenerational replicative aging of germ cells. The germ cell immortality function of Piwi occurs at multiple temperatures, is separable from its role in transgene silencing, and is not observed for strains that display high levels of transposition. Reduced daf-2/insulin/IGF-1 signaling, which extends somatic lifespan in a variety of species (Kenyon, 2010), restores germ cell immortality to prg-1 mutants by activating an endogenous RNA interference pathway that silences repetitive loci. Together our results place the stem cell self-renewal function of Piwi in the context of transgenerational replicative lifespan of germ cells, suggesting a heritable epigenetic factor that could regulate the rate of aging in stem cells.
RESULTS
Deficiency for prg-1 results in progressive sterility
To study the effects of Piwi on fertility in C. elegans, we backcrossed three alleles of prg-1 and four alleles of prg-2 (Batista et al., 2008; Wang and Reinke, 2008), thereby removing unlinked mutations and/or epigenetic effects of the parental backgrounds. In contrast to previous findings (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008), we did not generally observe strong defects in fertility at high temperature for outcrossed prg-1 mutants. Instead, slightly reduced brood sizes occurred for maternally depleted F3 prg-1 homozygotes at both 20°C and 25°C in comparison to N2 wild-type controls (Figure 1A), and a minority of prg-1 mutants displayed >80% embryonic lethality at 25°C (Figures 1A and S1A). We saw robust levels of fertility during propagation of prg-1 and prg-2 for 8 generations at either 20°C or 25°C based on Mortal Germline (Mrt) assays, where 6 L1 larvae were transferred to freshly seeded plates once per week (Ahmed and Hodgkin, 2000; Meier et al., 2006). No strain displayed even a moderate reduction in fertility during this period, based on complete consumption of the E. coli lawn (Ahmed and Hodgkin, 2000; Meier et al., 2006). Continued propagation of prg-1, but not prg-2, mutants resulted in drops in fertility and ultimately complete sterility (Figures 1B and S1B). This drop in fertility occurred both at 20°C and 25°C. Deficiency for three of four prg-2 alleles failed to exacerbate the fertility defects of prg-1 (Figure 1C). Although accelerated sterility was observed for prg-1; prg-2(ok1328) double mutants, this likely reflects an effect of a background mutation linked to ok1328. Thus loss of function of prg-1 leads to progressive loss of fertility over several generations, independent of prg-2.
Figure 1. Progressive sterility of prg-1 mutants.

(A) Levels of fertility for different prg-1 mutants at 20°C and 25°C (mean±s.d., n=10 lines per strain). Individuals from independently derived lines were singled and their progeny counted. (B) prg-1 exhibits progressive sterility at both 20°C and 25°C. Animals at 25°C appear more robust than siblings at 20°C (Mantel-Cox log-rank test, P=0.001 for tm872, P=0.030 for pk2298, P=0.144 for n4357) (n=12). (C) Progressive sterility of prg-1(tm872); prg-2 double mutants (n=5 strains per genotype). (D) Starvation extends transgenerational lifespan of prg-1 strains propagated at 25°C. Extension by starvation is dependent on daf-16.
We confirmed that prg-1 deficiency was the cause of progressive sterility in the above experiments with a rescuing transgene that expresses wild-type PRG-1 (Figure S1C) (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008). Some Argonaute proteins, including PRG-1, contain ‘Slicer’ domains that possess nuclease activity. We therefore tested a transgene that expresses Slicer-dead PRG-1 (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008), which also rescued the progressive sterility phenotype caused by prg-1 deficiency (Figure S1C). Consistently, Slicer nuclease activity is not required for piRNA-dependent genome silencing by PRG-1 (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008).
Deficiency for daf-2 suppresses progressive sterility of prg-1 mutants
Although prg-1 mutants have been previously reported to display sterility at 25°C (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008), we found that outcrossed prg-1 mutants became sterile more slowly at 25°C (48.4+/−4.3 generations to sterility) than at 20°C (24.9+/−2 generations to sterility; P=3.38e-5 Mantel-Cox log-rank test; n=40 strains per genotype) (Figure 1B). Thus, growth at high temperature doubled the transgenerational lifespan of prg-1 mutants (the mean number of generations that prg-1 mutant strains reproduce prior to becoming sterile). The term ‘transgenerational lifespan’ reflects the proliferative capacity of germ cells across generations, and was inspired by studies of ‘replicative lifespan’ that assess cellular aging in yeast and mammalian cells (Polymenis and Kennedy, 2012; Smelick and Ahmed, 2005). In contrast, ‘adult lifespan’ concerns aging that occurs in a single generation (Kenyon, 2010).
While studying the transgenerational lifespan of prg-1 mutants, we noticed that propagation at 25°C led to frequent starvation of the plates prior to transfer, which was rare at 20°C. We therefore repeated the assay ensuring that prg-1 strains did not starve at 25°C, and found that this eliminated the extension of transgenerational lifespan that was observed for prg-1 mutants that starve transiently (Figure 1D). Typically, C. elegans stocks are subjected to long periods of starvation and are infrequently outcrossed. We hypothesize that these conditions suppress the Mrt phenotype of prg-1 mutants and instead cause a distinct and possibly related epigenetic defect that is manifest as temperature-sensitive sterility (Batista et al., 2008; Wang and Reinke, 2008).
Many effects of starvation in C. elegans are triggered by activation of the DAF-16/FOXO transcription factor that promotes stress resistance and longevity (Kenyon, 2010; Lin et al., 1997; Ogg et al., 1997). We therefore constructed prg-1 daf-16 double mutants and found that their transgenerational lifespan was not extended by starvation (Figure 1D), implying that transient activation of DAF-16 in response to weekly bouts of starvation extends the transgenerational lifespan of prg-1 mutants. We next constitutively activated DAF-16 using three independent alleles of daf-2, which encodes the sole C. elegans homolog of mammalian insulin or IGF-1 receptors and negatively regulates DAF-16 (Kimura et al., 1997). We found that daf-2 mutations strongly suppressed the progressive sterility phenotype of prg-1. Remarkably, almost all prg-1; daf-2 double mutant strains could be propagated indefinitely (n=53/54 total) (Figures 2A and S2A).
Figure 2. daf-2 signaling can suppress fertility defects of prg-1 and modulates germline remodeling at sterility.

(A) Reduced daf-2 signaling suppresses prg-1 mediated progressive sterility. (B) lin-15B does not suppress transgenerational lifespan of prg-1 (n=30 strains per genotype). daf-16 and daf-18 are required for suppression of prg-1 by daf-2. Four prg-1 daf-16 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with daf-16 alleles mgDf50 or mu86 (n=10 strains per genotype). Four prg-1; daf-18 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with daf-18 alleles e1375 and ok480 (n=10 strains per genotype). For prg-1 daf-16; daf-2 triple mutants, prg-1(tm872) daf-16(mgDf50) and prg-1(n4357) daf-16(mu86) were each combined with three daf-2 alleles m41, e1368 and e1370 (n=10 strains scored per genotype) and examined for progressive sterility. Four prg-1; daf-2(e1368); daf-18 lines were constructed from the four allelic combinations of prg-1; daf-18 and examined for progressive sterility (n=10 strains scored per genotype). Data for all independent alleles was combined to show transgenerational lifespan for strains of the same genotype. (C) Lack of immediate sterility upon removal of daf-2 suggests suppression of the heritable epigenetic defect that causes sterility in prg-1 mutants. prg-1; daf-2(+/+) strains represent cross-progeny of late-generation prg-1; daf-2 double mutants where the daf-2 mutation has been removed. (D–E) Levels of spontaneous mutation assessed by reversion frequencies of unc-58 and unc-54. (F) Treatment of sterile late-generation prg-1 adults with daf-2 or age-1 RNAi restores fertility, and fertility can be maintained on these RNAi strains for at least 10 generations while RNAi is maintained.
daf-2 mutations promote stress resistance and dauer formation through DAF-16/FOXO (Kenyon, 2010). prg-1 daf-16; daf-2 triple mutants became progressively sterile, indicating that daf-2 deficiency suppresses the fertility defects of prg-1 by activating DAF-16 (Figure 2B). Transgenerational lifespan of prg-1 was reduced by ~30% for prg-1 daf-16 or prg-1 daf-16; daf-2 strains (P=5.05E-03 and 1.56E-03, respectively, Mantel-Cox log-rank test) (Figure 2B). We confirmed these observations using independent mutations in daf-18, which functions upstream of DAF-16 to promote longevity in response to reduced DAF-2 signaling (Ogg and Ruvkun, 1998), and found that prg-1; daf-18 double mutants also displayed shortened transgenerational lifespans (Figure 2B). Neither daf-16 nor daf-18 single mutants become sterile in Mortal Germline assays (Ahmed, 2006).
Reduced daf-2 activity is associated with an enhanced response to exogenous RNA interference and a soma-to-germline transformation (Curran et al., 2009; Wang and Ruvkun, 2004). However, these phenotypes also occur when lin-15B is deficient (Wang et al., 2005), and lin-15B did not suppress progressive sterility of prg-1 (Figure 2B).
Together our data reveal a specific response downstream of inactivation of daf-2 that allows animals to remain fertile in the absence of prg-1. As prg-1; daf-2 lines can be propagated indefinitely, we wondered whether these animals accumulate defects that cause them to become sterile immediately when daf-2 activity is restored - in other words whether daf-2 protects against accumulation of damage or simply allows animals to tolerate high levels of damage. To test this we crossed late-generation prg-1; daf-2 double mutants with early-generation prg-1 single mutant males and selected 16 prg-1 −/−; daf-2 +/+ lines descended from prg-1 −/−; daf-2 +/− F1 animals (Figure S2E). Though these lines maintained fertility for 10 generations, they became progressively sterile in a manner similar to prg-1 single mutants (Figure 2C). Thus, daf-2 deficiency directly prevents damage accumulation in prg-1 mutants.
Progressive sterility of prg-1 does not result from transposition
Having established that daf-2 represses the source of progressive sterility of prg-1 directly, we next wished to consider what the initial source of the damage might be. A conserved function of the Piwi/piRNA pathway is suppression of transposons (Juliano et al., 2011). The temperature-sensitive hybrid dysgenesis phenotype of Drosophila is caused by deficiency for a set of piRNAs, which elicits a form of sterility accompanied by high levels of transposon expression and activity (Kidwell et al., 1977). Thus, prg-1 mutants could become sterile as a consequence of transposition (Batista et al., 2008; Das et al., 2008). We carried out Comparative Genomic Hybridization (CGH) arrays to assay for increased transposon copy number in late-generation prg-1 mutants and found that indeed there was overall slightly increased transposon DNA in late-generation prg-1 compared to early-generation (Figures S2B and S2D), which was not observed for genes and simple repetitive regions (Figure S2C). However, several lines of evidence suggest that this is unlikely to explain the prg-1 fertility defects. First, inspection of many independent late-generation prg-1 strains failed to reveal frequent de novo mutations that cause visible phenotypes, which are readily observed for C. elegans strains with weak (10-fold) increases in the frequency of spontaneous mutation (Harris et al., 2006). To confirm this, forward mutation assays were conducted using unc-54(r293), which can be suppressed by mutation of any of seven smg genes, and using unc-58(e665), which can be suppressed by mutations in two loci (Harris et al., 2006). The frequency of forward mutation for prg-1 unc-54(r293) or prg-1; unc-58(e665) in either early-generation (F4) or late-generation strains very close to sterility (F24) was comparable to unc-54(r293) and unc-58(e665) single mutant controls (Figures 2D and E, Table S1). This rules out an increased spontaneous mutation frequency in prg-1 mutants. Second, RNA interference mutants rde-2 or mut-2, which are known to confer elevated levels of transposon mobility (Ketting and Plasterk, 2000; Tabara et al., 1999), did not become sterile when propagated at 20°C, despite causing ~6- to 12-fold increases in the frequency of spontaneous mutation (Figures 2D, 2E and Table S1). Third, we did not observe any evidence of chromosomal instability as measured by 4′,6-diamidino-2-phenylindole (DAPI) staining in late-generation prg-1 animals compared to either early-generation prg-1 or wild-type animals (Figure S1D) (Ahmed and Hodgkin, 2000).
Finally, if the prg-1 defects were caused by increased transposition or associated genomic instability we would not expect them to be reversible. We therefore treated late-generation sterile prg-1 adults with RNAi against either daf-2 or age-1, which also negatively regulates daf-16, and found that RNAi of either gene allowed some infertile adults to produce viable offspring again (Figure 2F). Further, fertility could be maintained for at least 10 additional generations by maintaining lines with RNAi treatment. The reversibility of the sterility phenotype makes it highly unlikely that sterility is caused by accumulation of deleterious transposon insertions. Our results instead suggest that the sterility phenotype of prg-1 mutants is likely due to accumulated epigenetic changes.
Transgene silencing is separable from germ cell immortality
PRG-1 can initiate silencing of foreign transgenes in C. elegans, which is then maintained by nuclear silencing proteins and by the Mutator-class secondary siRNA biogenesis protein MUT-7 (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012). We confirmed that Mutator-class genes rde-2 and mut-2 were required for transgene silencing (Figure S2F–K). Outcrossed mut-2, mut-7 and rde-2 mutants are not Mrt (Figure 5F), implying that silencing of foreign transgenes may not be linked to germ cell mortality of prg-1 mutants. Direct evidence for a distinction between transgene silencing and the Mrt phenotype of prg-1 was obtained using a silent transgene placed in a prg-1 mutant background (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012) and remained silent in sterile late-generation adults (Figure S2N–O).
Figure 5. Altered 22G-RNA frequencies and gene expression changes in prg-1 mutants.

(A) 22G-RNAs targeting transposons in prg-1 versus wild-type strains. (B) Increased 22G reads in late-generation prg-1 strains mapping to transposons are reduced in prg-1 mutants deficient for the Mutator pathway genes rde-2 or mut-7. (C) 22G-RNAs targeting tandem repeats in prg-1 versus wild-type strains. Note that it is easier to clearly identify transposon 22G-RNAs, because many permutations of each tandem repeat are found, so tandem repeat 22G-RNA data are almost certainly underestimated. (D) RT-PCR reveal increased expression of 174 mer tandem repeat for strains carrying the ypEx3 extrachromosomal array in comparison to sibling control strains lacking this array or prg-1 single mutant controls. (E) Repetitive extrachromosomal arrays containing CeRep59 and a 174 mer tandem repeats (ypEx3 array) or a histone locus cluster containing his-13, his-14, his-15, and his-16 genes (ypEx4 array) or a Helitron transposon (ypEx5 array) reveal that ypEx3 array accelerates the progressive sterility phenotype of prg-1. (F) rde-2 and ppw-1 are required for suppression of prg-1 by daf-2. Two ppw-1 prg-1 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with ppw-1(pk1425) (n=10 strains per genotype). Four ppw-1 prg-1; daf-2 triple mutants were studied where both ppw-1 prg-1 mutants were combined with daf-2 alleles e1370 or e1368 (n=5 strains per genotype). Two rde-2 prg-1 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with rde-2(ne221) (n=10 strains per genotype). Six rde-2 prg-1; daf-2 triple mutants were studied where both rde-2 prg-1 mutants were combined with daf-2 alleles e1370, e1368 and m41 (n=5 strains per genotype). (G) rde-2 is required for restoration of tandem silencing in prg-1 mutants by daf-2. (H) rbr-2 and spr-5 are required for suppression of prg-1 by daf-2. Four prg-1; rbr-2 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with rbr-2 alleles ok2544 or tm1231 (n=10 strains per genotype). Eight prg-1; daf-2; rbr-2 triple mutants were studied where each prg-1; rbr-2 mutant was combined with daf-2 alleles e1370 or e1368 (n=5 strains per genotype). Two prg-1 spr-5 double mutant strains were studied where prg-1 alleles tm872 and n4357 were combined with spr-5(by134) (n=10 strains per genotype). Three prg-1; daf-2; rbr-2 triple mutants were studied where each prg-1; rbr-2 mutant was combined with daf-2(e1370) and prg-1(n4357) spr-5 was combined with daf-2(e1368) (n=10 strains per genotype).
Disrupted silencing of repetitive loci in prg-1 mutants
PRG-1 interacts with ~16,000 21 nucleotide (nt) RNAs possessing 5′ uracil that represent the C. elegans piRNA repertoire and silence many segments of the genome (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008). PRG-1 has been implicated previously in small RNA-mediated silencing pathways (Batista et al., 2008; Das et al., 2008) and indeed can trigger transgenerational silencing that can persist in the absence of prg-1 (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012). Therefore to search for possible determinants of the epigenetic defects responsible for sterility, we examined the functional consequences of prg-1 deficiency on small RNA-mediated silencing pathways. We searched for changes in small RNAs or their targets that would become more severe over continued propagation and could be suppressed by deficiency in daf-2.
In Drosophila, piRNAs can be inherited through the female germline (Brennecke et al., 2008). We therefore asked first whether a progressive loss of piRNAs occurred during propagation of prg-1 mutants. We prepared small RNA libraries from wild-type animals and prg-1 mutant animals at different generations. We found that piRNAs are essentially absent from prg-1 strains at generations 4 and 8, as well as generation 12, which was close to sterility for both alleles examined (Figure 3A). As piRNA annotation is based on a genomic motif likely associated with piRNA biogenesis (Ruby et al., 2006), the few remaining small RNAs that fit our piRNA criteria are likely misannotated rather than persistent piRNA species. Progressive loss of piRNAs is therefore unlikely to explain the delay in loss of fertility in prg-1 animals.
Figure 3. piRNA loss affects expression of few genes targeted by piRNAs.
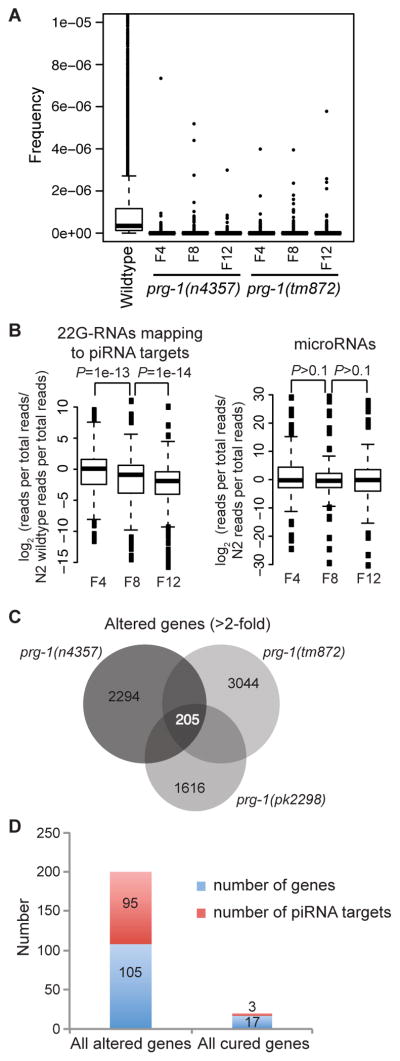
(A) Boxplots are based on read frequencies for 4,839 piRNAs sequenced in one or more libraries. Boxes indicate interquartile ranges, horizontal bars medians, whiskers extend to the most extreme data points with distance from the box no more than 1.5 times the interquartile range, crosses indicate outliers. (B) Boxplots showing reduced median levels of 22G-RNAs for piRNA targets in later generation prg-1 animals. Levels of microRNAs do not show the same progressive reduction. P values are for Wilcoxon signed rank tests. (C) Analysis of genes whose expression changed more than 2-fold in late- versus early-generation prg-1 strains reveals few common genes are altered for prg-1 alleles. (D) Few altered genes that are cured by daf-2 are predicted piRNA targets.
Another possible cause of transgenerational sterility might be progressive changes in gene expression due to loss of piRNA-mediated silencing. In C. elegans, 21 nt piRNAs silence their targets by engaging an endogenous secondary siRNA silencing pathway that utilizes so called 22G-RNAs that are 22 nt in length and possess a 5′ guanine nucleotide (Bagijn et al., 2012). For genes targeted by piRNAs, we observed a progressive reduction in the number of 22G-RNA reads, normalized to library size, mapping to these genes in later generation prg-1 animals (Figure 3B). No such progressive reduction was seen in microRNA levels. We tested the hypothesis that expression of 22G-RNA target genes might be improperly regulated by preparing RNA from early- and late-generation prg-1 strains and using cDNA created from this RNA to perform tiling microarrays that interrogate the entire C. elegans genome. Analysis of microarrays for three independent alleles of prg-1 showed little consistent change in the expression of genes targeted by 22G-RNAs (data not shown). Instead, we identified a set of 205 genes whose expression changed by more than 2-fold in late-generation prg-1 strains (Figure 3C), an intersection significantly greater than that predicted by 1000 simulations of random overlap (Z=35) and all but 3 of these genes changed in the same direction in all alleles, more than 10-fold greater than expected by chance (P<2e-16, χ2 test, two-tailed). However, the expression changes of only 20 of the 205 genes were significantly reduced by more than 1.4-fold (0.5 Log2 units) in late-generation prg-1; daf-2 mutants (P<0.05, Student’s T-test, two-sided, log scale) and only 3 were piRNA targets (Figures 3D and S3, Table S3), implying that daf-2 deficiency does not suppress the fertility defects of prg-1 mutants by restoring piRNA-dependent gene expression changes.
As mentioned above, piRNAs in a number of organisms, including C. elegans, have been shown to target transposons for silencing (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008; Juliano et al., 2011). Thus, the epigenetic defect of prg-1 germ cells could be due to derepression of repetitive genomic loci that are targeted by PRG-1 piRNAs. Using genome-wide tiling arrays, we observed an increase in the expression of a subset of transposons in late-generation prg-1 mutants (Figures 4A and 4B). The most up-regulated were the Mariner class transposons, including the previously characterized piRNA target Tc3 (Figure S4A) (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008). Intriguingly, we also observed increased expression of both simple repeat regions and tandem repeat tracts (hereafter referred to as simple repeats) across the genome in late generations for three different alleles of prg-1, with the overlap between upregulated repeat tracts highly statistically significant relative to random simulation. The statistical significance of this overlap in simple repeats was also significantly larger than for genes (Z=214 for simple repeats, Z=35 for genes) (Figures 3C and 4C). This was not accompanied by increased DNA copy number from these regions (Figure S2B–D). In contrast to the general lack of suppression of gene expression changes by daf-2 mutation, changes in expression of repetitive regions, including transposons and simple repeats, were all robustly suppressed for four different allelic combinations of prg-1; daf-2 (Figures 4D–I and S4D–E) (P<2e-16, two-sided paired T-test), including most of the 101 longest tandem repeat tracts found in the C. elegans genome (Figure 4I). Further, reduced levels of tandem repeat RNA were observed for independent repetitive loci for lines derived from sterile prg-1 mutant adults whose fertility was restored by daf-2 RNAi (Figure S4F). Thus, the expression of repetitive elements rather than the increased expression of genes might be a crucial factor in the acquisition of the sterility phenotype.
Figure 4. Derepression of repetitive elements in prg-1 mutants is cured by daf-2.

(A) Late-generation prg-1 mutants show a mean increase in transposon expression, compared to the expression of transposons in prg-1; daf-2 double mutants. (B) Density plot of the transposon expression changes shown in Figure 4A reveal increased expression of a subset of transposon sequences in late-generation prg-1 animals that is not seen in prg-1; daf-2 double mutants. (C) Simple repeats are upregulated in late-generation prg-1 mutants. (D) Simple repeats upregulated in prg-1 mutants are repressed in late generation prg-1; daf-2 mutants. (E–H) cDNA prepared from RNA was hybridized to microarrays revealed upregulation of tandem repeat tracts expression in late-generation prg-1 mutants but not in late generation prg-1; daf-2 mutants (E and G), as confirmed by RT-PCR analysis for late-generation wild-type as well as early-(E), and late-(L) generation mutant strains (F and H). F and H show expression of tandem repeats corresponding to E and G, respectively. (I) Genome-wide plots of 101 longest tandem repeat tracts defined by visually scanning the C. elegans genome in 70 kb sliding windows. Typically, tandem repeats display increased expression in late-generation prg-1(tm872) and prg-1(n4357) single mutants but silencing in prg-1 double mutants with daf-2 alleles e1368, e1370 or m41.
We asked whether progressive loss of 22G-RNAs might account for loss of silencing of transposons and tandem repeats. Overall levels of secondary 22G-RNAs mapping with up to two mismatches to transposon consensus sequences, normalized to the levels of a somatic microRNA, were reduced in early-generation prg-1 strains (P=0.0008, 1 sample T-test, two-sided) (Figures 5A and S4G). In later generations, 22G-RNAs targeting some transposons were further reduced, whereas 22G-RNAs for other transposon classes were restored to high levels, reflected by an apparently bimodal distribution of read differences relative to N2 wildtype (Figure 5A). Transposons with increased 22G-RNAs in late-generation prg-1 animals were enriched for the Tc4 family of transposons, whilst transposons with reduced 22G-RNAs were enriched for the Mariner family (P <0.05, Fisher’s Exact Test) (Figures S4B and S4C). Transposons showing increased 22G-RNAs in late-generation prg-1 animals were accompanied by increased 22G-RNA to 22A-RNA ratios (Figure S4H), suggesting that these increases are not due to degraded transposon RNA. Moreover, RNA levels of transposons showing increased 22G-RNAs tended to show smaller increases in late-generation prg-1 than those with reduced 22G-RNAs (P<0.05, two-tailed unpaired T-test) (Figure S4I), suggesting that the increased 22G-RNAs contributed to repression of their targets. The bimodal distribution was suppressed in double mutants lacking both prg-1 and either mut-7 or rde-2/mut-8, which encode Mutator/RNA interference proteins that target transposons for silencing; thus the increased small RNAs against transposons in late-generation prg-1 are dependent on mut-7 and rde-2/mut-8 (Figure 5B) (Ketting et al., 1999; Tabara et al., 1999). This suggests that in the absence of prg-1, an alternative silencing pathway dependent on the Mutator proteins is induced, which silences the Tc4 family of transposons in particular whilst leaving Mariner transposons with reduced levels of secondary siRNAs.
Outcrossed mut-7 and rde-2 Mutator mutants are not Mrt (Figure 5F), despite being required for general amplification of 22G-RNAs in response to exogenous and endogenous primary siRNAs (Zhang et al., 2011). By comparing the abundance of 22G-RNAs from the prg-1 and mut-7 single mutants with prg-1; mut-7 double mutants, the prg-1; mut-7 double mutants were much more similar to the mut-7 single mutant than to the prg-1 single mutant (Figure S5A). Importantly out of all the transposon consensus sequences, there were no transposons with more than 5 antisense 22G-RNA reads per million in mut-7 that had fewer than 5 reads per million in the prg-1 single mutant, showing that prg-1 is unlikely to operate in parallel to mut-7 at transposons. There were some isolated cases where there were fewer reads in prg-1; mut-7 than in mut-7; however, because these had a larger number of reads in prg-1, it is not straightforward to argue that loss of these small RNAs is related to the Mrt phenotype. Overall, the vast majority of PRG-1-dependent 22G-RNAs are also dependent on MUT-7 and are dispensable for germ cell immortality.
Several upregulated repeat tracts are also predicted targets of at least one piRNA (Tables S4 and S5). We therefore examined 22G-RNAs against simple repeats in prg-1 mutants. The number of 22G-RNAs against simple repeat regions was less in prg-1 mutants than in wild-type animals (P=0.002, 1 sample T-test, two-sided), and overall 22G-RNA levels decreased further in late-generation prg-1 animals (P=0.00026, 1 sample T-test, two-side) (Figure 5C). Furthermore, similarly to transposons, normalized 22G-RNA levels mapping to simple repeats were more widely distributed in late-generation prg-1 animals than in either N2 wild-type or early-generation prg-1 animals (P=6e-6 to wild-type and P= 8e-5 to early-generation prg-1, Kolmogorov-Smirnov test for different distributions) (Figures 5C and S5B). Again, increased 22G-RNAs were dependent on the Mutator pathway (Figure S5C). Simple repeats with increased or decreased 22G-RNA levels in late-generation prg-1 animals were highly upregulated compared to simple repeats with weak or no change in 22G-RNA levels (Figure S5D). Taken together with the analysis of transposons, these data imply that increased expression of repetitive regions of the genome in late-generation prg-1 mutants is accompanied by progressive dysfunction of 22G-RNAs targeting these regions, reflecting both loss of 22G-RNAs downstream of piRNAs, and potentially, upregulation of a prg-1-independent 22G-RNA pathway that can silence a subset of transposons and simple repeats.
We used RNAi to knock down a number of protein coding genes (Figure S5E) and transposons (Figure S5F) that were upregulated in late-generation prg-1 mutants, performing RNAi from early generations onwards, but none repressed the sterility phenotype of prg-1 mutants (P>0.191) (Figures S5E and S5F). We created three repetitive extrachromosomal arrays containing either histone loci, a tandem repeat CeRep59 or the Helitron transposon by microinjection of prg-1 mutants. Strikingly, an array that overexpressed CeRep59 (ypEx3) shortened transgenerational lifespan (P=2.06E-05), whereas Helitron transposon or histone locus arrays had no effect (P>0.29) (Figure 5D, 5E and S5G). Although extrachromosomal arrays can be silenced in the C. elegans germline by cosuppression (Dernburg et al., 2000; Ketting and Plasterk, 2000), RNA Fluorescence In Situ Hybridization revealed that prg-1 strains containing CeRep59 arrays expressed CeRep59 RNA at ~5-fold higher levels than prg-1 single mutant controls in both early embryos and throughout the animals including in germ cells (Figure S6C–R). We also found low levels of repetitive RNA expressed in wild-type embryos (Figure S6A–B), suggesting that repetitive loci are normally transiently expressed during development. These results imply that expression of repetitive loci contributes to the transgenerational sterility of prg-1 mutants.
A small RNA pathway restores germ cell immortality prg-1 mutants
Having established that progressive loss of 22G-RNAs against tandem repeats and transposons occurred in prg-1, we tested whether daf-2 might suppress increased repeat expression via siRNAs. We examined small RNA libraries from prg-1; daf-2 mutants at early- and late-generations. As for prg-1 single mutants, piRNAs were lost from both early- and late-generation prg-1; daf-2. We therefore examined protein-coding genes, transposons and simple repeats showing decreased small RNAs in late-generation prg-1 relative to early-generation for suppression of transgenerational decrease by daf-2. The number of suppressed genes was statistically significant for 22G-RNAs mapping to genes and simple repeats but not transposons, although individual examples of transposons in this category were found (Tables 1 and S6 to S8). In all cases, this suppression involved the Mutator pathway, as the majority of suppressed genes, transposons and repeats showed reduced 22G-RNA reads mapping to them in rde-2, prg-1; daf-2 triple mutants compared to early-generation prg-1 mutants (Table 1). In addition, sequencing of small RNAs from progeny of sterile prg-1 mutants treated with daf-2 RNAi revealed that 22G-RNAs targeting repetitive sequences were partially restored in comparison to late-generation prg-1 controls, though not to early-generation levels (P<1e-16 to late generation, two-tailed paired T-test) (Figure S5H). We therefore hypothesized that daf-2 may suppress activation of repetitive loci by upregulating an alternative prg-1-independent silencing pathway.
Table 1.
Effect of daf-2 on transgenerational alterations in 22G-RNA levels in prg-1.
| 2-fold down late prg-1 vs early prg-1 | Suppressed daf-2 | Significance (Fisher’s exact test) | Suppressed dependent on rde-2 | |
|---|---|---|---|---|
| Transposons | 54 | 4 | 1.00 | 4 |
| Genes | 356 | 71 | 5.80e-29 | 50 |
| Simple Repeats | 497 | 92 | 1.66e-12 | 56 |
We therefore tested whether the suppression of prg-1 fertility defects by daf-2 might be dependent on small RNAs. Deficiency for the rde-2 Mutator class gene abolished the ability of daf-2 mutations to ameliorate the germ cell immortality defects of prg-1 mutants (Figure 5F), and late-generation rde-2 prg-1; daf-2 strains expressed high levels of repetitive RNA (Figures 5G, S5J and S5K). Consistently, another Mutator class gene mut-7 was vital for suppession of prg-1 by daf-2, even though fertility was ameliorated for prg-1; mut-7 double mutants (Figure 5F). We also found that an Argonaute protein required for efficient germline RNAi, PPW-1 (Tijsterman et al., 2002), is necessary for suppression of prg-1 by daf-2 (Figure 5F). We conclude that an endogenous RNA interference pathway that requires RDE-2, MUT-7 and PPW-1 can restore germ cell immortality to prg-1 mutants (Figure 6).
Figure 6. Model of parallel small RNA silencing pathways that can repress transgenerational fertility defects.

(A) Wild-type PRG-1 maintains transgenerational fertility by silencing repetitive RNA expression, and functions separately to initiate silencing of foreign transgenes and most transposons. (B) Deficiency for prg-1 results in transgenerational desilencing of repetitive loci and sterility. Transposon and transgene silencing is maintained by Mutator proteins. (C) Increased DAF-16 signaling via daf-2 mutation suppresses progressive sterility and desilencing of repetitive loci when prg-1 is mutant. Mutations are indicated by a red X. Light color tints indicate pathway dysfunction for prg-1 mutants or low levels of DAF-16 activity in response to wild-type DAF-2 signaling.
Small RNA pathways can promote gene silencing by degrading RNA in the cytoplasm or silencing loci within the nucleus. We tested the hypothesis that the histone H3 lysine 4 (H3K4) demethylase RBR-2, which removes trimethyl H3K4 marks and promotes transcriptional silencing (Christensen et al., 2007), mediates the effects of transgenerational epigenetic marks that regulate somatic longevity in C. elegans (Greer et al., 2010). Further, reduced daf-2 signaling regulates rbr-2 expression (Lee et al., 2003). We found that rbr-2 is required for suppression of prg-1 by daf-2 mutation (Figure 5H). Late-generation prg-1; daf-2; rbr-2 strains expressed high levels of RNA from repetitive loci (Figures S5L–N), implying that RBR-2 demethylase suppresses the transgenerational fertility defects of prg-1 mutants by silencing these loci in response to reduced daf-2 signaling. We then tested a second demethylase, SPR-5, which removes dimethyl H3K4 marks. Although one allele of spr-5 has been reported to be Mrt at 20°C (Katz et al., 2009), we tested another null allele of spr-5, by134, which does not display fertility defects at 20°C. We then used spr-5 to confirm that H3K4 demethylation is required for suppression of prg-1 by daf-2 (Figure 5H).
Taken together therefore these data suggest that the transgenerational silencing defects of prg-1 can be suppressed by a small RNA-mediated genome silencing that is activated by reduced insulin/IGF-1 signaling (Figure 6).
DISCUSSION
Here we demonstrate that C. elegans prg-1 is required for germ cell immortality. This phenotype shows two clear distinctions from the role of Piwi proteins in promoting fertility in other organisms. First, prg-1 mutant animals do not become sterile immediately, and indeed maintain wild-type levels of fertility for several generations before becoming progressively sterile. Second, we show that the sterility of prg-1 animals is not due to increased transposition. Instead, our data supports a transgenerational epigenetic cause of sterility in prg-1 mutants.
A role for PRG-1 in transgenerational epigenetic maintenance of fertility in C. elegans connects to recent data showing that prg-1 acts upstream of epigenetic silencing of foreign transgenes in C. elegans germ cells (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012). In the case of transgenes however, the silent state is maintained independently of PRG-1 activity by proteins that mediate downstream 22G-RNA production, such as RDE-2, by nuclear RNAi factors, and by chromatin silencing proteins (Figure 6A–B) (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012). Several nuclear RNAi factors were recently reported to promote germ cell immortality (Buckley et al., 2012), and PRG-1 and associated piRNAs could function upstream of these proteins to direct silencing of endogenous nuclear loci. However, we show that PRG-1 is also required continuously for germ cell immortality, suggesting that ultimately prg-1 is indispensable for silencing of some endogenous loci. A second contrast with transgene silencing is that Mutator mutants such as rde-2 and mut-7, which display a strongly reduced secondary siRNA response, are wild-type for germ cell immortality at low temperatures (20°C). These mutants also maintain silencing of repetitive loci that become de-repressed in late-generation prg-1 mutants (Figures 5F, S5I), despite being essential for maintenance of transgene silencing (Figures S2F–K, Table S2) (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012). Thus, continuous initiation of silencing by PRG-1/piRNAs may be sufficient to promote germ cell immortality in the absence of an RDE-2-mediated secondary siRNA response. We present direct evidence for a distinction between transgene silencing and the Mrt phenotype of prg-1 by showing that transgene silencing is not disrupted in sterile late-generation prg-1 mutant adults (Figure S2N). We suggest that at least two classes of ‘non-self’ DNA exist: recently introduced foreign transgenes, whose permanent silencing can rapidly become independent of PRG-1 (Ashe et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012), and a distinct class of loci, possibly rapidly evolving tandem repeats or a recently introduced transposable element, whose long-term silencing requires the activity of PRG-1.
Despite the continuous requirement for PRG-1 in a wild-type background for maintenance of silencing of repetitive loci (Figures 4 and S4D–E), an alternative PRG-1-independent silencing pathway can be activated by reduced insulin signaling (Figure 2). Analysis of small RNA populations in a variety of prg-1 mutant backgrounds suggested that a small RNA silencing pathway could be the mechanism by which reduced insulin signaling suppresses the transgenerational fertility defects of prg-1 mutants (Figures 5A–C, S4G–I and S5B–D). This led us to define components of an endogenous small RNA silencing pathway that are required for daf-2 to suppress prg-1 (Figures 5F and 6C, Table S2). This small RNA pathway may act upstream of a chromatin-based silencing pathway involving rbr-2- and spr-5-mediated demethylation of histone H3 K4 (Figure 5H). Thus our results indicate that activation of a small RNA genome silencing pathway that protects germ cell immortality, in addition to its known role in initiating anti-aging gene expression (Balch et al., 2008; Kenyon, 2010), is a significant consequence of daf-2 deficiency. Piwi proteins promote silencing of transposable elements, transcriptional activation, imprinting, heterochromatin formation, and modulation of protein function via Heat Shock Proteins (Juliano et al., 2011; Rangan et al., 2011; Watanabe et al., 2011), and our results suggest that a genome silencing function of Piwi that is independent of suppression of many transposons and independent of most 22G effector silencing RNAs promotes transgenerational germ cell maintenance (Figure 6E).
We found that daf-16 and daf-18 mutations shortened the transgenerational lifespan of prg-1 mutants and prg-1; daf-2 double mutants (Figure 2B). Thus, basal levels of DAF-16 activity contribute to the transgenerational lifespan of prg-1 mutants, revealing an intriguing parallel with the established role for low levels of DAF-16 activity in promoting the adult lifespan of wildtype animals (Kenyon et al., 1993; Larsen et al., 1995). Although many mutations in the small RNA silencing pathway that functions downstream of DAF-16 to suppress deficiency for prg-1 also resulted in reduced transgenerational lifespan when combined with prg-1, this effect did not occur for all such mutations (Table S2). The reason for the shortened transgenerational lifespan of prg-1 daf-16 double mutants therefore remains uncertain.
Our observations defy a prediction of the antagonistic pleiotropy theory of aging, which suggests that prolonged lifespan might result in compromised fertility (Williams, 1957). Instead, some interventions that repress aging in somatic cells may be beneficial to germ cells. Whether the heritable epigenetic defects that result from prg-1 deficiency impact somatic lifespan, if these defects are related to the germline function of RBR-2 that can extend adult lifespan (Greer et al., 2011), and how DAF-16 regulates the small RNA pathway that suppresses deficiency for prg-1, are intriguing questions raised by this study.
Progressive sterility implies transgenerational accumulation of defects that could be relevant to proliferative aging of somatic cells. Our data suggests that epigenetic desilencing of transposons and tandem repeats could contribute to loss of germ cell immortality in prg-1 mutants. Drawing a parallel to human genetic diseases such as Huntington’s chorea, this implies that prg-1 is subject to “epigenetic anticipation” as each generation will inherit increased levels of repetitive RNA expression, which, combined with inefficient silencing, eventually causes failure of normal germ cell function. A fascinating prospect therefore is whether epigenetic anticipation might occur in human cells. It has recently been shown that repetitive segments of the genome become desilenced when mammalian cells undergo senescence (De Cecco et al., 2013) and in the aging-related disorder cancer (Ting et al., 2011; Zhu et al., 2011). Further, increased expression of Alu retrotransposons may contribute to adult-onset macular degeneration as well as proliferative aging of human stem cells grown in vitro (Kaneko et al., 2011; Wang et al., 2011). We speculate that Piwi-dependent regulation of repetitive rapidly evolving segments of genomes, such as transposons and tandem repeats, creates an epigenetic landscape in germ cells that is transmitted by human gametes, that could affect proliferative aging of somatic cells, and that could be modulated in future generations by repression of insulin/IGF-1-like signaling.
EXPERIMENTAL PROCEDURES
Germline Mortality assays
Worms were assessed for the Mortal Germline phenotype using the assay previously described (Ahmed and Hodgkin, 2000). Once per week, 6 L1 or L2 animals would be placed on fresh NGM plates seeded with OP50 E. coli bacteria. Each passage would be recorded and plates that yielded no additional L1 animals were marked as sterile. Mantel-Cox Log Rank Analysis was used to determine differences of transgenerational lifespan between strains.
Microarray data analysis
C. elegans tiling arrays normalized to gene models were used to compare gene expression changes in prg-1 and prg-1; daf-2. Repeat expression was analyzed by mapping the probes to repeat positions downloaded from the UCSC genome browser as described above. Control regions for each chromosome were generated using a custom script in R. First, a set of 1000 repeat sequences were sampled from the total complement of repeats on each chromosome (with replacement) and the length of these sequences stored. A random number generator was used to provide 1000 starting positions across the chromosome and these starting positions were paired with the lengths at random to calculate the end positions. Expression differences for these control regions were then calculated as for the repeats. Analysis of the significance of overlap between gene expression or repeat expression between alleles was also carried out using a custom script in R. A random sample of either genes or repeats was generated for each allele by sampling N members of the total set of either genes or repeats, where N is the number of altered genes or repeats for the allele in question. The overlap for the three random samples was then calculated and stored. The entire process was repeated 1000 times and a mean and standard deviation of random overlap computed. The size of the observed overlap could thus be compared to simulated random overlap using the Z statistic.
Extrachromosomal arrays
Primers were designed to amplify repetitive sequences directly from the genome. For an experimental array consisting of direct repeats, sequences from two CeRep59 loci (chrI:4281435, chrIII:7405366) and a 174-mer simple repeat (chrIV:6682640) were used. An additional experimental array consisting of a Helitron transposon sequence (chrIV:16880484). A control array was created using primers flanking a stretch of genomic sequence containing the H2A, H2B, H3 and H4 histone locus his-13, his-14, his-15, and his-16.
RNA Fluorescence In Situ Hybridization
Freshly outcrossed alleles of prg-1, n4357 and tm872, and prg-1 lines containing the extrachromosomal array ypEx3 were created, and RNA FISH was performed on F4 animals to visualize repetitive RNA expression. A DNA oligonucleotide probe coupled with a 5′ Cy5 flurophore was designed to detect RNA transcripts of CeRep59 on Chromosome I (located at 4281435-4294595 nt). The CeRep59 probe was tttctgaaggcagtaattct.
Supplementary Material
HIGHLIGHTS.
PRG-1/Piwi promotes germ cell immortality.
prg-1 mutants display “epigenetic anticipation” of repetitive RNA expression.
Reduced insulin signaling restores germ cell immortality to prg-1.
This feat is accomplished by activating a parallel nuclear RNA silencing pathway.
Acknowledgments
We thank J. Powell-Coffman for insight regarding suppression of sterility by daf-2 RNAi, D. Reiner, S. Alvares, A. Ashe for suggestions, S. Seong, T. Perdue, L. Leopold, J. Lieb for assistance. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. This work was supported by NIH grant GM083048 (S.A.), and by a Cancer Research UK Programme Grant (E.A.M.) and an ERC starting grant (E.A.M.).
Footnotes
ACCESSION NUMBERS
Small RNA sequencing and microarray data sets have been deposited at the Gene Expression Omnibus: GSE40569 - Expression analysis of Caenorhabditis elegans Bristol N2 prg-1 and Bristol N2 prg-1; daf-2 double mutant. GSE40572 - Transgenerational changes in small RNA profiles of C. elegans mutants lacking PRG-1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed S. Uncoupling of pathways that promote postmitotic life span and apoptosis from replicative immortality of Caenorhabditis elegans germ cells. Aging Cell. 2006;5:559–563. doi: 10.1111/j.1474-9726.2006.00244.x. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Hodgkin J. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature. 2000;403:159–164. doi: 10.1038/35003120. [DOI] [PubMed] [Google Scholar]
- Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development. 2007;134:2991–2999. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. piRNAs Can Trigger a Multigenerational Epigenetic Memory in the Germline of C. elegans. Cell. 2012;150:88–99. doi: 10.1016/j.cell.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick EM, Bouasker S, Lehrbach NJ, Simard MJ, Miska EA. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science. 2012;337:574–578. doi: 10.1126/science.1220952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science. 2008;322:1387–1392. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature. 2012;489:447–451. doi: 10.1038/nature11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128:1063–1076. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Curran SP, Wu X, Riedel CG, Ruvkun G. A soma-to-germline transformation in long-lived Caenorhabditis elegans mutants. Nature. 2009;459:1079–1084. doi: 10.1038/nature08106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013;12:247–256. doi: 10.1111/acel.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dernburg AF, Zalevsky J, Colaiacovo MP, Villeneuve AM. Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev. 2000;14:1578–1583. [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479:365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, Lowden M, Clejan I, Tzoneva M, Thomas JH, Hodgkin J, Ahmed S. Mutator phenotype of Caenorhabditis elegans DNA damage checkpoint mutants. Genetics. 2006;174:601–616. doi: 10.1534/genetics.106.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano C, Wang J, Lin H. Uniting germline and stem cells: the function of Piwi proteins and the piRNA pathway in diverse organisms. Annu Rev Genet. 2011;45:447–469. doi: 10.1146/annurev-genet-110410-132541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DJ, Edwards TM, Reinke V, Kelly WG. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell. 2009;137:308–320. doi: 10.1016/j.cell.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, van Luenen HG, Plasterk RH. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell. 1999;99:133–141. doi: 10.1016/s0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Plasterk RH. A genetic link between co-suppression and RNA interference in C. elegans. Nature. 2000;404:296–298. doi: 10.1038/35005113. [DOI] [PubMed] [Google Scholar]
- Kidwell MG, Kidwell JF, Sved JA. Hybrid Dysgenesis in DROSOPHILA MELANOGASTER: A Syndrome of Aberrant Traits Including Mutation, Sterility and Male Recombination. Genetics. 1977;86:813–833. doi: 10.1093/genetics/86.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Luteijn MJ, van Bergeijk P, Kaaij LJ, Almeida MV, Roovers EF, Berezikov E, Ketting RF. Extremely stable Piwi-induced gene silencing in Caenorhabditis elegans. Embo J. 2012;31:3422–3430. doi: 10.1038/emboj.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Clejan I, Liu Y, Lowden M, Gartner A, Hodgkin J, Ahmed S. trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet. 2006;2:e18. doi: 10.1371/journal.pgen.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Polymenis M, Kennedy BK. Chronological and replicative lifespan in yeast: do they meet in the middle? Cell Cycle. 2012;11:3531–3532. doi: 10.4161/cc.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Malone CD, Navarro C, Newbold SP, Hayes PS, Sachidanandam R, Hannon GJ, Lehmann R. piRNA production requires heterochromatin formation in Drosophila. Curr Biol. 2011;21:1373–1379. doi: 10.1016/j.cub.2011.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelick C, Ahmed S. Achieving immortality in the C. elegans germline. Ageing Res Rev. 2005;4:67–82. doi: 10.1016/j.arr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Tijsterman M, Okihara KL, Thijssen K, Plasterk RH. PPW-1, a PAZ/PIWI protein required for efficient germline RNAi, is defective in a natural isolate of C. elegans. Curr Biol. 2002;12:1535–1540. doi: 10.1016/s0960-9822(02)01110-7. [DOI] [PubMed] [Google Scholar]
- Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Kennedy S, Conte D, Jr, Kim JK, Gabel HW, Kamath RS, Mello CC, Ruvkun G. Somatic misexpression of germline P granules and enhanced RNA interference in retinoblastoma pathway mutants. Nature. 2005;436:593–597. doi: 10.1038/nature04010. [DOI] [PubMed] [Google Scholar]
- Wang D, Ruvkun G. Regulation of Caenorhabditis elegans RNA interference by the daf-2 insulin stress and longevity signaling pathway. Cold Spring Harb Symp Quant Biol. 2004;69:429–431. doi: 10.1101/sqb.2004.69.429. [DOI] [PubMed] [Google Scholar]
- Wang G, Reinke V. A C. elegans Piwi, PRG-1, regulates 21U-RNAs during spermatogenesis. Curr Biol. 2008;18:861–867. doi: 10.1016/j.cub.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Geesman GJ, Hostikka SL, Atallah M, Blackwell B, Lee E, Cook PJ, Pasaniuc B, Shariat G, Halperin E, et al. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle. 2011;10:3016–3030. doi: 10.4161/cc.10.17.17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011;332:848–852. doi: 10.1126/science.1203919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Xiao Y, Bedet C, Robert VJ, Simonet T, Dunkelbarger S, Rakotomalala C, Soete G, Korswagen HC, Strome S, Palladino F. Caenorhabditis elegans chromatin-associated proteins SET-2 and ASH-2 are differentially required for histone H3 Lys 4 methylation in embryos and adult germ cells. Proc Natl Acad Sci U S A. 2011;108:8305–8310. doi: 10.1073/pnas.1019290108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Montgomery TA, Gabel HW, Fischer SE, Phillips CM, Fahlgren N, Sullivan CM, Carrington JC, Ruvkun G. mut-16 and other mutator class genes modulate 22G and 26G siRNA pathways in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2011;108:1201–1208. doi: 10.1073/pnas.1018695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.