

Abstract
Background
Alcohol use disorders are often associated with lung disease. Alcohol exposure leads to the production of reactive oxygen species, lipid peroxidation, and formation of malondialdehyde (MDA) as well as induce the expression of cytochrome p450 2E1 (CYP2E1). Likewise, cigarette smoking can lead to lung lipid peroxidation and formation of MDA. MDA can bind to DNA forming MDA deoxyguanosine (M1dG) adducts, which have been implicated in alcohol-related cancers and cardiovascular disease. Because CYP2E1 regulates MDA production, and our previous studies have shown that alcohol and cigarette smoke can lead to MDA formation, we hypothesized that CYP2E1 would modulate M1dG adduct formation and single strand DNA damage in alcohol- and cigarette smoke-exposed lung cells and tissue.
Methods
Normal human bronchial epithelial cells (HBEC) were pre-treated with 10 μM DADS for 1h, and treated with 80 mM ethanol +/− 5% cigarette smoke extract (CSE) for 3 hrs for comet assay and 6 hrs for CYP2E1, MDA, and M1dG adduct assays. C57BL/6 mice were administered 20% ethanol ad libitum in drinking water for 8 wk and exposed to whole body cigarette smoke for 5 wk. Mice were also fed a CYP2E1 inhibitor, diallyl disulfide (DADS), at 1 μM/g of feed in their daily diet for 7 wk. Whole lung tissue homogenate was used for CYP2E1, MDA, and M1dG adduct assays.
Results
Ethanol exposure significantly increased HBEC olive tail moment. DADS pretreatment of HBEC attenuated this ethanol effect. Ethanol also induced MDA and M1dG adduct formation, which was also significantly reduced by DADS treatment. CSE +/− ethanol did not enhance these effects. In lung tissue homogenate of 8 wk alcohol-fed mice, MDA and M1dG adduct levels were significantly elevated in comparison to control mice and mice fed DADS while consuming alcohol. No increase in MDA and M1dG adduct formation was observed in 5 wk cigarette smoke-exposed mice.
Conclusions
These findings suggest that CYP2E1 plays a pivotal role in alcohol-induced M1dG adducts, and the use of DADS as dietary supplement can reverse the effects of alcohol on M1dG formation.
Keywords: Ethanol, Malondialdehyde, M1dG, CYP2E1, DADS
Introduction
On a daily basis, the lung is exposed to a wide range of oxidants such as dust particles, diesel exhaust, smog, cigarette smoke and alcohol (Gould et al., 2011). Alcohol consumption in humans is a serious health problem that may alter normal function and composition of lipid membranes (Yua et al., 2010) and is often associated with the increased occurrence of lung diseases like pneumonia and acute respiratory distress syndrome (Salaspuro and Salaspuro, 2004) and an increased cancer risk, suggesting a role for alcohol and its oxidative metabolites, acetaldehyde and malondialdehyde (MDA) (Kharbanda et al., 2002; Wyatt et al., 2005). Alcohol metabolism via the CYP2E1 metabolic pathway leads to the formation of ROS in addition to acetaldehyde, which reacts with cell membrane lipids leading to lipid peroxidation. This lipid peroxidation leads to the generation of reactive aldehydes such as MDA (Cederbaum, 2001). People who are alcohol dependent are more likely to be smokers than non-drinkers (Bobo and Husten, 2000) and are also more likely to die from smoking-related diseases as opposed to alcohol-based illness (Ellickson et al., 1992). Thousands of carcinogens and free radicals inhaled by cigarette smokers (Lykkesfeldt, 2007; Mermelstein, 2003) attach to the unsaturated fatty acid on cell membranes, leading to a chain reaction that generates lipid hydroperoxides, which finally decompose to MDA (Del Rio et al., 2005). MDA has been shown to be genotoxic and is capable of forming DNA adducts that are involved in tobacco and alcohol-related cancers and cardiovascular disease (Hoberg et al., 2004).
Because chemical carcinogenesis is a multistage process that requires the binding of a chemical carcinogen to DNA, thereby forming DNA adducts (Tang et al., 2013), reducing or preventing the formation of DNA adducts is essential and an important early step in carcinogenesis (Leuratti et al., 1998; Wang et al., 2013). MDA is one of the depurinating adducts that is highly electrophilic and preferentially attacks the most nucleophilic N7 position of guanosine on DNA (Pullman and Pullman, 1981). In spite of all this, MDA production and correlated DNA adduct (M1dG) formation in ethanol-exposed lung and/or derivative pulmonary cells have not been thoroughly investigated.
Diallyl disulfide (DADS) is an oil-soluble organosulfur compound obtained from garlic and is known to be effective against carcinogenesis and chemically induced toxicity in humans (Manivasagam et al., 2005; Song et al., 2009). In addition to this, it has also been shown to inhibit CYP2E1 metabolism (Ehnert et al., 2012; Shimada et al., 2006). Because alcohol metabolism through the CYP2E1 pathway is one of the important steps often involved in the formation of MDA, it would be promising to study the role of specific inhibitors of CYP2E1 on DNA damage, MDA production, and M1dG adduct formation. Therefore, we hypothesized that the co-exposure of alcohol and cigarette smoke would lead to high amounts of MDA-mediated DNA damage as measured by M1dG adduct levels in HBECs and mouse lung tissue. We also hypothesized that the co-administration of DADS along with alcohol and cigarette smoke would decrease the formation of M1dG adducts.
Methods
Cell Culture
Primary normal HBECs were cultured in bronchial epithelial cell growth medium (BEGM, Lonza, Allendale, NJ) supplemented with 2 μg/mL of amphotericin (MP Biomedicals LLC, Solon, OH). Cells were plated on Type I collagen (Vitrogen 100, Collagen Biomaterials, Palo Alto, CA) coated tissue culture dishes at 37°C in a humidified, 5% CO2 atmosphere. Cells were passaged once a wk at 1:3. Cells between the 2nd and 5th passage were used for experiments.
Cell Viability
No significant cell death or cytotoxicity occurred in HBECs treated with 80 mM alcohol, 5 % CSE and 10 μM DADS compared to control cells using lactate dehydrogenase [LDH] release assay (Thermo scientific, Rockford, IL)(data not shown). Previous studies have shown no significant cytotoxicity when bronchial epithelial cells are treated with ≤ 100 mM ethanol (Simet et al., 2012).
Cigarette Smoke Extract Preparation
Cigarette smoke extract (CSE) was prepared in accordance with Wyatt et al. 1998 (Wyatt et al., 1998) CSE was freshly prepared immediately before all experimental procedures. The cigarettes used were obtained from the University of Kentucky Tobacco-Health Research Division. A cigarette was connected to a peristaltic pump apparatus (Bentley Laboratories LLC, Edison, NJ), pyrolyzed, and the smoke from one cigarette was bubbled through 25 ml of sterile phosphate buffered saline (PBS; pH 7.4). One cigarette per 25 ml volume was used. The CSE in PBS was then sterile-filtered and diluted to 5% in BEGM (Lonza) as a percentage of the total volume.
Comet Assay
Comet assay was performed using a comet assay kit (Trevigen, Gaithersburg, MD) with slight modifications. Briefly, cells were cultured in 6-well plates, treated for 3 h with different conditions, and harvested. The cells were then pelleted and resuspended with cold PBS at 1 × 105 cells/ml. The cell suspension (50 μL) was mixed with 450 μl of low melting point agarose, and 50 μl of this mixture was pipetted over a sample area of comet slides. The slides were placed flat in a 4°C humidity chamber for 30 min, and later immersed in cold lysis solution provided in the kit and left overnight at 4°C. The slides were then transferred into alkaline unwinding solution (200 mM NaOH, 1 mM EDTA, pH 13) and incubated at room temperature for 20–60 min. The slides were then horizontally placed in an electrophoresis apparatus (Life Technologies Inc. Gaithersburg, MD) containing alkaline electrophoresis solution (200 mM NaOH, 1 mM EDTA pH 13) and run at 21 volt/cm for 30 min. Then the slides were fixed with 70% ethanol for 5 min, and were washed twice in distilled water, air-dried and stained with SYBR® gold staining solution (provided in the kit) for 30 min at 4°C. Cells were then viewed by epifluorescence microscopy (Nikon Eclipse E800, Melville, NY) and photographed with a digital camera (Nikon Digital Sight) under 4× magnifications. The measurement was performed using a public domain PC-image analysis program, CASP software, to analyze olive tail moment (OTM). An average of 70 cells were scored per sample.
Animal Exposure Model
All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Female C57BL/6 mice (8–10 wk old) were obtained from Jackson Laboratories (Bar Harbor, Maine), housed in-group cages, and fed commercial rodent chow and water ad libitum for 1 wk. The mice were randomly assigned to 8 treatment groups (sham, alcohol, smoke, alcohol + smoke, DADS control, alcohol + DADS, smoke + DADS, and alcohol + smoke + DADS). All animals were weighed weekly. Mice receiving DADS diet were fed DADS at a concentration of 1 μM/g of their feed and were fed the DADS diet throughout alcohol +/− smoke treatment periods. To ensure that the mice were consuming DADS, their chow was weighed twice weekly for 7 wks.
Alcohol feeding was performed as previously described in Wyatt et al. 2012 (Wyatt et al., 2012). Mice receiving alcohol were given increasing concentrations of alcohol in water over a 1-wk period until the target concentration of 20% was reached using the Meadows-Cook model (Song et al., 2002; Spitzer and Meadows, 1999). Mice in the alcohol group were given 10% ethanol (wt/vol) for 2 days, 15% ethanol (wt/vol) for 5 days, and 20% ethanol (wt/vol) for 7 wk. Mice in the matched control group were given water from the same source without alcohol. Cigarette smoke exposure was performed as previously described in McCaskill et al. 2011 (McCaskill et al., 2011) and Simet et al. 2010 (Simet et al., 2010). Briefly, cages containing C57BL/6 mice were placed in the exposure chamber of a Teague small animal whole body smoke exposure system (Model TE-10; Teague Enterprises, Davis, CA). Animals were exposed to a mixture of mainstream and side stream cigarette smoke via inhalation from 60 R1 reference cigarettes (Lexington, KY) at 150 mg/m3 total smoking particles for 3 hr/day, 5 days/wk, for up to 5 wk. Mice receiving cigarette smoke were gradually brought to their target exposure over a period of 1 wk. Mice were exposed to smoke from 20 cigarettes for day 1, 30 cigarettes for day 2, 40 cigarettes for day 3, 50 cigarettes for day 4 and 5 and 60 cigarettes from day 5 to 5 wk. Control animals were sham-exposed in chambers flowing room air.
CYP2E1 ELISA
CYP2E1 protein levels in the lung tissue homogenate were measured using a commercial ELISA kit (My Biosource, San Diego, CA). HBEC CYP2E1 protein was measured using a commercial ELISA (US Biological, Swampscott, MA) according to the manufacturer's instructions. Briefly, HBECs were pretreated with 10 μM DADS for 1 hr and were further treated with ethanol, CSE and the combination of ethanol and CSE for 6 hr. After 6 hr, the media was removed and cells washed with PBS. The cells were then harvested with protease inhibitor cocktail (Sigma, St Louis, MO) diluted (1:10) in lysis buffer (tris-HCl, ethylene glycol, tetra-acetic acid, magnesium chloride pH 7.4) and centrifuged at 233g at 4°C and sonicated (to disrupt cell membranes). Protein concentrations (mg/mL) were measured using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) to standardize the ELISA results.
ELISA for MDA
MDA levels in the lung tissue homogenate and HBEC were measured using a commercial ELISA kit (US Biologicals, Swampscott, MA) according to the manufacturer's instructions. Briefly, HBECs were pretreated with 10 μM DADS for 1 hr after which the cells were further treated with ethanol, CSE and the combination of ethanol and CSE for 6 hr. After 6 hr the media was removed and cells washed with PBS. Cells were then harvested with protease inhibitor cocktail, centrifuged at 4°C, sonicated, and protein measured as above before performing the ELISA.
ELISA for M1dG
Confluent HBECs were pretreated with 10 μM DADS for 1 hr and then further treated with ethanol, CSE and the combination of ethanol and CSE for 6 hr. After 6 hr the media was removed and washed with PBS. The cells were then harvested in lysis buffer containing protease inhibitor cocktail, centrifuged, and sonicated as described above before conducting the ELISA for M1dG. Briefly, polystyrene 96-well flat-bottomed plates (4 HBX; Thermo Scientific, Waltham, MA) were coated overnight at 4°C with 100 μl with samples (homogenized lung tissue or cell lysates in lysis buffer). Mouse anti-M1dG antibody was a kind gift of Dr. Lawrence Marnett (Vanderbilt University) and was conjugated to horseradish peroxidase (HRP) with a Lightning-LinkTM HRP Conjugation Kit (Innova Bioscience, Babraham, UK). Purified M1dG (Also kindly supplied by Dr. Lawrence Marnett) was added to mouse M1dG antibody conjugated with horseradish peroxidase (1:1000 in in PBS) to make final concentration of 40 ng/mL and incubated overnight in 4°C by gentle mixing in a shaker. After 24 hr the M1dG standard-antibody complex was further serially diluted (2 fold) with PBS for the standard curve. The plate, which was incubated overnight with 100 μL of sample, was further washed with buffer (0.5 % PBS tween) and then the serially diluted standard was added to the standard wells in the 96-well plate. Mouse M1dG antibody conjugated with HRP (1:1000 in PBS) was added to the sample wells and incubated for another 3 hrs at RT. After 3 hrs the plate was washed 4 times with wash buffer and developed with 100 μL tetramethylbenzidine substrate (Sigma, St Louis, MO) in the dark at room temperature before halting the color reaction with 8 M H2SO4. Plates were read at 450 nm using a plate reader (Molecular Devices, Sunnyvale, CA) to determine optical density.
Statistical analysis
All quantitative experiments were performed in triplicate. All data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA) and represented as mean ± standard error. Data were analyzed for statistical significance using one-way ANOVA followed by Tukey post hoc testing between each condition group. Significance was accepted at the 95% confidence interval (P <0.05).
Results
Effect of ethanol and cigarette smoke exposure on single strand DNA damage
Alcohol and cigarette smoke-induced single strand DNA damage was measured using COMET assay in primary HBECs (Fig. 1). Hydrogen peroxide was used as a positive control. Single strand DNA damage was measured using olive tail moment (OTM), which is the percentage of DNA in the tail and tail moment length from the center of the head to center of the tail. OTM increased significantly in the ethanol treatment group in comparison to the control group. There was also a significant increase in OTM when the cells were exposed to 5% cigarette smoke extract (CSE). Diallyl disulfide (DADS) treatment significantly reduced OTM when cells were treated with 10 μM DADS in combination with 80 mM ethanol. Similar results were also seen when cells were treated with ethanol and 5% CSE in combination with DADS but no such effect was seen when cells were treated with CSE in presence of DADS.
Fig. 1. Effect of ethanol exposure on single strand DNA damage measured by comet assay in HBECs.

Monolayers of human bronchial epithelial cells (HBECs) were pretreated for 1 hr with diallyl disulfide (DADS; 10 μM) and further treated for 3 hr with ethanol (EtOH; 80 mM), cigarette smoke extract (CSE; 5%) and co-exposure condition. Hydrogen peroxide (H202; 0.1 mM) was used as positive control. Exposure to ethanol caused significant DNA damage, which was reduced significantly by DADS exposure. Values represent mean ± S.E.M. of (100–150) cells scored. Experiment was performed 3 independent times.
Effect of ethanol and cigarette smoke exposure on CYP2E1 levels in HBEC and C57BL/6 mice whole lung tissue homogenate
Alcohol is known to induce CYP2E1 levels, therefore we measured the CYP2E1 levels in HBECs and mouse lung tissue homogenate of C57BL/6 mice fed with ethanol ad libitum in water and exposed to whole body cigarette smoke. In addition, some mice were fed a DADS diet. We found a significant increase in the CYP2E1 levels in the ethanol exposed HBEC in comparison to control which was not observed in mice exposed to 5% CSE. As expected, there was no significant difference between ethanol only exposed and cigarette smoked and ethanol co-exposed mice on CYP2E1 levels. But when cells were treated with DADS in combination with ethanol, there was a significant reduction in the CYP2E1 level (Fig. 2A). Likewise, we observed a similar result in mice fed ethanol and exposed to cigarette smoke along with DADS diet (Fig. 2B).
Fig. 2. Effect of ethanol and cigarette smoke on CYP2E1 levels in HBECs and C57BL/6 mice whole lung tissue homogenate.

Human bronchial epithelial cells (HBECs) lysate and crude homogenates of mouse lung tissue were assayed using a sandwich ELISA. HBECs were pretreated for 1 hr with diallyl disulfide (DADS; 10 μM) and further exposed with ethanol (EtOH; 80 mM), cigarette smoke extract (CSE; 5%) and co-exposure condition for 6h and assayed using sandwich ELISA for CYP2E1 levels (A). Mice consumed EtOH ad libitum in water and were exposed to whole body cigarette smoke. Exposure to ethanol significantly induced CYP2E1 levels, which were reduced significantly by DADS supplement (B). Values represent mean ± S.E.M. of n=4 mice per condition and n=3 independent experiments for HBECs.
MDA levels in HBECs and lung tissue exposed to ethanol, cigarette smoke, and diallyl disulfide
We measured MDA, a marker of lipid peroxidation in HBECs and mouse lung tissue homogenate of C57BL/6 mice fed with ethanol ad libitum in water and exposed to whole body cigarette smoke, using a sandwich ELISA (Fig. 3A). We observed a significant increase in MDA levels when cells were treated with 80 mM ethanol for 6 h. There was no significant increase in MDA levels when cells were exposed to 5% CSE alone. There was a slight upward trend seen in MDA levels when co-exposed with ethanol and CSE, but the increase was not significant. Treatment with 10 μM DADS lowered the MDA level significantly in ethanol-treated cells. This reduction was not observed in ethanol and cigarette smoke extract co-treatment. In mouse lung tissue homogenate (Fig. 3B), there was a significant increase in MDA levels in mice fed ethanol. We also observed a significant increase in MDA levels in mice fed ethanol and exposed to cigarette smoke. DADS supplement significantly lowered MDA levels in ethanol-fed mice. A similar effect was observed in mice fed ethanol and exposed to cigarette smoke when given DADS supplement. No such effect was seen on cigarette smoke only exposed mice.
Fig. 3. Effect of ethanol and cigarette smoke on MDA levels in HBECs and C57BL/6 mice whole lung tissue homogenate.

Lysate of human bronchial epithelial cells (HBECs) were pretreated for 1 hr with diallyl disulfide (DADS; 10 μM) and further exposed to ethanol (EtOH; 80 mM), cigarette smoke extract (CSE; 5%) and co-exposure condition for 6h and crude homogenates of mouse lung tissue were assayed for malondialdehyde (MDA) levels using a sandwich ELISA. Ethanol alone significantly increased MDA levels. Exposure to ethanol and cigarette smoke significantly increased MDA levels in tissue homogenate, which was reduced significantly by DADS supplement. Values represent mean ± S.E.M. of n=4 mice and n=3 independent experiments for HBECs.
M1dG levels in HBEC and lung tissue exposed to ethanol, cigarette smoke, and DADS
HBECs treated with ethanol exhibited significantly high M1dG adduct level in comparison to control (Fig. 4A). A similar result was observed when cells were treated with ethanol and 5% CSE, but we did not see any synergistic increase in the adduct levels. Interestingly, there was no significant increase in the M1dG adduct levels when cells were exposed to CSE alone. Treatment with 10 μM DADS along with ethanol significantly reduced the adduct levels. A similar result was observed when cells were co-exposed to ethanol and 5% CSE along with DADS. In mouse lung tissue homogenate (Fig. 4B) there was a significant increase in adduct level in mice fed ethanol ad libitum in water. We also observed a significant increase in adduct level in mice fed with ethanol and exposed to whole body cigarette smoke. Again, no synergy was observed. However, co-exposed mice fed DADS in their diet had a significant reduction in the adduct level in their lung tissue. A significant reduction in adduct level was also seen when ethanol-fed mice were given DADS in their diet.
Fig. 4. Effect of ethanol and cigarette smoke on M1dG levels in HBECs and C57BL/6 mice whole lung tissue homogenate.

Lysate of human bronchial epithelial cells (HBECs) were pretreated for 1 hr with diallyl disulfide (DADS; 10 μM) and further exposed to ethanol (EtOH; 80 mM), cigarette smoke extract (CSE; 5%) and co-exposure condition for 6h and crude homogenates of mouse lung tissue were assayed using indirect ELISA for M1dG levels. Ethanol alone and in combination with cigarette smoke significantly increased M1dG levels. Ethanol exposure significantly increased M1dG levels, which were reduced significantly by DADS supplement. Values represent mean ± S.E.M. of n=4 mice and of n=3 independent experiments for HBECs.
Discussion
In recent years researchers have focused on the effects of alcohol consumption and smoking on the lungs, but few studies have been conducted on the role of different types aldehydes formed in the lung tissue. Alcohol may act as a solvent and enhance the penetration of tobacco-related carcinogenic compounds in the pulmonary epithelium lining environment (Pöschl and Seitz, 2004). Additionally, a “recycling” effect leading to repeated exposure of high local concentrations of alcohol is often observed during alcohol ingestion where alcohol gets vaporized while moving into the conducting airways and gets deposited back into the airway to be released again into the airways during exhalation (Sisson, 2007). Reactive oxygen species has been often suggested as the main contributor to oxidative stress injury on pulmonary health (Cigremis et al., 2004). Alcohol is primarily converted to acetaldehyde by both alcohol dehydrogenase (ADH) and the CYP2E1 pathway (Tuma and Casey, 2003). Aldehydes such as acetaldehyde and MDA formed as a result of alcohol metabolism have been shown to form adducts to both proteins and nucleotides by many studies. A stable hybrid adduct known as MDA-acetaldehyde (MAA) adduct is formed when acetaldehyde and MDA react together with nucleophilic proteins (Tuma, 2002). Specifically, the by-product of the reaction of MDA and DNA, which has been shown to be mutagenic in bacterial and mammalian cells, is the pyrimido[1,2-a]purin-10-(3H) one nucleoside (M1dG) derived from deoxyguanosine (Wang et al., 2004; Sun et al., 2004). This endogenous DNA adduct plays an important role in the etiology of human cancer and is often derived from oxidative stress, lipid peroxidation, or other endogenous processes (Leuratti et al., 1998; Wang et al., 2013). M1dG can lead to mispairing due to apurinic DNA sites when replicated in vitro by multiple different DNA polymerases (Akingbade et al., 2012; Wang and Liehr, 1995).
Co-exposure to both alcohol and cigarette smoke leads to very high concentrations of both acetaldehyde and MDA favoring the formation of MAA-adducted proteins in the lungs of mice (McCaskill et al., 2011). However, in the current study, we propose a model (Fig. 5) in which CYP2E1-mediated metabolism of alcohol alone is the major pathway for the formation of M1dG DNA adducts in the lung. CYP2E1-mediated ethanol metabolism generates ROS, which can further lead to lipid peroxidation generating MDA, which can attach to the guanine base of DNA to form the M1dG adduct. Inhibition of CYP2E1 leads to a reduction in both MDA and M1dG adduct formation. Studies have shown an association of CYP2E1-mediated alcohol metabolism and adduct formation in the liver (Swaminathan et al., 2013), juxtaposed with pulmonary CYP2E1 induction by carcinogens from pyrolyzed tobacco (Osna et al., 2007; French et al., 1997).
Fig. 5. Proposed model of M1dG adduct formation.
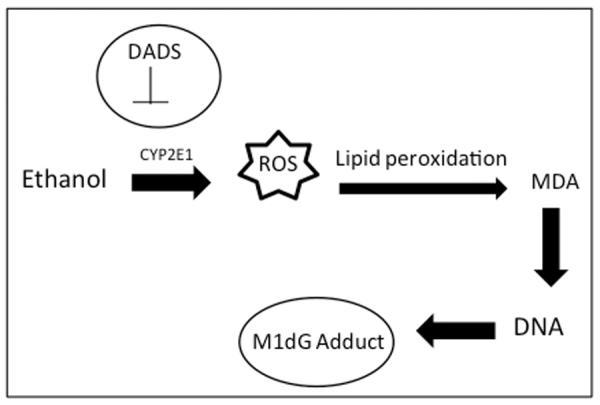
CYP2E1 mediated metabolism of alcohol leads to generation of reactive oxygen species (ROS). ROS enhances lipid peroxidation leading to the generation of malondialdehyde (MDA), which can adduct to the guanosine base of DNA to form a M1dG adduct. Inhibition of CYP2E1 by diallyl disulfide (DADS) leads to reduction in both MDA levels and M1dG adduct formation.
Our research showed an increase in the single strand DNA damage in the HBEC when exposed to ethanol, but the highest DNA damage was seen in the co-exposure group. A similar result was observed in human intestinal epithelial Caco-2 cells (Park et al., 2012) where cigarette smoke also led to a significant increase in DNA damage. Our result is consistent with studies done by Lui et al. in HBECs showing cigarette smoke extract-induced DNA damage (Liu et al., 2005). These findings suggest that a CYP2E1-independent effect on DNA damage occurs in response to cigarette smoke, likely in response to the known levels of oxidants contained in pyrolyzed tobacco. Our study also provides novel information about a specific malondialdehyde-mediated DNA adduct (M1dG) formed in the bronchial epithelial cells and lung tissue. High levels of M1dG adduct was measured in the alcohol-exposed group in comparison the smoke exposed group. This is consistent with results of our previous studies showing high level of MDA in the BALF fluid of alcohol fed mice in comparison to smoke exposed mice (McCaskill et al., 2011). Burning of tobacco resulted in production of high acetaldehyde and less MDA in comparison to alcohol only-exposed mice (McCaskill et al., 2011). In contrast to previous studies, there was no synergistic effect of alcohol and cigarette smoke on M1dG formation as previously in MAA adducted protein studies. Previous studies have shown that cigarette smoke and alcohol co-exposure is required to make sufficient acetaldehyde and MDA concentrations to generate MAA-adducted protein in lung (Wyatt et al., 2012). In our study, we have found that exposure to alcohol singularly results in sufficient MDA concentrations for M1dG adduct formation, suggesting that the pathway for the formation of these two adducts is likely different.
Our study also investigated the role of CYP2E1 in alcohol-mediated DNA damage, MDA and M1dG formation in cells and lung tissue. Consistent with previous studies, ethanol led to significant increase in CYP2E1 levels in both mouse tissue and HBEC (Runge et al., 2001; Shimada et al., 2006). Further inhibition of CYP2E1 with DADS led to the significant decrease in the single strand DNA damage, which was further supported by a decrease in MDA and M1dG adduct formation in both HBECs and mouse lung tissue. This result is in accordance with published results showing decreased oxidative stress and less MDA adduct formation by inhibition of CYP2E1 in human hepatoma VL-17A cell line and human hepatocytes by DADS (Swaminathan et al., 2013; Shimada et al., 2006). These data provide evidence that CYP2E1 plays an important role in alcohol mediated oxidative stress and M1dG adduct formation.
Conclusion
In summary, alcohol consumption can lead to generation of reactive oxygen species resulting in lipid peroxidation and production of various gene toxic aldehydes such as MDA which can further lead to formation of various DNA (M1dG) adducts in lung tissue through a CYP2E1-mediated pathway. Heavy drinking of alcohol may lead to DNA damage suggesting a pathogenic role of MDA-derived DNA adducts in carcinogenesis, which can be reduced by the dietary consumption of DADS.
Acknowledgements
The Authors wish to thank Ms. Lisa Chudomelka for expert editing and manuscript preparation as well as Dr. Lawrence J. Marnett (Vanderbilt University) for his generous gift of M1dG and M1dG antibody and Wan Tin Lin, an exchange student from Taiwan for her help during the development of the M1dG assay.
This material is the result of work supported with resources and the use of facilities at the Omaha VA Medical Center, Omaha, NE (Department of Veterans Affairs [VA Merit Review I01 BX000728] to TAW). This work was supported by NIH-NIAAA (R01AA017993 and R01AA017993-S1) to TAW.
Abbreviations
- MDA
Malondialdehyde
- ROS
Reactive oxygen species
- M1dG
Malondialdehyde deoxyguanosine
- DADS
Diallyl disulfide
- CYP2E1
Cytochrome P450 2E1
- LDH
Lactate dehydrogenase
- CSE
Cigarette smoke extract
- BEGM
Bronchial epithelial cell growth medium
- HBECs
Human Bronchial Epithelial Cells
- MAA
Malondialdehyde Acetaldehyde
- CASP
Comet Assay Software Project
- OTM
Olive Tail Moment
References
- Akingbade D, Kingsley PJ, Shuck SC, Cooper T, Carnahan R, Szekely J, Marnett LJ. Selection of Monoclonal Antibodies Against 6-oxo-M1dG and Their Use in an LC-MS/MS Assay for the Presence of 6-oxo-M1dG in Vivo. Chem Res Toxicol. 2012;25:454–461. doi: 10.1021/tx200494h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobo JK, Husten C. Sociocultural influences on smoking and drinking. Alcohol Research and Health. 2000;24:225–232. [PMC free article] [PubMed] [Google Scholar]
- Cederbaum AI. Introduction—serial review: alcohol, oxidative stress and cell injury. Free Radical Biology and Medicine. 2001;31:1524–1526. doi: 10.1016/s0891-5849(01)00741-9. [DOI] [PubMed] [Google Scholar]
- Cigremis Y, Turkoz Y, Akgoz M, Sozmen M. The effects of chronic exposure to ethanol and cigarette smoke on the level of reduced glutathione and malondialdehyde in rat kidney. Urol Res. 2004;32:213–218. doi: 10.1007/s00240-004-0406-x. [DOI] [PubMed] [Google Scholar]
- Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutrition, metabolism and cardiovascular diseases. 2005;15:316–328. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Ehnert S, Braun K, Buchholz A, Freude T, Egana J, Schenck T, Schyschka L, Neumaier M, Döbele S, Stöckle U. Diallyl-disulphide is the effective ingredient of garlic oil that protects primary human osteoblasts from damage due to cigarette smoke. Food Chem. 2012;132:724–729. [Google Scholar]
- Ellickson PL, Hays RD, Bell RM. Stepping through the drug use sequence: longitudinal scalogram analysis of initiation and regular use. J Abnorm Psychol. 1992;101:441–451. doi: 10.1037//0021-843x.101.3.441. [DOI] [PubMed] [Google Scholar]
- French SW, Morimoto M, Reitz RC, Koop D, Klopfenstein B, Estes K, Clot P, Ingelman-Sundberg M, Albano E. Lipid peroxidation, CYP2E1 and arachidonic acid metabolism in alcoholic liver disease in rats. J Nutr. 1997;127:907S–911S. doi: 10.1093/jn/127.5.907S. [DOI] [PubMed] [Google Scholar]
- Gould NS, Min E, Gauthier S, Martin RJ, Day BJ. Lung glutathione adaptive responses to cigarette smoke exposure. Respir Res. 2011;12:133. doi: 10.1186/1465-9921-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg A, Otteneder M, Marnett LJ, Poulsen HE. Measurement of the malondialdehyde– 2'-deoxyguanosine adduct in human urine by immuno-extraction and liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry. Journal of mass spectrometry. 2004;39:38–42. doi: 10.1002/jms.547. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Shubert KA, Wyatt TA, Sorrell MF, Tuma DJ. Effect of malondialdehyde–acetaldehyde–protein adducts on the protein kinase C-dependent secretion of urokinase-type plasminogen activator in hepatic stellate cells. Biochem Pharmacol. 2002;63:553–562. doi: 10.1016/s0006-2952(01)00883-8. [DOI] [PubMed] [Google Scholar]
- Leuratti C, Singh R, Lagneau C, Farmer PB, Plastaras JP, Marnett LJ, Shuker D. Determination of malondialdehyde-induced DNA damage in human tissues using an immunoslot blot assay. Carcinogenesis. 1998;19:1919–1924. doi: 10.1093/carcin/19.11.1919. [DOI] [PubMed] [Google Scholar]
- Liu X, Conner H, Kobayashi T, Kim H, Wen F, Abe S, Fang Q, Wang X, Hashimoto M, Bitterman P, Rennard SI. Cigarette Smoke Extract Induces DNA Damage but Not Apoptosis in Human Bronchial Epithelial Cells. American Journal of Respiratory Cell and Molecular Biology. 2005;33:121–129. doi: 10.1165/rcmb.2003-0341OC. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt J. Malondialdehyde as biomarker of oxidative damage to lipids caused by smoking. Clinica chimica acta. 2007;380:50–58. doi: 10.1016/j.cca.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Manivasagam T, Subramanian P, Suthakar G, Essa MM. The chemopreventive effect of diallyl disulphide on N-nitrosodiethylamine induced heptocarcinogenesis. J Appl Biomedicine. 2005;3:187–191. [Google Scholar]
- McCaskill ML, Kharbanda KK, Tuma DJ, Reynolds JD, DeVasure JM, Sisson JH, Wyatt TA. Hybrid malondialdehyde and acetaldehyde protein adducts form in the lungs of mice exposed to alcohol and cigarette smoke. Alcoholism: Clinical and Experimental Research. 2011;35:1106–1113. doi: 10.1111/j.1530-0277.2011.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein R. Teen smoking cessation. Tob Control. 2003;12:i25–i34. doi: 10.1136/tc.12.suppl_1.i25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, White RL, Todero S, Mc Vicker BL, Thiele GM, Clemens DL, Tuma DJ, Donohue TM. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology. 2007;45:53–61. doi: 10.1002/hep.21442. [DOI] [PubMed] [Google Scholar]
- Park SC, Lim J, Jeen YT, Keum B, Seo YS, Kim YS, Lee SJ, Lee HS, Chun HJ, Um SH. Ethanol-induced DNA damage and repair-related molecules in human intestinal epithelial Caco-2 cells. Molecular Medicine Reports. 2012;5:1027–1032. doi: 10.3892/mmr.2012.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöschl G, Seitz HK. Alcohol and cancer. Alcohol and alcoholism. 2004;39:155–165. doi: 10.1093/alcalc/agh057. [DOI] [PubMed] [Google Scholar]
- Pullman A, Pullman B. Molecular electrostatic potential of the nucleic acids. Q Rev Biophys. 1981;14:289–380. doi: 10.1017/s0033583500002341. [DOI] [PubMed] [Google Scholar]
- Runge DM, Stock TW, Lehmann T, Taege C, Bernauer U, Stolz DB, Hofmann S, Foth H. Expression of cytochrome P450 2E1 in normal human bronchial epithelial cells and activation by ethanol in culture. Arch Toxicol. 2001;75:335–345. doi: 10.1007/s002040100248. [DOI] [PubMed] [Google Scholar]
- Salaspuro V, Salaspuro M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. International journal of cancer. 2004;111:480–483. doi: 10.1002/ijc.20293. [DOI] [PubMed] [Google Scholar]
- Shimada M, Liu L, Nussler N, Jonas S, Langrehr JM, Ogawa T, Kaminishi M, Neuhaus P, Nussler AK. Human hepatocytes are protected from ethanol-induced cytotoxicity by DADS via CYP2E1 inhibition. Toxicol Lett. 2006;163:242–249. doi: 10.1016/j.toxlet.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Simet SM, Sisson JH, Pavlik JA, DeVasure JM, Boyer C, Liu X, Kawasaki S, Sharp JG, Rennard SI, Wyatt TA. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. American journal of respiratory cell and molecular biology. 2010;43:635. doi: 10.1165/rcmb.2009-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simet S, Wyatt TA, DeVasure J, Yanov D, Allen-Gipson D, Sisson JH. Alcohol Increases the Permeability of Airway Epithelial Tight Junctions in Beas2B and NHBE Cells. Alcoholism: Clinical and Experimental Research. 2012;36:432–442. doi: 10.1111/j.1530-0277.2011.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41:293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Lee SK, Kim KM, Park SE, Park S, Kim KH, Ahn SC, Park YC. Molecular mechanism of diallyl disulfide in cell cycle arrest and apoptosis in HCT-116 colon cancer cells. J Biochem Mol Toxicol. 2009;23:71–79. doi: 10.1002/jbt.20266. [DOI] [PubMed] [Google Scholar]
- Song K, Coleman RA, Zhu X, Alber C, Ballas ZK, Waldschmidt TJ, Cook RT. Chronic ethanol consumption by mice results in activated splenic T cells. J Leukoc Biol. 2002;72:1109–1116. [PubMed] [Google Scholar]
- Spitzer JH, Meadows GG. Modulation of perforin, granzyme A, and granzyme B in murine natural killer (NK), IL2 stimulated NK, and lymphokine-activated killer cells by alcohol consumption. Cell Immunol. 1999;194:205–212. doi: 10.1006/cimm.1999.1511. [DOI] [PubMed] [Google Scholar]
- Sun X, Nair J, Bartsch H. A Modified Immuno-Enriched 32P-Postlabeling Method for Analyzing the Malondialdehyde-Deoxyguanosine Adduct, 3-(2-Deoxy-β-d-erythropentofuranosyl)-pyrimido [1, 2-α] purin-10 (3 H) one in Human Tissue Samples. Chem Res Toxicol. 2004;17:268–272. doi: 10.1021/tx034183p. [DOI] [PubMed] [Google Scholar]
- Swaminathan K, Clemens DL, Dey A. Inhibition of CYP2E1 leads to decreased malondialdehyde-acetaldehyde adduct formation in VL-17A cells under chronic alcohol exposure. Life Sci. 2013 doi: 10.1016/j.lfs.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Tang Y, Kassie F, Qian X, Ansha B, Turesky RJ. DNA Adduct Formation of 2-Amino-9H-pyrido [2, 3-b] indole and 2-Amino-3, 4-dimethylimidazo [4, 5-f] quinoline in Mouse Liver and Extrahepatic Tissues During a Subchronic Feeding Study. Toxicological Sciences. 2013;133:248–258. doi: 10.1093/toxsci/kft077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radical Biology and Medicine. 2002;32:303–308. doi: 10.1016/s0891-5849(01)00742-0. [DOI] [PubMed] [Google Scholar]
- Tuma J, Casey CA. Dangerous byproducts of alcohol breakdown-focus on adducts. Alcohol Research and Health. 2003;27:285–290. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Marnett LJ, Harris TM, Rizzo CJ. A novel synthesis of malondialdehyde adducts of deoxyguanosine, deoxyadenosine, and deoxycytidine. Chem Res Toxicol. 2004;17:144–149. doi: 10.1021/tx034174g. [DOI] [PubMed] [Google Scholar]
- Wang M, Peng L, Jensen CJ, Deng S, West BJ. Food Science & Nutrition. 2013. Noni juice reduces lipid peroxidation–derived DNA adducts in heavy smokers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Liehr JG. Lipid hydroperoxide-induced endogenous DNA adducts in hamsters: possible mechanism of lipid hydroperoxide-mediated carcinogenesis. Arch Biochem Biophys. 1995;316:38–46. doi: 10.1006/abbi.1995.1007. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Kharbanda KK, McCaskill ML, Tuma DJ, Yanov D, DeVasure J, Sisson JH. Malondialdehyde–acetaldehyde-adducted protein inhalation causes lung injury. Alcohol. 2012;46:51–59. doi: 10.1016/j.alcohol.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Kharbanda KK, Tuma DJ, Sisson JH, Spurzem JR. Malondialdehyde–acetaldehyde adducts decrease bronchial epithelial wound repair. Alcohol. 2005;36:31–40. doi: 10.1016/j.alcohol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol. 1998;275:L827–35. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]
- Yua H, Oyama T, Isse T, Kitagawa K, Pham T, Tanaka M, Kawamoto T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact. 2010;188:367–375. doi: 10.1016/j.cbi.2010.08.005. [DOI] [PubMed] [Google Scholar]