

Abstract
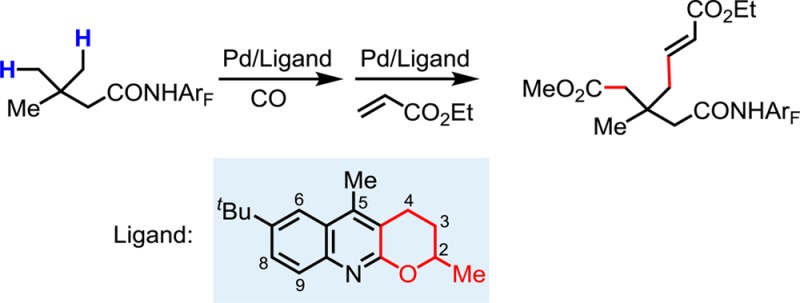
Monoselective γ-C–H olefination and carbonylation of aliphatic acids has been accomplished by using a combination of a quinoline-based ligand and a weakly coordinating amide directing group. The reaction provides a new route for constructing richly functionalized all-carbon quaternary carbon centers at the β-position of aliphatic acids.
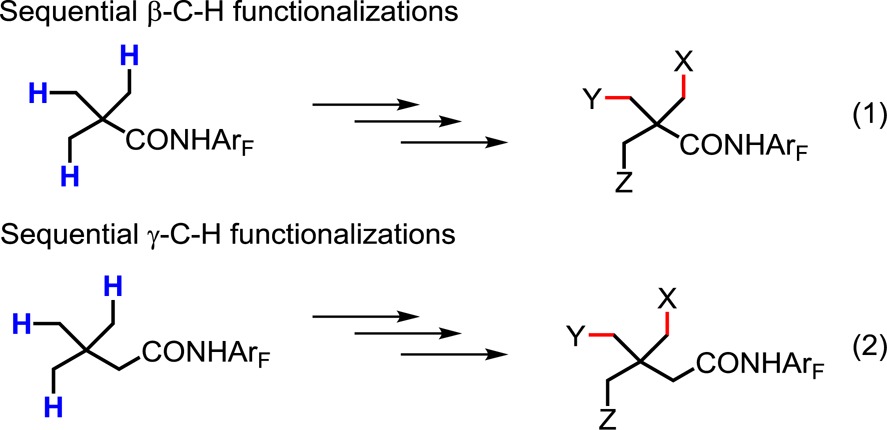
Among the many potential applications of C–H activation reactions in synthesis, functionalizing abundant, yet underutilized starting materials at strategic positions to construct common and synthetically enabling synthons has emerged as an attractive, achievable goal.1 In this context, a wide range of sp2 C–H activation reactions of benzoic acids,2 phenyl acetic acids and hydrocinnamic acids3 have been developed. These transformations have greatly enriched the diversity of structurally limited but readily available bulky chemicals as building blocks. In parallel, we are also investigating the possibility of establishing a platform for the construction of α-quaternary carbon centers from pivalic acid, the success of which will be critically dependent on the continued development of diverse racemic and asymmetric β-C–H functionalizations (eq 1).4 However, to extend this approach toward the construction of β-quaternary carbon centers—another major class of versatile synthons—γ-C–H functionalizations also need to be fully investigated and developed (eq 2). Specifically, it would be advantageous to install functional groups at the γ-position that are amenable for further transformations in order to construct synthetically useful intermediates. Herein we report our initial progress toward this goal through the monoselective γ-C–H olefination and -carbonylation of a variety of β-quaternary amides. We found that the use of a newly developed quinoline-based ligand was essential to allow for these unprecedented transformations.
 |
1 |
β-C–H functionalizations of aliphatic acids using directed metalation have been extensively investigated and reported over the past decade.4,5 In contrast, however, corresponding γ-C–H functionalizations remain largely underdeveloped.6,7 Furthermore, the development of sp3 C–H olefination reactions in general remains a challenge, with relatively few examples having been reported in recent years.8 This is not surprising considering the known difficulties in developing the Heck reaction using alkyl halides.9 Thus, in order to establish a versatile platform for constructing β-quaternary carbon centers via γ-C–H activation, we sought to address these specific challenges. Encouraged by the recent observation that pyridine- or quinoline-based ligands can promote the β-C–H arylation of aliphatic amides,10 we surmised that further optimization of these ligands could enable an unprecedented γ-C–H functionalization .
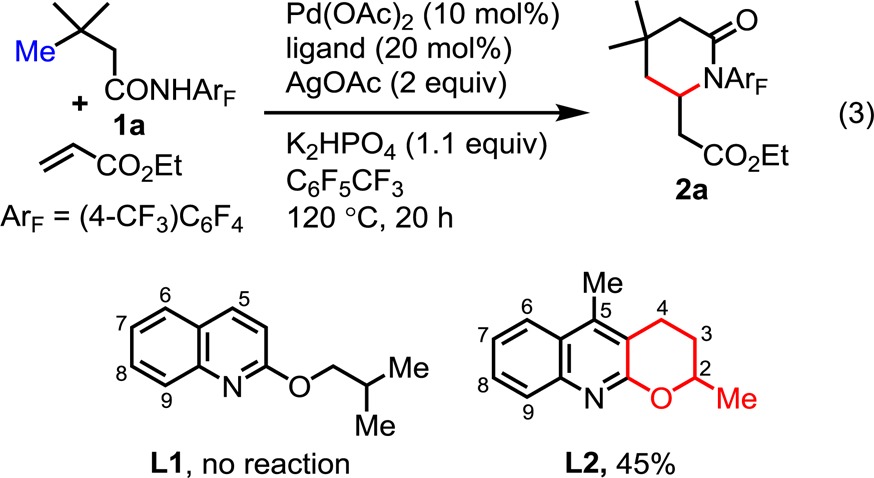
Our initial studies were guided by reaction parameters that were found to be optimal for β-sp3 C–H olefinations.8a Unfortunately, substrate 1a (derived from 3,3-dimethylbutanoic acid) failed to react with ethyl acrylate under these conditions. To our dismay, we found that tuning of various reaction parameters proved largely unsuccessful. We next tested a series of pyridine- and quinoline-based ligands, including 2-alkoxylquinoline L1, which was found to be extremely effective for promoting the β-arylation of sp3 C–H bonds.10 However, these efforts were also met without success (eq 3).
Following these fruitless attempts, we decided to focus on the development of new ligands, beginning with tricyclic ligand L2, which contains a dihydropyran ring. We reasoned that by restraining the conformational freedom of the 2-alkoxy motif, one of the oxygen lone pairs would be constrained to a plane parallel to the π-system of the quinoline ring.11 This would in turn result in a more electron-rich ligand, potentially affecting the reactivity of the Pd(II) catalyst. As such, extensive screening of oxidants and bases was carried out with L2 as our ligand (see Supporting Information). We were pleased to find that C–H olefination of amide 1a with ethyl acrylate in the presence of 10 mol % Pd(OAc)2, 20 mol % L2, 2.0 equiv of AgOAc, and 1.1 equiv of K2HPO4 in C6F5CF3 provided the desired lactam 2a (formed after subsequent intramolecular hetero-Michael addition) in 45% yield (eq 3). However, further tuning of the reaction parameters at this stage failed to improve the yield.
 |
3 |
To investigate the impact of ligand structure on reaction efficiency, we revisited the use of pyridine- and quinoline-based ligands L3–L6 under these newly established conditions. The consistently poor yields obtained with these ligands suggest that the pyran structural motif is crucial for the observed reactivity with L2 (Table 1). We therefore prepared analogues L7–L15 and investigated their use as ligands for this reaction. Taken together with the results obtained with ligand L2, ligands L7–L9 suggest that incorporation of electron-donating groups at C-7 and C-9 of the quinoline ring decreases the effectiveness of the ligand. In contrast, the yield was improved to 56% when a methoxy group was positioned at C-8 in L9. Addition of the electron-withdrawing trifluoromethyl or fluoro group (L10–L12) had significant detrimental effects on the observed reactivity. Mindful of the potential coordination of the methoxy moiety with the Pd(II) center, we focused on the introduction of noncoordinative electron-donating substituents (L13–L15). Gratifyingly, we found that the use of ligand L15, bearing a tert-butyl group at the C-7 position, afforded lactam 2a in 62% yield. Substitution of the dihydropyran ring with dihydrofuran decreased the yield (L16, 40%) indicating that the orientation of the lone pair on the oxygen is vital. Additionally, we also found that increased ligand loadings (40 mol %) improved the yield to 71%. Through further screening, it was discovered that by performing the reaction under an oxygen atmosphere and introducing 10 mol % co-oxidant TEMPO4d and 2 equiv of NaHCO3 we could increase the yield of 2a to 95% (see Supporting Information). We also tested other amide directing groups derived from different amines including aniline, albeit leading to a significant decrease in yields (see Supporting Information).
Table 1. Ligand Developmenta.
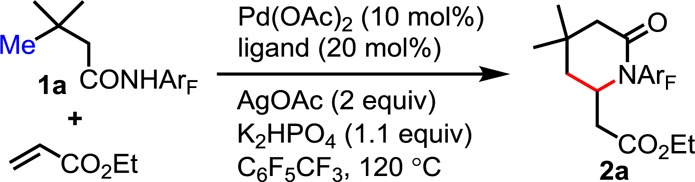
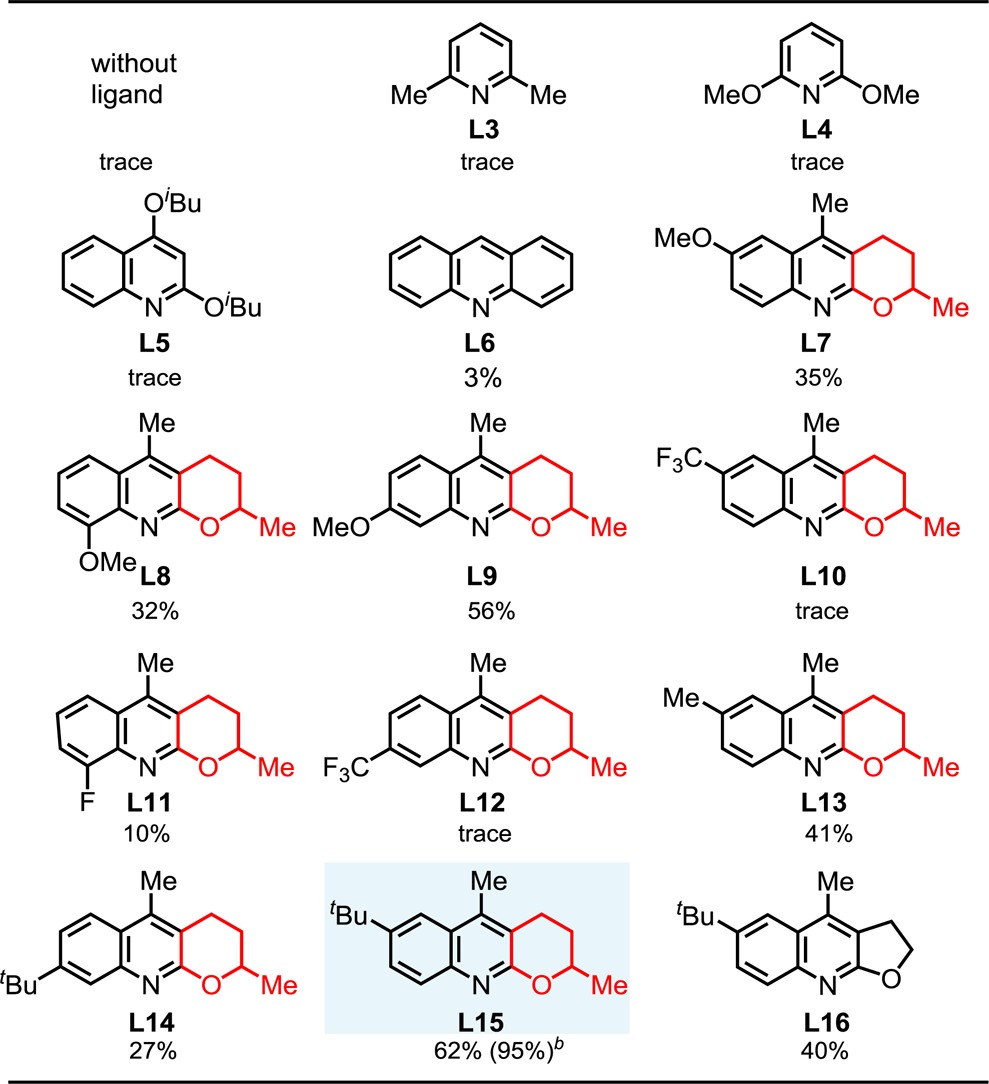
Reaction conditions: 0.1 mmol of substrate, 50 μL of ethyl acrylate, 0.01 mmol of Pd(OAc)2 (10 mol %), 0.02 mmol of Ligand, 0.2 mmol of AgOAc, 0.11 mmol of K2HPO4, 1 mL of C6F5CF3, 50 mL sealed tube, 120 °C (oil), 20 h. Yield was determined by the 1H NMR spectroscopy using CH2Br2 as the internal standard.
40 mol % L15, 10 mol % TEMPO, O2 (1 atm), and 0.2 mmol of NaHCO3 were used.
With these optimized conditions in hand, we proceeded to survey the scope of the reaction with substrates derived from β-quaternary center-bearing aliphatic amides (Table 2). Replacing one or two methyl groups with ethyl substituents decreased the isolated yields from 87% to 68% and 70%, respectively (2a–c). Substrates incorporating benzyl and cyclopentyl functional groups at the β-position are olefinated to give the desired lactams in moderate yields (2d, 2e). Both γ- and δ-methoxy groups as well as γ-esters are tolerated, leading to moderate and synthetically useful yields (2f–h). Interestingly, cyclic substrates work well and provide the desired spiral lactams in 66–83% yield (2i–l). To our satisfaction, we found that both tetrahydropyran and piperidine structural motifs are also compatible with this olefination protocol (2k, 2l).
Table 2. Scope of Substratesa.
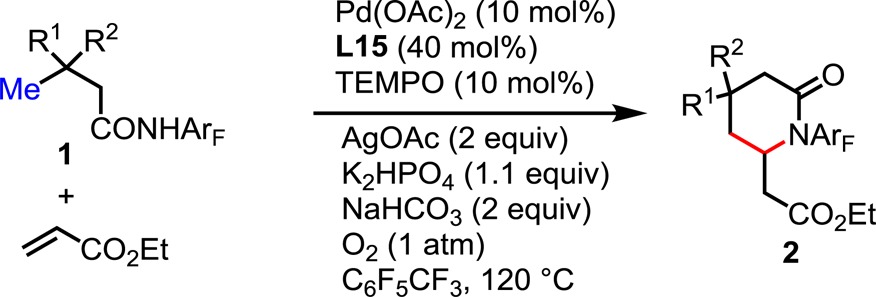
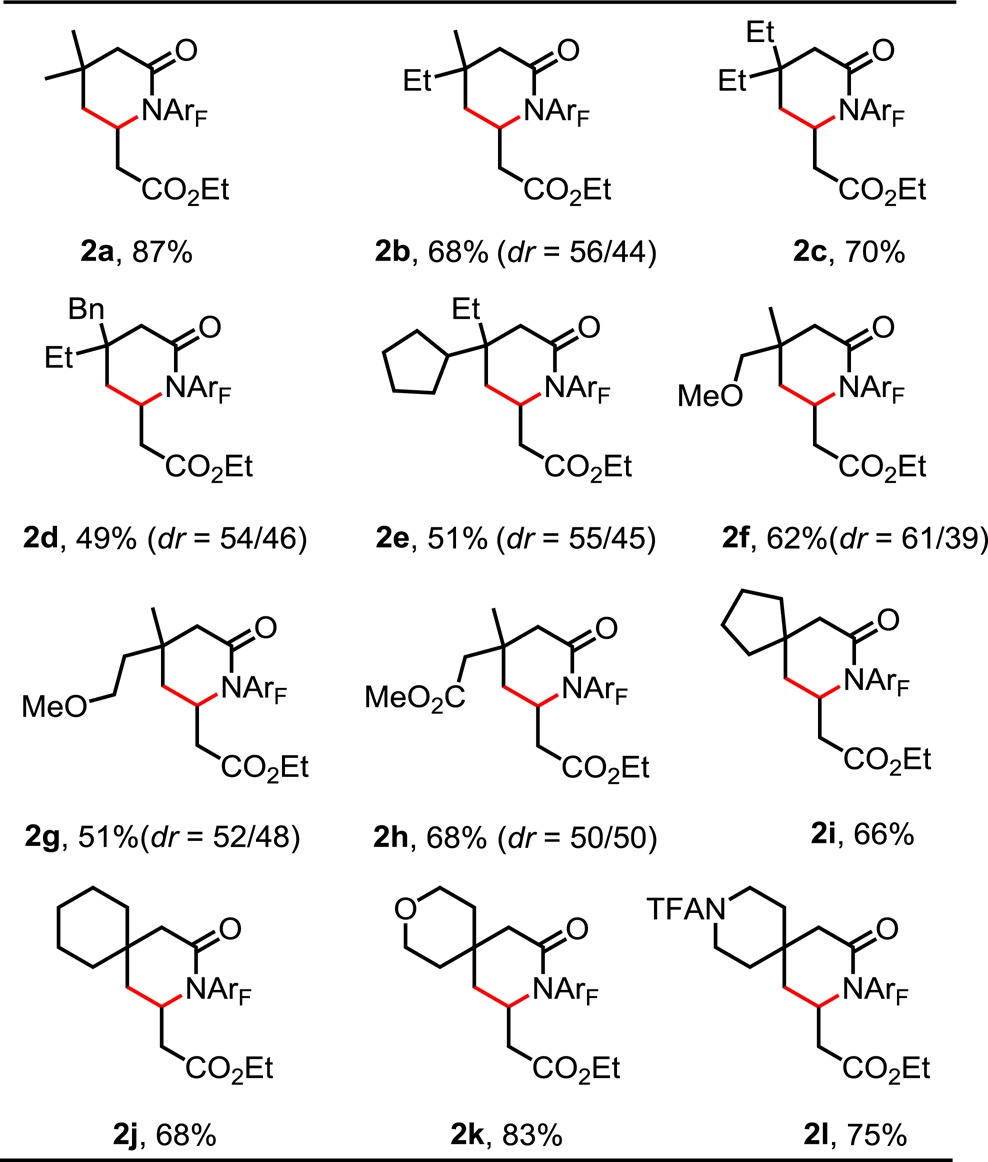
Reaction conditions: 0.1 mmol of substrate, 50 μL of ethyl acrylate, 0.01 mmol of Pd(OAc)2 (10 mol %), 0.04 mmol of Ligand, 0.01 mmol of TEMPO, 0.2 mmol of AgOAc, 0.11 mmol of K2HPO4 and 0.2 mmol of NaHCO3, 1 mL of C6F5CF3, O2 (1 atm), 50 mL sealed tube, 120 °C (oil), 20 h. Isolated yield.
We also investigated the use of a variety of activated olefins as coupling partners (Table 3). Olefination of 1a with benzyl acrylate yielded 2aa in 77% yield. α,β-Unsaturated ketones and nitriles, however, proved less efficient. Thus, the catalyst loading was increased to 15 mol % Pd(OAc)2 in order to improve the yields (2ab–d, 64–73% yield).
Table 3. Scope of Olefinsa.
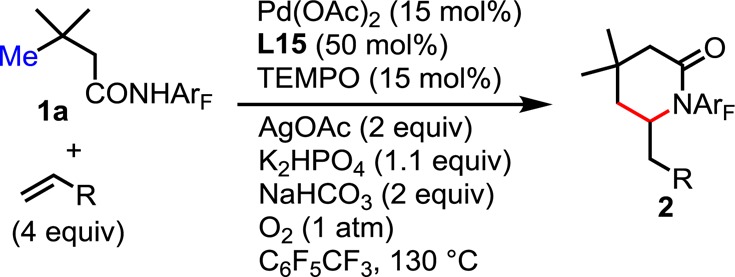
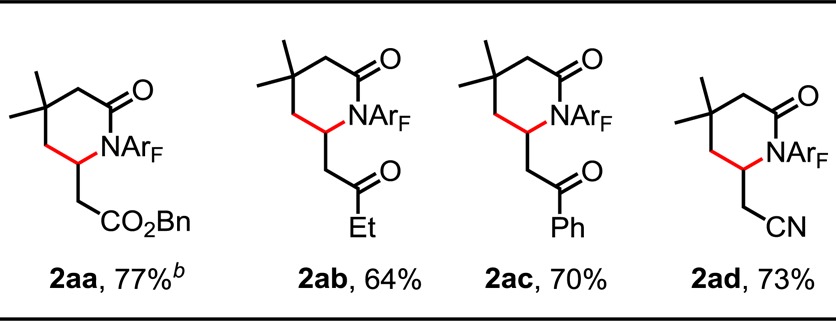
Reaction conditions: 0.1 mmol of substrate, 0.4 mmol of alkenes, 15 mol % Pd(OAc)2, 50 mol % Ligand, 0.015 mmol of TEMPO, 0.2 mmol of AgOAc, 0.11 mmol of K2HPO4 and 0.2 mmol of NaHCO3, 1 mL of C6F5CF3, O2 (1 atm), 50 mL sealed tube, 130 °C (oil), 20 h. Isolated yield.
10 mol % Pd(OAc)2 and 40 mol % Ligand used.
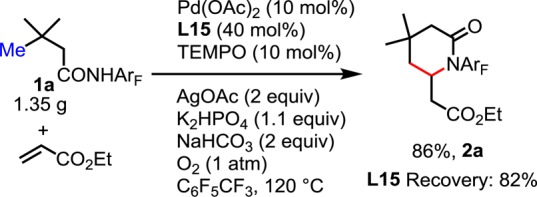
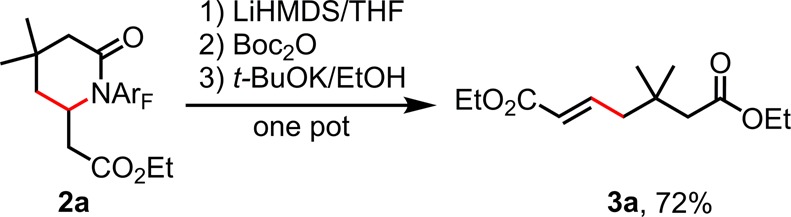
We also prepared lactam 2a on gram scale in order to demonstrate the robustness of this newly developed transformation (Scheme 1). Furthermore, to render this reaction even more synthetically useful, we developed a one-pot deprotonation/ring-opening procedure to convert 2a into γ-olefinated ester 3a, which can be used as a synthon for a variety of further elaborations (Scheme 2).
Scheme 1. Gram-Scale Synthesis.
Scheme 2. Deprotection.
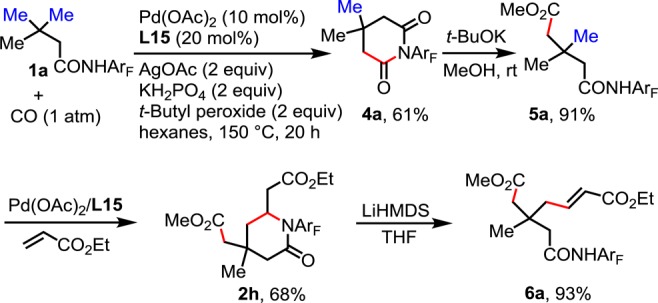
Although this method is currently limited to the monofunctionalization of all-carbon β-quaternary carbon centers, we envision that sequential C–H functionalizations at the γ-carbon would provide a novel disconnection for the construction of richly functionalized all-carbon β-quaternary carbon centers. To this end, we developed a γ-carbonylation protocol utilizing ligand L15 (for ligand screening and optimization of conditions, see Supporting Information). The use of oxidant tert-butyl peroxide increased the NMR yield from 37% to 67%. The reaction was also successfully performed in 1 g scale to give 4a in 61% isolated yield (Scheme 3). The monoselectivity was ensured through intermediate formation of imide 4a, which is then converted to amide 5a, a suitable substrate for our newly developed olefination reaction.4d5a is subsequently subjected to ethyl acrylate in the presence of Pd(II) and ligand L15 to yield lactam 2h, which can in turn be opened to provide amide 6a. Omission of the ester forming step in the ring-opening process ensured formation of the amide such that the four carbons on the quaternary center would be distinct.
Scheme 3. Sequential C–H Functionalizations.
In summary, we have developed the first example of γ-C(sp3)–H olefination, which provides a novel disconnection for the preparation of γ-olefinated aliphatic acids. The use of a newly developed quinoline-based ligand is crucial for realizing this transformation. This ligand also enables a sequential and monoselective γ-carbonylation/olefination procedure to afford highly functionalized all-carbon quaternary centers.
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support.
Supporting Information Available
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Engle K. M.; Yu J.-Q. J. Org. Chem. 2013, 78, 8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miura M.; Tsuda T.; Satoh T.; Pivsa-Art S.; Nomura M. J. Org. Chem. 1998, 63, 5211. [Google Scholar]; b Chiong H. A.; Pham Q.-N.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 9879. [DOI] [PubMed] [Google Scholar]; c Mei T.-S.; Giri R.; Maugel N.; Yu J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215. [DOI] [PubMed] [Google Scholar]
- a Wang D.-H.; Engle K. M.; Shi B.-F.; Yu J.-Q. Science 2010, 327, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Leow D.; Li G.; Mei T.-S.; Yu J.-Q. Nature 2012, 486, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Giri R.; Liang J.; Lei J.-G.; Li J.-J.; Wang D.-H.; Chen X.; Naggar I. C.; Guo C.; Foxman B. M.; Yu J.-Q. Angew. Chem., Int. Ed. 2005, 44, 7420. [DOI] [PubMed] [Google Scholar]; b Giri R.; Maugel N.; Li J.-J.; Wang D.-H.; Breazzano S. P.; Saunders L. B.; Yu J.-Q. J. Am. Chem. Soc. 2007, 129, 3510. [DOI] [PubMed] [Google Scholar]; c Wang D.-H.; Wasa M.; Giri R.; Yu J.-Q. J. Am. Chem. Soc. 2008, 130, 7190. [DOI] [PubMed] [Google Scholar]; d Yoo E. J.; Wasa M.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 17378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zaitsev V. G.; Shabashov D.; Daugulis O. J. Am. Chem. Soc. 2005, 127, 13154. [DOI] [PubMed] [Google Scholar]; b Shabashov D.; Daugulis O. J. Am. Chem. Soc. 2010, 132, 3965. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Reddy B. V. S.; Reddy L. R.; Corey E. J. Org. Lett. 2006, 8, 3391. [DOI] [PubMed] [Google Scholar]; d Zhang S.-Y.; Li Q.; He G.; Nack W. A.; Chen G. J. Am. Chem. Soc. 2013, 135, 12135. [DOI] [PubMed] [Google Scholar]; e Chen F.-J.; Zhao S.; Hu F.; Chen K.; Zhang Q.; Zhang S.-Q.; Shi B.-F. Chem. Sci. 2013, 4, 4187. [Google Scholar]; f Zhou L.; Lu W. Org. Lett. 2014, 16, 508. [DOI] [PubMed] [Google Scholar]; g Hasegawa N.; Charra V.; Inoue S.; Fukumoto Y.; Chatani N. J. Am. Chem. Soc. 2011, 133, 8070. [DOI] [PubMed] [Google Scholar]; h Aihara Y.; Chatani N. J. Am. Chem. Soc. 2014, 136, 898. [DOI] [PubMed] [Google Scholar]; i Wu X.; Zhao Y.; Ge H. J. Am. Chem. Soc. 2014, 136, 1789. [DOI] [PubMed] [Google Scholar]; j Shang R.; Ilies L.; Matsumoto A.; Nakamura E. J. Am. Chem. Soc. 2013, 135, 6030. [DOI] [PubMed] [Google Scholar]
- He G.; Zhang S.-Y.; Nack W. A.; Li Q.; Chen G. Angew. Chem., Int. Ed. 2013, 52, 11124. [DOI] [PubMed] [Google Scholar]
- For arylation of sp3 C–H bonds of amine derivatives via six-membered palladacycles:; a Nadres E. T.; Daugulis O. J. Am. Chem. Soc. 2012, 134, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nadres E. T.; Santos G. I. F.; Shabashow D.; Daugulis O. J. Org. Chem. 2013, 78, 9689. [DOI] [PMC free article] [PubMed] [Google Scholar]; c He G.; Zhao Y.; Zhang S.; Lu C.; Chen G. J. Am. Chem. Soc. 2012, 134, 3. [DOI] [PubMed] [Google Scholar]
- a Wasa M.; Engle K. M.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 3680. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stowers K. J.; Fortner K. C.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a rare example of intramolecular olefination of unactivated, β-hydrogen-containing alkyl halides:Firmansjah L.; Fu G. C. J. Am. Chem. Soc. 2007, 129, 11340. [DOI] [PubMed] [Google Scholar]
- a Wasa M.; Chan K. S. L.; Zhang X.-G.; He J.; Miura M.; Yu J.-Q. J. Am. Chem. Soc. 2012, 134, 18570. [DOI] [PMC free article] [PubMed] [Google Scholar]; b He J.; Li S.; Deng Y.; Fu H.; Laforteza B. N.; Spangler J. E.; Homs A.; Yu J.-Q. Science 2014, 343, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For insightful discussions on the conformation of 2-methoxypyridine:Chein R.-J.; Corey E. J. Org. Lett. 2010, 12, 132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.