

ABSTRACT
Microbial activities in soils, such as (incomplete) denitrification, represent major sources of nitrous oxide (N2O), a potent greenhouse gas. The key enzyme for mitigating N2O emissions is NosZ, which catalyzes N2O reduction to N2. We recently described “atypical” functional NosZ proteins encoded by both denitrifiers and nondenitrifiers, which were missed in previous environmental surveys (R. A. Sanford et al., Proc. Natl. Acad. Sci. U. S. A. 109:19709–19714, 2012, doi:10.1073/pnas.1211238109). Here, we analyzed the abundance and diversity of both nosZ types in whole-genome shotgun metagenomes from sandy and silty loam agricultural soils that typify the U.S. Midwest corn belt. First, different search algorithms and parameters for detecting nosZ metagenomic reads were evaluated based on in silico-generated (mock) metagenomes. Using the derived cutoffs, 71 distinct alleles (95% amino acid identity level) encoding typical or atypical NosZ proteins were detected in both soil types. Remarkably, more than 70% of the total nosZ reads in both soils were classified as atypical, emphasizing that prior surveys underestimated nosZ abundance. Approximately 15% of the total nosZ reads were taxonomically related to Anaeromyxobacter, which was the most abundant genus encoding atypical NosZ-type proteins in both soil types. Further analyses revealed that atypical nosZ genes outnumbered typical nosZ genes in most publicly available soil metagenomes, underscoring their potential role in mediating N2O consumption in soils. Therefore, this study provides a bioinformatics strategy to reliably detect target genes in complex short-read metagenomes and suggests that the analysis of both typical and atypical nosZ sequences is required to understand and predict N2O flux in soils.
IMPORTANCE
Nitrous oxide (N2O) is a potent greenhouse gas with ozone layer destruction potential. Microbial activities control both the production and the consumption of N2O, i.e., its conversion to innocuous dinitrogen gas (N2). Until recently, consumption of N2O was attributed to bacteria encoding “typical” nitrous oxide reductase (NosZ). However, recent phylogenetic and physiological studies have shown that previously uncharacterized, functional, “atypical” NosZ proteins are encoded in genomes of diverse bacterial groups. The present study revealed that atypical nosZ genes outnumbered their typical counterparts, highlighting their potential role in N2O consumption in soils and possibly other environments. These findings advance our understanding of the diversity of microbes and functional genes involved in the nitrogen cycle and provide the means (e.g., gene sequences) to study N2O fluxes to the atmosphere and associated climate change.
INTRODUCTION
In recent years, anthropogenic emissions of greenhouse gases have received increasing attention because of their contribution to global warming (1, 2). Prominent among these gases is nitrous oxide (N2O) (3), which also contributes to ozone depletion (4, 5). The anthropogenic fixation of dinitrogen (N2), by means of the Haber-Bosch process, has led to the overuse of synthetic nitrogen-based fertilizers in agriculture (1, 6). As a consequence of the increased nitrogen (N) content of soils, atmospheric N2O concentrations have risen about 20% relative to preindustrial-era levels (2). N2O emissions are largely the result of bacterial pathways controlling the nitrogen cycle. In particular, N2O is generated primarily as a product of incomplete classic denitrification (i.e., NO3− reduction to N2O via NO2− and NO) and secondarily as a by-product of dissimilatory nitrate reduction to ammonia (DNRA) and oxidation of ammonium to nitrite (nitrification) (7, 8). Besides bacterial activities, abiotic processes and fungal denitrification are thought to be sources of N2O (9, 10). Model predictions of N2O consumption in terrestrial environments focus primarily on the N2O-to-N2 reduction step, presently attributed to classical denitrifiers possessing nitrous oxide reductase (NosZ) (7).
Our previous work has revealed the existence of two phylogenetically distinct NosZ clades, one encompassing typical Z-type NosZ proteins, which are commonly found in the Alpha-, Beta-, and Gammaproteobacteria, and the other encompassing atypical NosZ proteins present in diverse organisms representing different phyla. Further analysis of sequenced genomes revealed that most of the typical nosZ genes are found in bacteria capable of complete denitrification (i.e., encoding all the enzymes for converting NO3−/NO2− to N2), whereas atypical nosZ genes are found in bacteria with more-diverse N metabolism, including those performing DNRA and missing the NO-generating nitrite reductase genes nirK and nirS (11, 12). Notably, atypical NosZ proteins have been shown to function as nitrous oxide reductases in several bacteria, such as Wolinella succinogenes (13, 14), Geobacillus thermodenitrificans (15), the soil isolate Anaeromyxobacter dehalogenans (11), and several Bacillus species isolated from soils (16, 17).
Examination of the potential of microbial communities to reduce N2O to N2 has traditionally been performed by evaluating nosZ gene and/or transcript presence or abundance by PCR (18, 19). Primers targeting nosZ genes, however, were designed according to characterized typical nosZ gene sequences and therefore missed the bulk of divergent atypical genes (11, 12). Furthermore, measured N2O emissions from soils were frequently lower than predictions based on (typical) NosZ transcript abundance and dynamics (20, 21). Therefore, it is likely that atypical NosZ abundance accounts, at least in part, for the discrepancy between predicted and observed N2O fluxes.
To circumvent the limitations and explore the total natural diversity of nosZ genes in the environment, we analyzed short-read metagenomic data sets from various soils and locations. Even though metagenomics can provide a relatively unbiased, PCR-independent view of the diversity and abundance of individual genes present in a sample, several technical challenges must first be addressed. For instance, in metagenomes of highly diverse microbial communities, such as those obtained from soils, the rates of false positives and false negatives when using similarity searches to detect individual genes in assembled contigs or unassembled short reads has not been rigorously evaluated, with the probable exception of error rates assessed for the purpose of taxonomic classification, i.e., assigning a sequence to a taxon without necessarily evaluating its potential function and sequence diversity (22, 23). Cutoffs that might minimize the number of false-positive matches have not been determined for short-read metagenomes; instead, arbitrary, predetermined cutoffs based on E values (i.e., the likelihood of finding a match by chance) represent the common practice (24).
The objective of the present study was to analyze the diversity and abundance of both typical and atypical nosZ genes in soils with contrasting physicochemical properties. To this end, we first developed a strategy based on similarity searches to determine appropriate cutoffs for accurately detecting metagenomic nosZ fragments by analyzing in silico metagenomes of known sequence composition. Subsequently, we applied this strategy and derived cutoffs to detect nosZ reads in metagenomes from two agricultural soils in the U.S. Midwest that have been subjects of an ongoing multiyear study to assess nitrogen cycling processes, as well as in publicly available metagenomes from various soil ecosystems. Our metagenomic, PCR-independent approach provided a comprehensive and quantitative examination of the diversity and abundance of both typical and atypical nosZ genes in soils.
RESULTS
Evaluating search algorithms and cutoffs for detecting nosZ genes in metagenomes.
To determine the best algorithm and parameters for detecting nosZ reads in 100-bp-long read metagenomes, a reference database of manually verified nosZ genes that preclustered at 95% sequence identity was queried against two such in silico-generated data sets, libraries I (representing the whole genome of 122 NosZ-encoding organisms) and II (representing the whole genome of 1,081 bacteria, including those in library I). From a receiver operating characteristic (ROC) curve analysis of true- and false-positive rates, the most appropriate bit score cutoff values were 107 and 52.2 for the BLASTn and BLASTx searches, respectively. These bit scores provided sensitivities (the fraction of correctly classified positive BLAST matches) of 100% and 98.4% for BLASTn and BLASTx, respectively, and a specificity (the fraction of correctly classified negative BLAST matches) of 99.9% for both algorithms. A hidden Markov model (HMM) search resulted in a sensitivity of 46% and a specificity of 99.9% (Table 1). Additional HMM searches, using models that included more typical and atypical NosZ sequences (built from reference sequences in Table S1 in the supplemental material) improved the number of nosZ reads retrieved from both in silico libraries (~56%). Irrespective of the HMM model employed, a lower fraction of nosZ reads was captured when the HMM was used than when BLAST searches were performed. Most of the reads missed by the HMM-based approach lacked highly conserved amino acid residues, and this accounted for the lower performance of HMM searches, consistent with expectations (HMM models rely heavily on conserved residues). Therefore, remaining analyses were performed using the BLAST algorithms.
TABLE 1 .
Comparison of BLASTn, BLASTx, and HMMER algorithms for retrieving typical and atypical nosZ reads from the in silico libraries I and II
| Method | In silico library | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| BLASTn | I | 100.0 | 99.9 |
| II | 100.0 | 99.9 | |
| BLASTx | I | 98.4 | 99.9 |
| II | 98.4 | 99.9 | |
| hmmsearch (protein) | I | 48.9 | 99.9 |
| II | 46.0 | 99.9 |
To test the limitations in retrieving metagenomic nosZ reads, single typical and atypical representative NosZ protein or nucleotide sequences were independently queried against library II. Although BLASTn had a specificity similar to that of BLASTx, the latter algorithm was able to capture 735% and 270% more reads (i.e., reads annotated as nosZ with a bit score greater or equal to the calculated cutoff for true positives) of the typical and the atypical references, respectively. Therefore, BLASTx was used in the remaining analyses. Using typical NosZ from Bradyrhizobium japonicum strain USDA 110 as a single reference, reads derived from 74 out of 127 different alleles encoding NosZ were captured and found to be enriched in nosZ sequences closely related to the reference sequence. In contrast, reads for only 32 out of 127 alleles encoding NosZ were captured when atypical NosZ from Anaeromyxobacter sp. strain Fw 109-5 was used as a reference in the analysis (Fig. 1, right panel). This atypical NosZ reference does not exhibit sequence identity to other target sequences in the 54 to 82% amino acid identity range, and thus, the lack of moderately related sequences among the target sequences accounts for the results obtained (Fig. 1, left panel). More importantly, a linear relationship was observed between the fraction of total reads detected and the level of divergence between the full-length reference and target sequences (Fig. 1), where 50% or more of the reads were detected when the two sequences shared more than 64 or 68% sequence identity for typical or atypical NosZ reference sequences, respectively.
FIG 1 .

Fraction of nosZ reads recovered from an in silico data set as a function of their relatedness to the reference query sequence. nosZ reads were retrieved from in silico-generated library II using Bradyrhizobium japonicum strain USDA 110 to represent typical NosZ (left) or Anaeromyxobacter sp. strain Fw 109-5 to represent atypical NosZ (right) reference sequences in a BLASTx search. The average bit score value (y axis) for the fraction of nosZ reads recovered (circle size) was plotted against the percentage of identity between each reference sequence and the full-length NosZ sequences from which the retrieved (matching) reads originated (target sequence). The linear relationships observed between the fraction of reads detected and the percentage of identity between the full-length target and references sequences were calculated as follows: y = 0.464x + 40.73 (R2 = 0.90) for typical sequences and y = 0.527x + 42.19 (R2 = 0.89) for atypical sequences, where y is the percentage of identity between the target and full-length reference sequences and x is the fraction of reads retrieved.
Further examination of the reads recruited along the typical NosZ reference (Fig. 2) showed that the true-positive matches (i.e., reads derived from nosZ genes with a bit score greater than 52.2) were evenly distributed along the NosZ reference sequence. The N terminus of the reference sequence (1 to 60 amino acid positions) was rarely covered by either true- or false-positive matches, suggesting that this part of the gene should be avoided when assessing nosZ abundance in metagenomes.
FIG 2 .
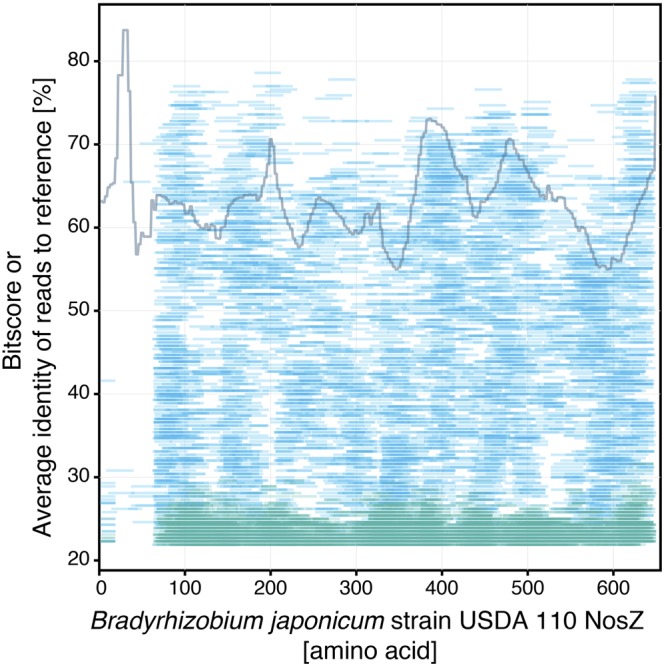
Coverage of matching nosZ reads from library II along the Bradyrhizobium japonicum NosZ reference sequence. Reads from library II matching the Bradyrhizobium japonicum strain USDA 110 NosZ reference are plotted according to their bit score values. Blue and green lines represent reads originating from nosZ genes and other genes, respectively. The solid gray line represents the average percentage of identity of nosZ reads (blue lines) matching the NosZ reference in a 3-amino-acid window.
Abundance of nosZ genes in sandy and silty soils.
A characterization of the taxa, coverage, and assembly statistics of the two soil metagenomes is described in the supplemental material. Both soil metagenomes were queried against a 95%-identical preclustered set of reference NosZ sequences. All matches having a bit score greater than the calculated cutoff determined based on the in silico library analysis were identified as nosZ reads and classified as typical or atypical depending on their best match. Atypical nosZ reads were clearly the most abundant, comprising 72.9% and 89.6% of the total nosZ reads found in the sand and silt loam soil metagenomes, respectively (Fig. 3). Further, 97% of the nosZ reads found in both soil metagenomes (4,929 and 7,280, total, in the sandy and silty loam soils, respectively) were recruited by 72 of the 105 NosZ reference sequences, revealing that most of the diversity covered by the references was represented in both soils (Table S2). In addition, both soil samples showed similar estimated absolute abundances for nosZ reads: ~1.4 × 10−5 and 2.1 × 10−5 reads of all reads for the Havana sand and Urbana silt loam, respectively (Fig. 3). The ratio of nosZ reads to single-copy housekeeping gene reads indicated that approximately 16% of the soil bacterial genomes harbored a nosZ gene (Table S3).
FIG 3 .
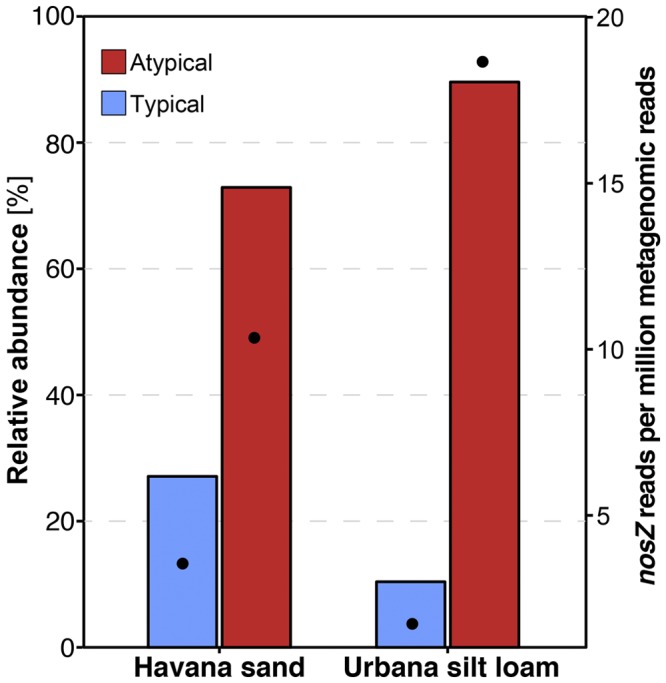
Relative abundances for typical and atypical nosZ genes in Havana sand and Urbana silt loam soil metagenomes. All metagenomic nosZ reads from soil were classified as typical or atypical according to their best match against a reference database of NosZ sequences, and the calculated relative abundances of the two gene types are shown (primary y axis, bars). The absolute abundance, i.e., number of identified nosZ reads per million of all reads in each metagenome is also shown (secondary y axis, dots).
Phylogenetic analysis of the atypical nosZ reads showed that closely related genes found in members of the Anaeromyxobacter, Gemmatimonas, Opitutus, and Hydrogenobacter genera were the most abundant in both soil samples (Fig. 4). Additionally, less abundant genera, such as Bradyrhizobium and Rhodopseudomonas, both known to harbor typical nosZ genes, were also present in both soils. Remarkably, atypical nosZ reads affiliated with Anaeromyxobacter represented 12.7% and 15.2% of the total nosZ reads found in both sand and silt loam soils, respectively. The most abundant typical nosZ reads were assigned to the Ralstonia (3.2%), Thiobacillus (3.1%), Bradyrhizobium (1.6%), and Rhodopseudomonas (1.7%) genera (Table S2).
FIG 4 .

Phylogenetic affiliations for the five most abundant genera harboring typical and atypical nosZ genes in Havana sand and Urbana silt loam soil metagenomes. Metagenomic reads were assigned to a genus based on their best match against a reference database of NosZ sequences, and the normalized number of reads (based on the size of the data sets) assigned to each genus is shown for Havana sand (A) and Urbana silt loam (B) soils. Red and blue bars represent atypical and typical genes, respectively.
nosZ diversity and abundance in other soil metagenomes.
In general, atypical nosZ reads were more abundant than other reads in the soil metagenomes evaluated (Fig. 5). The frozen deep-soil permafrost metagenomes (core 1 sample in reference 25) showed a greater abundance for typical nosZ reads (~80% of total NosZ); however, atypical nosZ reads predominated in the upper or active layer (~74% of total nosZ sequences). Interestingly, after induced thawing, the microbial communities at both depths showed a small increase in the relative abundance of atypical nosZ reads. Furthermore, except with the boreal forest biome, several biomes studied by Fierer and colleagues (26), including tropical forest, polar and hot desert, arctic tundra, and temperate grassland, showed higher abundances of atypical than of typical nosZ reads.
FIG 5 .

Relative abundances of typical and atypical nosZ sequences in various soil ecosystems. nosZ reads were retrieved from available Illumina short-read metagenomes by following the same approach used with the Illinois soils reported in this study. The bars represent the probability of finding typical (blue) or atypical (red) sequences as a proxy for their relative abundances, and the error bars represent the variance of the sample mean for each soil metagenome. Data sets were obtained from Mackelprang et al. (25) (library I), Fierer et al. (26) (library II), and Luo et al. (40) (library III).
DISCUSSION
Importance of atypical NosZ.
The discovery of functional atypical NosZ proteins has opened the possibility that a much larger number of microorganisms with previously unaccounted N2O-reducing potential contribute to lessening the N2O emissions to the atmosphere than previously expected (12). The abundance and diversity of atypical nosZ genes were likely missed in previous PCR-based surveys because typical nosZ sequences presented the basis for primer design (11, 12) and the two nosZ types share only 60.9% ± 8.2% nucleotide identity, on average. In the present PCR-independent metagenome analysis, atypical NosZ sequences were more abundant (>73% of total nosZ reads) than their typical counterparts, not only in two agricultural soils differing in physicochemical properties representative of many regions in the Midwest United States but also in soils from distant geographic locations representing a variety of habitats. Our results were also consistent with the widespread presence of atypical nosZ genes, previously hypothesized based on the number of genomes found to encode atypical NosZ proteins among the available genome sequences (11). Therefore, these findings reveal an unexpectedly high potential for N2O reduction mediated by atypical NosZ in a variety of soil habitats.
It is important to note that our study, being solely based on DNA sequences, evaluated N2O reduction potential as opposed to the specific in situ activity of NosZ enzymes, typical or atypical. Since negative (purifying) selection efficiently removes unused genes from genomes in microbial populations, the high abundance of atypical nosZ sequences found in different soil samples underpins their functional and/or ecological potential (e.g., Fig. 5). Given also that N2O reduction is the only known biochemical function carried by NosZ (11), our results collectively suggest that atypical NosZ proteins are as important, if not more important, than their typical counterparts in controlling N2O fluxes in soils and likely other environments. Our study also provided the means (e.g., gene sequences for primer design and a bioinformatics strategy) to facilitate future studies of the effect of environmental conditions on NosZ activity and dynamics toward a more predictive understanding of the nitrogen cycle.
The most abundant nosZ genes in the agricultural soils studied here are affiliated with the Anaeromyxobacter genus (Fig. 4). Members of this genus are widely distributed in soils with different physical and chemical characteristics as well as soils from a variety of geographic locations (11, 27, 28). The high abundance of nosZ genes affiliated with members of the nondenitrifying Anaeromyxobacter genus is consistent with recent PCR surveys that employed primers targeting atypical nosZ sequences (11, 12) and A. dehalogenans 16S rRNA gene sequences (11). Further, a high phylogenetic congruence between typical nosZ and 16S rRNA gene phylogenies was previously reported (29, 30). Therefore, abundant atypical nosZ metagenomic sequences (Fig. 4 and Table S2) that have distant matches to homologs of known NosZ-encoding taxa may be harbored by novel taxa. The sequences reported here should facilitate the identification of new taxa, expanding our understanding of phylogenetic diversity in NosZ-encoding soil organisms. The majority of the abundant atypical nosZ reads that were assignable to known taxa were found in potentially nondenitrifying genomes of genera such as Anaeromyxobacter, Ignavibacterium, Opitutus, Dyadobacter, and Gemmatimonas, which were overlooked in previous PCR surveys targeting typical nosZ genes. Therefore, the inclusion of these unaccounted N2O reducers in future environmental studies may help bridge the gap between measured N2O emissions and denitrification potential based solely on typical nosZ gene or nosZ transcript measurements.
Our results for the Illinois soils are based on a composite sample comprised of equal DNA quantities extracted from multiple subsamples, intended to capture spatial heterogeneity at each field site (at both centimeter depths and meter landscape scales). Since the soils were taken at a single time point, it is likely that some of the trends reported here for these soils (e.g., abundance of specific NosZ-encoding taxa, average coverage of metagenomes, etc.) might differ temporally, given that agricultural soils typically receive seasonal management inputs. Nonetheless, the high abundance of atypical nosZ sequences found in these agricultural soil metagenomes and in soils from different locations and of diverse physicochemical characteristics (e.g., Fig. 5) emphasize their potential importance for nitrogen cycling.
A bioinformatics methodology to detect target genes.
Our evaluation of in silico-generated data sets of known species and gene composition showed that both BLASTn and BLASTx algorithms represent reliable means of detecting reads encoding nosZ (or other target) genes, albeit with their own strengths and limitations. The selection of the most appropriate algorithm should consider the computational resources available. For example, BLASTx is more computationally demanding than BLASTn but can capture more-divergent sequences if more distantly related sequences/organisms are targeted. However, BLASTn similarity searches are less affected by frameshift-introducing sequencing errors, which might be significant in short-read data even after stringent quality read trimming. Frameshift correction tools such as FrameBot (31), HMM-Frame (32), and FragGeneScan (33) are available to correct these sequencing artifacts and also predict protein-coding regions in short reads.
The low performance of the profile-based approach (HMM) versus that of BLASTx (Table 1) is presumably attributable to the lower sensitivity of the former with (i) short sequences, (ii) sequencing errors creating frameshifts, and (iii) a reduced fraction of highly conserved amino acid residues specific to the protein of interest, as suggested previously (32, 34). Regardless of these limitations, HMM-based searches are preferable when targeting distantly related homologs and using full-length sequences (e.g., targeting complete gene sequences recovered in assembled contigs).
Recommendations for the study of other genes.
The aforementioned approach based on in silico metagenomes, the BLAST algorithm, and ROC analysis can be modified for other functional genes of interest. Special attention should be given to conserved domains in the target gene or protein that are shared with other nontarget genes/proteins. As shown in Fig. 2, no false-positive matches were observed for the B. japonicum strain USDA 110 NosZ reference sequence for bit score values above the calculated cutoff. The latter finding indicates that no high-identity domains or motifs are shared with other non-NosZ sequences. Other genes may deviate from this pattern, and a case-by-case evaluation (e.g., Fig. S1) is recommended. Our approach, when modified to use sliding windows along the sequence of the reference gene, can also determine appropriate cutoffs for different regions of the sequence and identify regions that represent reliable targets for further analyses (e.g., low abundance of false-positive matches and PCR primer design).
In silico data sets simulating different error rates, insert sizes, and coverage can easily be constructed to mimic different short-read sequencing technologies or methodologies. Nonetheless, simulating the diversity and variable abundances of individual taxa of real soil metagenomes remains challenging (e.g., our in silico data sets had substantially less diversity than the real metagenomes used in the study). The expansion of in silico library I by 13.6 million reads from 959 sequenced genomes not containing a nosZ gene (i.e., in silico library II) did not increase the number of false-positive matches obtained for nosZ (Table 1), suggesting that a small number of, if any, false-positive matches should be expected for real metagenomes. In addition, having a comprehensive and well-curated set of protein or gene reference sequences is a key requirement for robust assessment of the best cutoffs and parameters to effectively retrieve reads encoding the gene(s) of interest.
In conclusion, we developed a bioinformatics approach for the detection of target genes in short-read metagenomes. This methodology can be extended to the study of any other genes or proteins of interest. The high abundance of the previously unaccounted atypical nosZ genes in the soil samples suggests that nondenitrifiers and denitrifiers that harbor atypical nosZ genes may contribute more than previously thought to the reduction of N2O to innocuous N2 gas.
MATERIALS AND METHODS
Samples, DNA extraction, and sequencing.
In November 2011, agricultural soil samples were collected from two sites with long histories of commercial corn and soybean production in the U.S. Midwest corn belt: (i) Havana, IL (93% sand; 7% clay; lat 40.296, long −89.944; elevation, 150 m), and (ii) Urbana, IL (21% sand; 69% silt; 10% clay; lat 40.075, long −88.242; elevation, 222 m). In order to provide a metagenome representative of the total soil profile and minimize the effect of sample heterogeneity, soil was collected as three replicate cores (2.5 cm by 30 cm) taken at three locations 30 m apart within each field plot (9 cores, total, per field), with each core partitioned into four depths (0 to 5 cm, 5 to 10 cm, 10 to 20 cm, and 20 to 30 cm). Soil physicochemical characteristics were measured at each depth (A&L Laboratories, Ft. Wayne, IN) (Table S4). DNA was extracted from ~0.5 g of soil from each fraction according to a previously described phenol-chloroform extraction and purification protocol (35), and approximately equal quantities of DNA from each fraction based on agarose gel quantification were pooled to create one composite sample for each soil type. The Illumina TruSeq and Nextera DNA library preparation protocols were used for the Havana sand and Urbana silt loam samples, respectively. Sequencing of composite DNA samples was performed using the Illumina HiSeq 2000 platform, resulting in 38.4 and 40.2 Gbp of 100-bp long paired-end reads for the Havana sand and Urbana silt loam samples, respectively.
Sequence processing.
An in-house Python script (available at http://enve-omics.gatech.edu) was used for quality trimming of raw Illumina reads as described previously (36). In brief, this script trims from both the 5′ and the 3′ end of a sequence using an average Phred score threshold of 20 in 3-bp-long windows and discards resulting sequences shorter than 50 bp (Table S5). The same trimming strategy was applied to publicly available metagenomes for consistency. All BLAST+ (37) and HMMER (38) analyses were based on both single reads, when the corresponding sister read was not available or discarded after the trimming step, and pair-end reads.
In silico libraries and cutoff calculation.
An in-house Python script was used to generate in silico libraries from available complete genomes in the NCBI database as of April 2013 (2,355 sequenced genomes) as described previously (36). Briefly, this script simulates an Illumina run by generating 100-bp paired-end reads with a sequencing error of 0.5%, an insert size of 500 bp, and a user-defined coverage of 3-fold. The script also reports the coordinates of the genome from which the reads were generated, so that gene encoding information for each read is available (based on NCBI protein table files or ptt files). The first in silico-generated data set (library I) was built based on seven bacterial plasmids and 115 chromosomes previously determined to bear a nosZ gene (12). In silico library II was constructed using the 122 DNA sequences from library I and an additional 959 sequenced chromosomes that did not encode NosZ (confirmed independently by BLASTp analysis). Libraries I and II had a total of 7,460 NosZ-encoding reads and were ~14 and ~136 million reads in size, respectively.
BLAST analyses were performed using the BLAST+ 2.2.7 release with the following settings: a word size of 7, a penalty of −2, no dust, an E value cutoff of 0.001, and an x-drop gap of 150. BLASTx settings were no SEG program, an E value cutoff of 0.001, and a word size of 3. Previously described nosZ nucleotide or protein references from complete genomes were clustered at 95% sequence identity, and the longest representative sequence from each cluster was used to construct a reference database, consisting of 54 typical, 47 atypical, and 4 halophilic archaeal representative reference sequences. All reads originating from a nosZ gene, whether located on a chromosome or a plasmid, that matched a nucleotide or protein sequence reference were classified as true positives. Reads not originating from a nosZ reference sequence that matched a reference sequence were classified as false positives. The numbers of true and false positives obtained from each algorithm (performance) were evaluated by the receiver operating characteristic (ROC) curve using the R pROC library (39). The bit score cutoff that maximized performance was calculated as the line that maximized the distance to the identity line (i.e., the nondiscriminatory diagonal line where sensitivity and specificity are equal) according to the Youden method for a partial area under the curve (pAUC) between 90% and 100% of specificity (39) (see Fig. S1 for a flow chart of the approach).
A hidden Markov model (HMM) based on the sequences of six functionally characterized NosZ proteins (Bradyrhizobium japonicum USDA 110 27375426, Wolinella succinogenes 46934822, Paracoccus denitrificans 2833444, Achromobacter cycloclastes 37538302, and Anaeromyxobacter dehalogenans 2CP-C 86158824) was built with HMMER 3.0 and used to query translated reads from libraries I and II for nosZ matches based on the hmmsearch algorithm (38) (Table 1). ROC analyses were not performed for HMM searches due to the high sensitivity and specificity obtained after each search.
Detecting nosZ reads in metagenomes.
Publicly available metagenomes from Alaskan permafrost (25), soil biomes (26), and soils exposed to a decade of warming (40) were downloaded from the DOE Joint Genome Institute (http://www.jgi.doe.gov), MG-RAST (http://metagenomics.anl.gov), and NCBI Sequence Read Archive Web servers, respectively. NosZ-encoding reads (or nosZ reads for simplicity) in the above-named metagenomes were identified by BLAST searches against the NosZ reference sequences clustered by 95% identity (Table S1) and classified as typical or atypical NosZ sequences based on their best match. To account for differences in the numbers of sequences among the publicly available metagenomes, the presence or absence of each type of nosZ read was represented as a binomial distribution for each metagenome. Assuming independence in the presence or absence of each type of nosZ gene in each soil sample, the probability of finding either type was calculated from the frequency of nosZ reads detected in each metagenome (i.e., a probability closer to 1 implies a higher abundance for the corresponding type of nosZ gene in the metagenome). To account for differences in the numbers of reads for each metagenome, the standard deviation of the sample mean was calculated for each distribution.
Fractions of genomes containing a nosZ gene.
To estimate the fractions of the microbial populations in the soil community with nosZ genes in their genomes, the following approach was used. Sequences of three single-copy housekeeping genes (dnaK, recA, and rpoB) were used as references to query each metagenome. The reference set for each housekeeping gene included sequences from 30 different bacterial species (denoted by an asterisk in Table S1) that also contained a typical or an atypical nosZ gene (i.e., half of the species in the set harbored a typical nosZ and the other half an atypical gene). The total number of matches obtained in a BLASTn search (settings were as follows: no dust, a word size of 7, a penalty of −2, a maximum number of target sequences of 1, an x-drop of 150, and an E value of 0.001) for each set of housekeeping and nosZ genes was normalized by the average length of the query (reference) sequences. The fraction of the microbial community harboring nosZ genes was calculated as the ratio of the normalized number of nosZ reads to the number of reads assigned to each of the housekeeping genes (assuming one nosZ gene copy per genome, which is the case for >97% of the analyzed genomes in Table S1; see also Table S3).
Both the Havana sand and Urbana silt loam metagenomes are available under accession numbers SRR1152189 and SRR1153387 in the Sequence Read Archive server.
SUPPLEMENTAL MATERIAL
Supplemental results. Download
Supplemental materials and methods. Download
Flow chart for calculating gene-specific bit score cutoffs. Download
Soil metagenomic library quality and domain-level composition based on taxonomic affiliation of protein-encoding reads. Download
Taxonomic characterization of the metagenomes based on the recovered 16S rRNA gene reads. Download
Genomes used to generate in silico libraries I and II.
List of NosZ reference sequences representing more than 0.1% of total nosZ reads in soil metagenomes.
Fraction of the soil microbial community encoding NosZ proteins.
Physicochemical properties of the agricultural soils used in the study.
Agricultural soil metagenomes and assembly statistics.
ACKNOWLEDGMENTS
This research was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, Genomic Science Program (award DE-SC0006662). L.H.O. was supported by a Chilean Fulbright-Conicyt doctoral scholarship.
We thank Michael Weigand for helpful discussions and suggestions.
Footnotes
Citation Orellana LH, Rodriguez-R LM, Higgins S, Chee-Sanford JC, Sanford R, Ritalahti K, Löffler FE, Konstantinidis KT. 2014. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. mBio 5(3):e01193-14. doi:10.1128/mBio.01193-14.
REFERENCES
- 1. Canfield DE, Glazer AN, Falkowski PG. 2010. The evolution and future of Earth’s nitrogen cycle. Science 330:192–196. 10.1126/science.1186120 [DOI] [PubMed] [Google Scholar]
- 2. Montzka SA, Dlugokencky EJ, Butler JH. 2011. Non-CO2 greenhouse gases and climate change. Nature 476:43–50. 10.1038/nature10322 [DOI] [PubMed] [Google Scholar]
- 3. Forster P, Ramaswamy V, Artaxo P, Berntsen T, Betts R, Fahey DW, Haywood J, Lean J, Lowe DC, Myhre G, Nganga J, Prinn R, Raga G, Schultz M, Van Dorland R. 2007. Changes in atmospheric constituents and in radiative forcing, p 129–234 In Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL. (ed), Climate change 2007: the physical science basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 4. Ravishankara AR, Daniel JS, Portmann RW. 2009. Nitrous Oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326:123–125. 10.1126/science.1176985 [DOI] [PubMed] [Google Scholar]
- 5. Portmann RW, Daniel JS, Ravishankara AR. 2012. Stratospheric ozone depletion due to nitrous oxide: influences of other gases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1256–1264. 10.1098/rstb.2011.0377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reay DS, Davidson EA, Smith KA, Smith P, Melillo JM, Dentener F, Crutzen PJ. 2012. Global agriculture and nitrous oxide emissions. Nat. Clim. Chang. 2:410–416. 10.1038/nclimate1458 [DOI] [Google Scholar]
- 7. Zumft WG, Kroneck PM. 2007. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by bacteria and archaea. Adv. Microb. Physiol. 52:107–227. 10.1016/S0065-2911(06)52003-X [DOI] [PubMed] [Google Scholar]
- 8. Morales SE, Cosart T, Holben WE. 2010. Bacterial gene abundances as indicators of greenhouse gas emission in soils. ISME J 4:799–808. 10.1038/ismej.2010.8 [DOI] [PubMed] [Google Scholar]
- 9. Laughlin RJ, Stevens RJ. 2002. Evidence for fungal dominance of denitrification and codenitrification in a grassland soil. Soils Sci. Soc. J. 66:1540. 10.2136/sssaj2002.1540 [DOI] [Google Scholar]
- 10. Cooper DC, Picardal FW, Schimmelmann A, Coby AJ, Cooper DC, Picardal FW, Schimmelmann A, Coby AJ. 2003. Chemical and biological interactions during nitrate and goethite reduction by Shewanella putrefaciens 200. Appl. Environ. Microbiol. 69:3517–3525. 10.1128/AEM.69.6.3517-3525.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanford RA, Wagner DD, Wu Q, Chee-Sanford JC, Thomas SH, Cruz-García C, Rodríguez G, Massol-Deyá A, Krishnani KK, Ritalahti KM, Nissen S, Konstantinidis KT, Löffler FE. 2012. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. U. S. A. 109:19709–19714. 10.1073/pnas.1211238109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones CM, Graf DR, Bru D, Philippot L, Hallin S. 2013. The unaccounted yet abundant nitrous oxide-reducing microbial community: a potential nitrous oxide sink. ISME J. 7:417–426. 10.1038/ismej.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Payne WJ, Grant MA, Shapleigh J, Hoffman P. 1982. Nitrogen oxide reduction in Wolinella succinogenes and Campylobacter species. J. Bacteriol. 152:915–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simon J, Einsle O, Kroneck PM, Zumft WG. 2004. The unprecedented nos gene cluster of Wolinella succinogenes encodes a novel respiratory electron transfer pathway to cytochrome c nitrous oxide reductase. FEBS Lett. 569:7–12. 10.1016/j.febslet.2004.05.060 [DOI] [PubMed] [Google Scholar]
- 15. Liu X, Gao C, Zhang A, Jin P, Wang L, Feng L. 2008. The nos gene cluster from gram-positive bacterium Geobacillus thermodenitrificans NG80-2 and functional characterization of the recombinant NosZ. FEMS Microbiol. Lett. 289:46–52. 10.1111/j.1574-6968.2008.01362.x [DOI] [PubMed] [Google Scholar]
- 16. Jones CM, Welsh A, Throbäck IN, Dörsch P, Bakken LR, Hallin S. 2011. Phenotypic and genotypic heterogeneity among closely related soil-borne N2—and N2O-producing Bacillus isolates harboring the nosZ gene. FEMS Microbiol. Ecol. 76:541–552. 10.1111/j.1574-6941.2011.01071.x [DOI] [PubMed] [Google Scholar]
- 17. Mania D, Heylen K, van Spanning RJ, Frostegård A. 8 April 2014. The nitrate-ammonifying and nosZ carrying bacterium Bacillus vireti is a potent source and sink for nitric and nitrous oxides under high nitrate conditions. Environ. Microbiol. 10.1111/1462-2920.12478 [DOI] [PubMed] [Google Scholar]
- 18. Scala DJ, Kerkhof LJ. 1998. Nitrous oxide reductase (nosZ) gene-specific PCR primers for detection of denitrifiers and three nosZ genes from marine sediments. FEMS Microbiol. Lett. 162:61–68. 10.1111/j.1574-6968.1998.tb12979.x [DOI] [PubMed] [Google Scholar]
- 19. Henry S, Bru D, Stres B, Hallet S, Philippot L. 2006. Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl. Environ. Microbiol. 72:5181–5189. 10.1128/AEM.00231-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cuhel J, Simek M, Laughlin RJ, Bru D, Chèneby D, Watson CJ, Philippot L. 2010. Insights into the effect of soil pH on N(2)O and N(2) emissions and denitrifier community size and activity. Appl. Environ. Microbiol. 76:1870–1878. 10.1128/AEM.02484-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henderson SL, Dandie CE, Patten CL, Zebarth BJ, Burton DL, Trevors JT, Goyer C. 2010. Changes in denitrifier abundance, denitrification gene mRNA levels, nitrous oxide emissions, and denitrification in anoxic soil microcosms amended with glucose and plant residues. Appl. Environ. Microbiol. 76:2155–2164. 10.1128/AEM.02993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gerlach W, Stoye J. 2011. Taxonomic classification of metagenomic shotgun sequences with CARMA3. Nucleic Acids Res. 39:e91. 10.1093/nar/gkr225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. 2008. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 72:557–578. 10.1128/MMBR.00009-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ, Rubin EM, Jansson JK. 2011. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480:368–371. 10.1038/nature10576 [DOI] [PubMed] [Google Scholar]
- 26. Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. 2012. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. U. S. A. 109:21390–21395. 10.1073/pnas.1215210110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanford RA, Cole JR, Tiedje JM. 2002. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl. Environ. Microbiol. 68:893–900. 10.1128/AEM.68.2.893-900.2002 PubMed; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petrie L, North NN, Dollhopf SL, Balkwill DL, Kostka JE. 2003. Enumeration and characterization of iron(III)-reducing microbial communities from acidic subsurface sediments contaminated with uranium. Appl. Environ. Microbiol. 69:7467–7479. 10.1128/AEM.69.12.7467-7479.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones CM, Stres B, Rosenquist M, Hallin S. 2008. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol. Biol. Evol. 25:1955–1966. 10.1093/molbev/msn146 [DOI] [PubMed] [Google Scholar]
- 30. Palmer K, Drake HL, Horn MA. 2009. Genome-derived criteria for assigning environmental narG and nosZ sequences to operational taxonomic units of nitrate reducers. Appl. Environ. Microbiol. 75:5170–5174. 10.1128/AEM.00254-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Q, Quensen JF, Fish JA, Lee TK, Sun Y, Tiedje JM, Cole JR. 2013. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio 4(5):e00592–13. 10.1128/mBio.00592-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Sun Y. 2011. HMM-FRAME: accurate protein domain classification for metagenomic sequences containing frameshift errors. BMC Bioinformatics 12:198. 10.1186/1471-2105-12-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191. 10.1093/nar/gkq747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Sun Y. 2012. Metadomain: a profile HMM-based protein domain classification tool for short sequences, p 271–282 Biocomputing 2012. 10.1142/9789814366496_0026 [DOI] [PubMed] [Google Scholar]
- 35. Welsh A, Chee-Sanford JC, Connor LM, Löffler FE, Sanford RA. 2014. Refined NrfA phylogeny improves PCR-based nrfA gene detection. Appl. Environ. Microbiol. 80:2110–2119. 10.1128/AEM.03443-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. 2012. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 6:898–901. 10.1038/ismej.2011.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eddy SR. 2011. Accelerated profile HMM Searches. PLoS Comput. Biol. 7:e1002195. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, Müller M. 2011. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12:77. 10.1186/1471-2105-12-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo C, Rodriguez-R LM, Johnston ER, Wu L, Cheng L, Xue K, Tu Q, Deng Y, He Z, Shi JZ, Yuan MM, Sherry Ra, Li D, Luo Y, Schuur EaG, Chain P, Tiedje JM, Zhou J, Konstantinidis KT. 2014. Soil microbial community responses to a decade of warming as revealed by comparative metagenomics. Appl. Environ. Microbiol. 80:1777–1786. 10.1128/AEM.03712-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental results. Download
Supplemental materials and methods. Download
Flow chart for calculating gene-specific bit score cutoffs. Download
Soil metagenomic library quality and domain-level composition based on taxonomic affiliation of protein-encoding reads. Download
Taxonomic characterization of the metagenomes based on the recovered 16S rRNA gene reads. Download
Genomes used to generate in silico libraries I and II.
List of NosZ reference sequences representing more than 0.1% of total nosZ reads in soil metagenomes.
Fraction of the soil microbial community encoding NosZ proteins.
Physicochemical properties of the agricultural soils used in the study.
Agricultural soil metagenomes and assembly statistics.