

Abstract
Prions are proteinaceous infectious agents responsible for the transmission of prion diseases. The lack of a procedure for cultivating prions in the laboratory has been a major limitation to the study of the unorthodox nature of this infectious agent and the molecular mechanism by which the normal prion protein (PrPC) is converted into the abnormal isoform (PrPSc). Protein misfolding cyclic amplification (PMCA ), described in detail in this protocol, is a simple, fast and efficient methodology to mimic prion replication in the test tube. PMCA involves incubating materials containing minute amounts of infectious prions with an excess of PrPC and boosting the conversion by cycles of sonication to fragment the converting units, thereby leading to accelerated prion replication. PMCA is able to detect the equivalent of a single molecule of infectious PrPSc and propagate prions that maintain high infectivity, strain properties and species specificity. A single PMCA assay takes little more than 3 d to replicate a large amount of prions, which could take years in an in vivo situation. Since its invention 10 years ago, PMCA has helped to answer fundamental questions about this intriguing infectious agent and has been broadly applied in research areas that include the food industry, blood bank safety and human and veterinary disease diagnosis.
INTRODUCTION
Prions and transmissible spongiform encephalopathies
Transmissible spongiform encephalopathies (TSEs) or prion diseases are a group of etiologically diverse neurological disorders, which include several pathological conditions in animals and humans1,2. These diseases are characterized by long incubation periods, which are followed by a short symptomatic stage in which there is a progressive and severe degeneration of brain function. The zoonotic potential of TSEs has been evident since the bovine spongiform encephalopathy outbreaks during the 1980s and the 1990s (refs. 3,4). The human version of bovine spongiform encephalopathy, termed variant Creutzfeldt-Jakob disease, has been reported to be transmitted from human to human by blood transfusions, even when blood donors are years from developing a clinically detectable pathology5–7. Owing to these long incubation periods, the number of people who could be silently incubating this disease is unknown.
Extensive experimentation and clinical data suggest that TSEs are caused by the accumulation of an abnormal form of the prion protein, PrPSc, which is generated at the expense of a normally produced protein, PrPC (refs. 1 and 8). PrPSc is also the main or sole component of the infectious agent. The brain accumulation of PrPSc is thought to be the cause of cell death, brain inflammation and spongiform degeneration typically observed in the brains of affected individuals9. Although PrPSc also accumulates in other tissues, such as spleen, lymph nodes, muscle and kidney, the deleterious effects of this protein are restricted mainly to the brain, probably because of the high concentrations of PrPC in neurons.
The development of the PMCA assay
The PMCA procedure was first described 10 years ago10. During its short life, PMCA has experienced several changes and improvements, although the core principle of the technology remains the same: to mimic in vitro the PrPC to PrPSc conversion that features in vivo prion replication. Although the molecular mechanism of prion replication is not completely understood, a large body of evidence indicates that it involves the template-induced conversion of endogenous PrPC into PrPSc catalyzed by tiny quantities of exogenous PrPSc (refs. 1 and 11). PrPSc is a polymer of variable size that can form large fibrillar aggregates, resembling amyloid fibrils observed in Alzheimer’s disease and other amyloid-related disorders12,13. The comparison of prion conversion with the process of amyloid formation provides a model for the PrPC to PrPSc conversion, in which the pathological protein may act as a seed recruiting molecules of PrPC, inducing and stabilizing their mis-folding by incorporation into the growing oligomer11,12,14,15. In this way, the PrPSc polymer is elongated at the ends as new molecules of PrPC are converted and incorporated. The rate-limiting process in the seeding-nucleation mechanism of prion replication is the fragmentation of the polymer to release more units to catalyze the templated conversion. During PMCA, minute amounts of PrPSc are incubated with an abundant source of PrPC in the test tube to induce the growth of PrPSc polymers (Fig. 1). Thereafter, the samples are subjected to ultrasound in order to break down the polymers, thus multiplying the number of seeds for conversion in a new cycle of incubation11. Hence, after each cycle, the number of seeds is increased in an exponential manner (Fig. 1). The cyclic nature of PMCA and the strong power of amplification are equivalent to the widely used PCR technique for DNA amplification. One of the important improvements to PMCA after its original development was automation16,17, making the technique suitable for high throughput and applicable to basic scientific research and industrial practices. Another improvement was the addition of beads, which improve the efficiency of amplification and reduce intersample variability18–20 by aiding the homogenous dispersion of the ultrasound waves in the tube.
Figure 1.
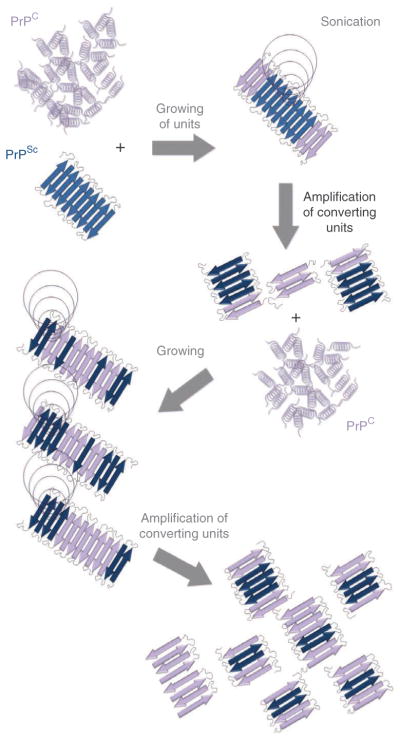
Schematic diagram for protein misfolding cyclic amplification. PMCA is based on the assumption that prion replication occurs by a seeding/nucleation model, in which PrPSc seeds bind and convert PrPC by incorporating the protein into the polymer. In PMCA, PrPSc and PrPC are mixed and incubated, allowing the misfolding of PrPC, which permits the incorporation and growth of PrPSc aggregates. After incubation, samples are submitted to sonication in order to fragment PrPSc polymers, thereby generating new free ends suitable for continued prion replication. This process is cyclically repeated in order to produce an exponential amplification of PrPC conversion.
PMCA: a decade of ‘cultivating’ prions in the test tube
The versatility and efficiency of PMCA enables multiple applications in many fields, both for basic science and for translational research. It can be applied to understanding the unique prion biology, development of ultrasensitive prion detection assays, screening for molecules to interfere with prion formation and investigating the presence of prions in biological and environmental samples.
PMCA has been used to investigate the nature of the infectious agent and its intricate mechanism of propagation, including the probable involvement of cellular cofactors. In vitro–amplified PrPSc has been shown to have biochemical and structural properties identical to those of brain-derived PrPSc (ref. 16). Furthermore, PMCA-generated prions have been shown to be infectious in various species of wild-type animals16,21–23, providing strong support to the hypothesis that the infectious agent in TSEs is composed exclusively of protein. Infectious material has been generated by PMCA using brain extracts, cell lysates, highly purified PrPC and even recombinant PrP produced in bacteria16,24,25. The studies of the minimal components needed to sustain prion replication by PMCA have highlighted the involvement of cellular cofactors in prion conversion24,26–28. At present, the minimal recipe for producing PrPSc with an infectivity titer equivalent to brain-derived prions consists of using lipids, polyanions (mostly RNA) and an adequate PrPC substrate27. PMCA has also been shown to reproduce complex processes characteristic of prion transmission, including strain diversity21, species barrier, strain adaptation and strain memory22,23,29. Finally, extensive PMCA cycling has enabled the de novo generation of infectious material (i.e., without the addition of pre-existing PrPSc), mimicking the spontaneous appearance of prions in sporadic forms of TSEs24,25,30.
An obvious application of PMCA is in the detection of small amounts of prions in biological samples as a means of biochemical diagnosis for disease. PrPSc is so far the only validated and specific marker for TSEs31. The search for other markers has been mostly unsuccessful, although several neuronal, astrocytic and glial proteins that are elevated in sporadic Creutzfeldt-Jakob disease (such as 14-3-3, Tau, S-100, ubiquitin, erythroid differentiation-related factor, Grp58 and so on) have been identified31, some of which are also altered in biological fluids. The biggest problem facing a diagnostic test based on PrPSc detection is the very low concentration of this protein in tissues other than brain and in preclinical disease stages, when diagnosis is most useful and needed31. Most of the efforts to develop a diagnostic system for prion diseases have been focused on increasing the sensitivity of the current detection methods. PMCA offers the opportunity to enhance existing methods by amplifying the amount of PrPSc in the sample. Indeed, PMCA has been shown to detect prions in body fluids, including blood, urine and cerebrospinal fluid32–35, even at presymptomatic disease stages36. In fact, PMCA has been reported to be sensitive enough to detect the equivalent of a single infectious prion molecule17. In addition to its application in prion diagnosis, an ultrasensitive prion detection technique is also very useful for investigating the possibility that diverse samples and materials might be contaminated with prions, such as animal tissues for consumption, organs for transplant, blood for transfusion, environmental samples (water, grass, soil), raw materials used to produce medical products (serum, urine), surgical instruments and so on. The use of PMCA for prion detection in these samples will help to minimize further spreading of disease and enable regulatory authorities to legislate the safe use of these materials. A recent development that should be useful for this application is quantitative PMCA (Box 1; see also Supplementary Figs. 1–4), which enables not only the detection of minute quantities of prions but also the estimation of the concentration of PrPSc present in the sample37.
Box 1. Quantitative PMCA (qPMCA) to estimate PrPSc quantity in different tissues and biological fluids ● TIMING 323 h (for a four-round assay).
Standard PMCA is a qualitative technique that permits the detection of very small quantities of PrPSc by cyclic amplification of its misfolding. To provide a quantitative estimation of PrPSc concentration in tissues and fluids, we have developed qPMCA, which consists of measuring the extent of PMCA cycling required to detect known concentrations of PrPSc (ref. 37). By extrapolating the number of PMCA cycles and rounds needed to detect PrPSc in a given tissue or fluid with this calibration curve, a rough estimation of the PrPSc initially present in the sample can be achieved.
PROCEDURE
Preparation of PrPsc standard ● TIMING 4 h
-
1
Take an aliquot of 500 μl of a 10% (wt/vol) brain homogenate from a terminally ill prion-infected animal (prepared as explained in Steps 7–9 of the main PROCEDURE) and mix with 500 μl of 20% (wt/vol) Sarkosyl prepared in PBS plus PI in an ultracentrifuge tube. Mix well by pipetting.
-
2
Centrifuge the sample at ~146,000g (46,200 r.p.m.) in a Beckman-Coulter ultracentrifuge for 1 h at 4 °C.
-
3
Carefully discard the supernatant by pouring it down the opposite side of the tube from the pellet and add 1 ml of PBS containing PI to the pellet.
▲ CRITICAL STEP The pellet should not be disrupted, as some PrPSc could be lost in this process. The addition of the PBS in this step is to remove Sarkosyl traces that could affect the PMCA procedure.
-
4
Centrifuge the sample at ~146,000g (46,200 r.p.m.) in a Beckman-Coulter ultracentrifuge for 30 min at 4 °C.
-
5
Remove the supernatant carefully by pouring it down the opposite side of the tube from the pellet. Let the tube dry by placing it upside down on a clean adsorbent surface (paper towel).
-
6
Add 300 μl of PBS containing PI and resuspend the pellet. Transfer it to a 1.5-ml Eppendorf tube.
-
7
Wash the bottom of the ultracentrifuge tube from step 5 by pipetting it with 200 μl of PBS containing PI. Remove all volume remaining in the tube and transfer to the 1.5-ml Eppendorf tube from step 6. You should end up with a volume similar to the starting sample (500 μl).
-
8
Place the tube on ice and resuspend the pellet using a Bandelin sonicator at 0.1 s pulses for 20 s at 40% power. The process should be repeated twice in order to ensure a complete disruption of the pellet.
! CAUTION We strongly recommend performing this step in a closed chamber in order to avoid the spread of sample to the environment. We do not recommend using a water bath sonicator, as pellets obtained by this method are hard to disrupt.
■ PAUSE POINT Resulting materials can be maintained for at least 3 months at - 20 °C until use.
Estimation of the concentration Of Prpsc standard ● TIMING 19 h
-
9
Take 90 μl of the resulting material and mix with 10 μl of protein denaturing buffer. Mix well by pipetting and vortexing.
-
10
Heat the sample for 10 min at 95–100 °C.
▲ CRITICAL STEP To ensure a complete denaturation for further characterization of the PrPSc standard, the material should be completely homogeneous at this point.
▲ CRITICAL STEP As the objective of this part of the protocol is to prepare a standard for quantification, any loss of volume by evaporation should be prevented or corrected in the calculation.
-
11
Let the sample cool down by placing it at room temperature for 3–5 min.
-
12
Add 13 μl of NP-40, 13 μl of G7 buffer and 3 μl of PNGase F to the sample.
▲ CRITICAL STEP The sample should not be hot, as it could inhibit PNGase F activity.
-
13
Incubate the sample overnight at 37 °C using an Eppendorf thermomixer at 450 r.p.m.
-
14
Add 60 μl of LDS protein loading buffer and heat the sample for 10 min at 95–100 °C.
-
15
Perform a western blot analysis (as described in Steps 17–29 of the main PROCEDURE) of the samples at different dilutions (dilution factor of 2) with known amounts of recombinant protein from the same species. Measure the amount of PrPSc in the sample by densitometry using a BioRad dark chamber according to the manufacturer’s instructions (Supplementary Fig. 4a).
qPMCA procedure ● TIMING depends on the number of PMCA cycles and rounds used
-
16
Prepare the samples to be analyzed (e.g., spleen, blood, muscle, urine) as described in Steps 1–6 of the main PROCEDURE.
▲ CRITICAL STEP To prevent a differential effect of various tissue components on PMCA, all samples should be treated in the same way in order to remove potential interferences in PMCA. When the sample is a liquid (urine, plasma and so on), the starting point is 500 μl of the sample without homogenization. Solid tissues must be well homogenized before starting the procedure.
-
17
Resuspend each pellet in 100 μl of NBH substrate by pipetting; follow this with two sonication pulses in a Misonix 4000 sonicator at 250–260 W of power for 20 s.
-
18
Run the procedure for sPMCA and detect the product by western blot analysis as indicated in Steps 16–29 of the main PROCEDURE, using brain-derived PrPSc precipitated with Sarkosyl as a standard. The standard sample should be tested in tenfold dilution factors (Supplementary Fig. 4b). We strongly recommend testing at least 16 tenfold dilutions of the control sample. Experiments should be performed in quintuplicate.
▲ CRITICAL STEP Standard sample should be run every time a qPMCA is performed. Do not rely on previous calibration curves to estimate PrPSc concentration in tissues.
▲ CRITICAL STEP We strongly advise the use of many negative controls (tubes with NBH substrate without any inocula) in order to detect possible false-positive results due to contamination.
? TROUBLESHOOTING
-
19
Generate a PrPSc detection curve by identifying the PMCA round where a clear PrP27-30 appears in a specific dilution (Supplementary Fig. 4b). At least three independent sets of sPMCA (preferably five) should be analyzed to elaborate this curve. Consider as positive signals only those showing densitometric values of 10% over the background37.
EXPECTED RESULTS
Expected results of PMCA amplification using a Sarkosyl-treated PrPSc inoculum are observed in Supplementary Figure 4b. Express the results in terms of the quantity (in g or mol) of PrPSc used to begin the reaction, which is estimated by comparison with the western blot signal of known concentrations of recombinant PrP (Supplementary Fig. 4a). When compared with the brain dilution, it is often clear that the PMCA amplification efficiency observed in Sarkosyl-treated samples is lower than that of untreated samples. However, Sarkosyl treatment of the standard inoculum is absolutely necessary for an appropriate quantification, as the tissue samples to be analyzed must either be treated in this way to remove components that could affect PMCA (such as blood) or concentrated (owing to low PrPSc content, as in urine, plasma and so on). PrP27-30 content in a given tissue or in fluids should be estimated by extrapolation to the calibration curve generated by the standard inoculum. For more details regarding qPMCA, check ref. 37.
PMCA has also been used as a screening assay to identify molecules that inhibit PrPC to PrPSc conversion38, and to develop strategies for removing and decontaminating prions from biological samples20,39. Since the first report, PMCA has been used extensively throughout the world for multiple applications; more than 100 articles have been published by more than 20 different research groups that have used PMCA for diverse applications.
Experimental design
PMCA consists of cycles of accelerated prion replication in a cell-free system. The specimen to be analyzed (containing or suspected to contain infectious prions; e.g., tissue homogenates or body fluids) is mixed with an excess of PrPC (substrate for the amplification reaction). Samples are placed in an automatic sonicator that is programmed to perform cycles of incubation/sonication. If necessary, several rounds of PMCA can be carried out in order to enhance detection by simply diluting the sample into fresh substrate. PMCA is not a PrPSc detection technique but a preamplification step before detection. As such, PMCA is compatible with any procedure for detecting PrPSc. After the process is completed, the extent of prion replication can be determined by any procedure that is able to measure PrPSc biochemically (for example, western blotting, ELISA, infectivity bioassay and conformational-dependent immunoassay). Here we describe the use of western blot analysis after proteinase K (PK) treatment, as this is the most widely used methodology for PrPSc detection. In the following procedure, we describe the standard PMCA methodology for achieving seeded prion replication of PrPSc using complete brain extracts from homologous species (Fig. 2). PMCA may also be used to produce prions de novo without the addition of pregenerated PrPSc (ref. 30; Box 2). This permits mimicking the spontaneous generation of prions that feature the sporadic origin of most forms of human TSEs. In the Supplementary Methods, we describe the procedures for performing PMCA using purified components or recombinant PrPC, which is important for studying the mechanism of prion replication and the cellular factors implicated in this process. An example of the results obtained using this method is given in Supplementary Figure 1.
Figure 2.

PMCA flowchart. A conventional PMCA assay is usually composed of four main stages: substrate preparation, inoculum preparation, PMCA procedure and visualization of the extent of prion formation. After mixing PMCA substrate and inoculum, samples are submitted to incubation/sonication cycles that will allow an exponential generation of infectious prions. After the PMCA reaction is completed, samples are digested with PK and visualized by western blotting. Modifications to this technique have been used to replicate prions from different strains/species. PMCA has been adapted for many different applications, including PrPSc detection in body fluids and tissues and interspecies prion replication. PROCEDURE Steps are indicated in purple boxes with the step number(s) in parentheses. Critical steps are labeled in blue and Troubleshooting steps in green.
Box 2. De novo formation of infectious prions ● TIMING ~1,200 h (for ten rounds).
De novo formation of infectious prions by PMCA refers to the process by which PrPSc is produced after extensive PMCA cycling in a reaction started in the absence of PrPSc inocula24,30. In our experience, spontaneous appearance of PrPSc does not occur under standard PMCA conditions (not more than seven rounds of sPMCA)30. However, we and other groups have shown that after extensive PMCA cycling, de novo formation of PrPSc may be observed at a low frequency24,25,30,49; this may represent a process analogous to the spontaneous appearance of PrPSc in sporadic forms of disease. In our experience, the frequency of de novo PrPSc depends on the species being analyzed30. To increase the chances of de novo PrPSc formation, we have devised a protocol consisting of extended cycles between various rounds of PMCA.
PROCEDURE ● TIMING variable; for 10 rounds of PMCA, 1,200–1,250 h
-
Prepare NBH substrate as described in Steps 1–6 of the main PROCEDURE.
▲ CRITICAL STEP Whenever possible, all materials, reagents and equipment should be prion-free before starting the procedure. If not, ensure that all working areas are clean before starting this procedure. This can be done by extensively cleaning the surfaces with 70% (vol/vol) ethanol and then covering them with new adsorbent sheets. This applies to the working surface inside a biosafety hood as well as to normal laboratory spaces. All these materials should be discarded and replaced after use. If possible, clean the surfaces with 2 N NaOH after alcohol treatment to get rid of previous prion-derived material. Remember to properly rinse out NaOH from equipment and instruments. If possible, carry out the experiments in a prion-free laboratory. As contamination can be an important confounding issue for de novo prion formation, the measures described to avoid contamination must be strictly enforced (see Troubleshooting for Step 15 in the main PROCEDURE). We recommend performing all experimental procedures in a laminar flow biosafety cabinet to further minimize chances of contamination.
-
Fill at least ten 0.2-ml PCR tubes with 100 μl of 10% (wt/vol) hamster NBH substrate. Close the tubes carefully, avoiding spills (the use of clean forceps is recommended), and place them into the sonicator rack around the center.
? TROUBLESHOOTING
-
Place the rack on top of the horn, ensuring that the water in the horn covers the liquid content of the 0.2-ml PCR tubes.
▲ CRITICAL STEP If any part of the sonicator apparatus (including the horn and rack) has been used before, thoroughly wash it with 2 N NaOH to avoid cross-contamination with PrPSc from previous reactions. Ensure that the NaOH is removed by rinsing after decontaminating the surfaces to avoid alkali-based inactivation of prions.
-
Start the PMCA amplification by programming cycles of 29 min 40 s of incubation and 20 s of sonication. Perform the procedure at 37 °C.
▲ CRITICAL STEP The sonication level should reach an average potency of ~250 W over several cycles.
Leave the sample in the sonicator until 240 PMCA cycles have been performed.
Take the same number of new and clean 0.2-ml PCR tubes as used in Step 1. Fill each with 90 μl of 10% (wt/vol) NBH substrate. Add 10 μl of a reaction mixture from the previous PMCA round to one of the new and clean tubes. Repeat so that each reaction from the previous PMCA round has been diluted 1:10 into a new tube. Mix well.
Repeat Steps 4–6 above at least ten times.
-
For analysis, follow Steps 17–29 of the PROCEDURE.
? TROUBLESHOOTING
EXPECTED RESULTS
A low rate of de novo PrPSc formation can usually be observed after 7–10 rounds of extended PMCA, depending on the background PrP sequence (species)30. This material should produce a TSE-like pathology upon intracerebral inoculation in wild-type animals. In our experience, it is often observed that de novo PrPSc corresponds to novel prion strains, which can be determined by infectivity bioassay. It is currently unknown whether the spontaneous appearance of PrPSc units occurs through seeding from either minute amounts of infectious prions present in healthy animal brains or a stochastic induction of PrPC misfolding. For that reason, the more tubes tested, the better chances of getting spontaneous PrPSc-positive samples. For additional information regarding the de novo generation of prions, see ref. 30.
Various sources of materials can be used as inoculum to trigger PMCA, including tissue homogenates (brain, spleen, skeletal muscles, kidney and others), biological fluids (blood, urine, cerebrospinal fluid and others), secretory and excretory samples (saliva, feces and others), environmental materials (soil, grass, water) and solid surfaces (metal wires, wood, pieces of paper and others). For space constraints, the procedure describes the use of brain homogenate as inoculum, as it is the most common source of prions. Even when you are attempting to detect PrPSc in other samples or materials, we recommend using infected brain homogenate at various dilutions as a positive control for the PMCA reaction. The procedures for PMCA amplification using samples other than brain homogenate as inocula are similar, except that they often involve steps that aim to concentrate the sample or to remove components that can interfere with PMCA (such as blood or urine). We refer the reader to the following references for amplification protocols for other samples32–34,37,40–43.
MATERIALS
REAGENTS
Substrate. To perform the procedure given below, you will need 6- to 8-week-old wild-type animals or transgenic mice expressing the respective prion protein. The specific substrate for the procedure is a noninfected brain homogenate (NBH) from the same animal species of the prion- containing inoculum, prepared as described in Steps 1–6 of the PROCEDURE. The advantage of using transgenic mice over other large animals is that they can be made to express larger quantities of the protein, and tissue can be collected immediately after killing and after proper perfusion ! CAUTION All experiments involving live animals must conform to all relevant governmental and institutional ethics regulations.
Inoculum. To follow the procedure given below, you will need terminally ill prion-infected animals to provide the inoculum. The specific inoculum for the procedure given below is brain homogenates from terminally ill prion-infected animals, prepared as described in Steps 7–9 of the PROCEDURE ! CAUTION All experiments involving live animals must conform to all relevant governmental and institutional ethics regulations.
Phosphate-buffered saline (PBS, 10×) without calcium and magnesium (MP Biomedicals, cat. no. 1960454)
NaCl (Fisher Scientific, cat. no. S271)
PBS (1×) without calcium and magnesium (MP Biomedicals, cat. no. 1860454)
EDTA (0.5 M stock solution, pH 8.0; Promega, cat. no. V4231)
Triton X-100 (Sigma-Aldrich, cat. no. T8787)
Complete protease inhibitor cocktail (PI, Tablets; Roche Diagnostics, cat. no. 11 697 498 001)
Proteinase K (PK) (Sigma-Aldrich, cat. no. P2308)
Tris base (Fisher Scientific, cat. no. BP152)
Glycine (Fisher Bioreagents, cat. no. BP381)
LDS sample buffer (4×) NuPAGE (Invitrogen, cat. no. NP0007)
NuPAGE MES SDS running buffer (20×; Invitrogen, cat. no. NP0002)
NUPAGE Bis/Tris gels, 4–12% (wt/vol), 1.5 mm (Invitrogen, cat. no. NP0335BOX)
Nitrocellulose membrane, Hybond ECL, 0.45 μm, 30 cm × 3 m, version LRPNK/95/8 (GE Healthcare, cat. no. RPN303D)
Monoclonal antibody anti-PrP mAb 6D11 (Covance, cat. no. SIG-39810) or 3F4 (Covance, cat. no. SIG-39600)
ECL anti-mouse IgG, horseradish peroxidase–linked whole antibody (from sheep) (GE Healthcare, cat. no. NA931V)
Amersham ECL Plus western blotting dectection system (GE Healthcare, cat. no. RPN2132)
Sarkosyl (Fisher Bioreagents, cat. no. BP234)
Tween-20 (Acros, cat. no. 23336-0010)
Liquid nitrogen
Protein denaturing buffer (New England Biolabs, cat. no. B1704S)
NP-40 solution (10% (vol/vol); New England Biolabs, cat. no. B2704S)
PNGase F and G7 buffer (New England Biolabs, cat. no. P0704L)
Nanopure water
EQUIPMENT
Potter-Elvehjem tissue grinder (10 ml; Wheaton Science Products, cat. no. 08-414-14A)
Potter-Elvehjem tissue grinder (55 ml; Wheaton Science Products, cat. no. 08-414-16M)
Thermo Strip PCR tubes (0.2 ml; Thermo Scientific, cat. no. AB-0266) ▲ CRITICAL In our experience, not all 0.2-ml PCR tubes give an efficient PMCA amplification. We have observed that the thickness of the tube walls seems to have a considerable effect on the amplification yield. We strongly recommend using the Thermo Scientific tubes or testing empirically the efficiency of different tubes using an appropriate positive control.
Microcentrifuge tubes, 1.5 ml
Zirconia/silica beads (1 mm; BioSpec Products, cat. no. 11079110z) or Teflon beads (2.38 mm, McMaster-Carr or 0.75-1 mm, Roth)
Sample vial (2.5 ml, Fisher Scientific, cat. no. 03-338-1B)
Microcentrifuge tubes, 0.5-ml conical screw cap tube (Fisher Scientific, cat. no. 02-681-334)
Microcentrifuge tube caps, screw caps with O-rings (Fisher Scientific, cat. no. 02-681-368)
Syringe, 50 ml
Stainless steel surgical scissors
Stainless steel forceps
Misonix 4000 sonicator (Qsonica)
Allegra 25R centrifuge (Beckman-Coulter)
L8-70M ultracentrifuge (Beckman-Coulter)
70.1 Ti rotor (Beckman-Coulter)
Ultracentrifuge tubes (Beckman-Coulter, cat. no. 355630)
Falcon–type conical tubes, 50 and 15 ml
Freezer unit, − 20 °C
Freezer unit, − 80 °C
EI2 general purpose incubator (Sheldon Lab)
Thermomixer (Eppendorf)
Isotemp digital dry bath incubator (Fisher Scientific, cat. no. 11-718-2Q)
Tube thermal probe (Physitemp, Model BAT-12)
Trans-blot chamber (Bio-Rad, cat. no. 170-3946)
Bandelin Sonoplus sonicator (Bandelin Electronic)
EpiChemi Benchtop Darkroom (UVP)
REAGENT SETUP
Perfusion buffer (PB)
This buffer contains 5 mM EDTA prepared in 1× PBS. For a 2-liter preparation, mix 200 ml of 10× PBS, 20 ml of 0.5 M EDTA and 1,780 ml of nanopure water. PB can be stored at 4 °C for up to 6 months.
Conversion buffer (CB)
This buffer contains 150 mM NaCl and 1% (vol/vol) Triton X-100 prepared in PBS. For a 500-ml preparation, mix 4.4 g of NaCl, 5 ml of Triton X-100 and 485 ml of 1× PBS. Keep the CB stock at 4 °C and discard it after 6 months. Add one tablet of PI to 50 ml of conversion buffer immediately before preparing brain homogenates. A pH value between 7.0 and 7.3 should be obtained. For amplification of some prions (e.g., variant Creutzfeldt-Jakob Disease), addition of digitonin (at a final concentration of 0.05% (wt/vol)) to the conversion buffer may increase the efficiency of PMCA. PI Dissolve one tablet of PI in 50 ml of PBS. Freshly prepare and discard after use.
EQUIPMENT SETUP
Sonicator
Set the sonicator at the appropriate setting to reach a 250–260 W potency for the 263K hamster prion strain. For other species/strains, sonication power should be optimized experimentally, but in general it should be lower than the one used for the 263K strain. Sonication cycles should be set at 20 s, and incubation cycles should be set at 29 min 40 s (i.e., 29:40), respectively. Place the sonication horn inside a dry incubator set it at 37 °C. Fill the sonicator horn with 200 ml of distilled water. Tubes must not touch the sonicator surface. Ensure that the water level does not change, and replenish the water when evaporation occurs.
Tube racks
Commercially available tube racks used for Misonix 4000 sonicators should not be loaded to more than 60% of their capacity. Tubes close to the horn’s walls might show lower amplification efficiency compared with those placed in the center18. However, assuming the sonicator is working well and conditions are optimal, we have not seen marked experimental variability with respect to tube positioning on the sonicator plate17.
PROCEDURE
Substrate preparation ● TIMING 4 h
-
1|
By CO2 inhalation, euthanize 6- to 8-week-old wild-type animals or transgenic mice expressing the prion protein of interest.
! CAUTION You should have all proper permits and animal protocols approved before starting the experiments. Check with your local animal welfare committee for appropriate guidelines.
-
2|
Perfuse the animal with PB by using cardiac puncture (60 ml for hamster and 20 ml for mouse).
▲ CRITICAL STEP Some blood components can negatively affect the prion conversion process32. After perfusion, a blood-‘clean’ brain should be obtained, as shown in Supplementary Figure 2.
▲ CRITICAL STEP In the case of larger animals such as cattle, sheep, goats, cervids and others, wherein perfusion before tissue dissection is difficult or impossible, we recommend removing the entire brain as quickly as possible in order to reduce postmortem degradation. Subsequently, wash it immediately with cold (4 °C) 5 mM EDTA prepared in PBS in order to remove excess blood. Alternatively, we recommend using brains from transgenic mice overexpressing PrPC of the desired species. On the basis of in vivo experiments, we recommend using young adult animal brains as substrate.
? TROUBLESHOOTING
-
3|
Remove the brain and quickly rinse in PB. The material can be used immediately or frozen in liquid nitrogen and stored at − 80 °C until use.
■PAUSE POINT Store the brain at − 80 °C until use. Material can be stored indefinitely; when needed, allow it to defrost at room temperature (~22 °C).
-
4|
Homogenize the brain in CB at 10% (wt/vol) using a Potter homogenizer on ice. The highest in vitro PrPC → PrPSc conversion is obtained using 7.5–10% (wt/vol) NBH substrate; however, PMCA also works with a 2.5–5% NBH substrate, although it results in a lower (proportional) rate of amplification.
▲ CRITICAL STEP The homogenization process must be done on ice in order to avoid PrPC degradation. The final substrate preparation should be turbid, with small visible membrane fragments still present.
-
5|
After homogenization, tissue debris should be removed by centrifuging the brain homogenate at 805g at 4 °C for 45 s using an Allegra 25R centrifuge (or an equivalent).
▲ CRITICAL STEP This centrifugation step is only done to remove large debris. Care has to be taken to avoid using a higher speed or longer centrifugation than recommended, as this could reduce the amount of PrPC and/or conversion factor required for optimal amplification.
-
6|
Remove the supernatant and discard the pellet. Place the supernatant on ice. Mix it well by vortexing and aliquot the supernatant (NBH) into 1.5-ml RNAse-free microcentrifuge tubes. The material can be used immediately or frozen in liquid nitrogen and stored at − 80 °C until use.
▲ CRITICAL STEP Aliquots should be prepared quickly in order to avoid degradation. We strongly recommend thawing NBH substrate aliquots only once. Freeze-thaw cycles should be avoided, because they markedly decrease the amplification efficiency. Storing PMCA substrates at –20 °C clearly decreases the quality of the substrate.
■ PAUSE POINT Homogenates can be stored indefinitely at − 80 °C until use.
Inoculum preparation ● TIMING 2 h
-
7|
Euthanize a terminally ill, prion-infected animal by CO2 inhalation.
! CAUTION Depending on the material under study, this procedure should be performed in a facility with an adequate biosafety level. Follow appropriate biosafety laboratory protocols; check with your local biosafety officer for appropriate guidelines.
-
8|
Remove the brain and homogenize it at 10% (wt/vol) in PBS containing PI.
-
9|
Centrifuge the brain homogenate at 805g at 4 °C for 45 s. Discard the pellet. For long-term storage, PrPSc-containing preparations can be stored either at − 80 or − 20 °C.
▲ CRITICAL STEP Because most PrPSc corresponds to large aggregates that are bound to membrane fractions, centrifugation at higher speeds should be avoided in order to reduce the risk of losing infectious material in the pellet.
▲ CRITICAL STEP Some of the PrPSc concentration procedures (e.g., phosphotungstic acid precipitation or immuno-precipitation) decrease substantially the efficiency of PMCA amplification, and thus are not recommended.
■PAUSE POINT Infected brain homogenate can be stored indefinitely either at − 80 or − 20 °C.
pMca procedure ● TIMING variable; ~75 h per round of standard cycles
-
10|
Add three Teflon, four silica or five glass beads into 0.2-ml PCR tubes. PMCA also works without the addition of beads, but as described by the Baskakov, Castilla and Beekes groups, beads improve the efficiency of amplification and reduce intersample variability18–20.
▲ CRITICAL STEP In our experience, not all 0.2-ml PCR tubes give an efficient PMCA amplification. See MATERIALS for further details.
? TROUBLESHOOTING
-
11|
Mix PrPSc-containing brain homogenate (inocula) with NBH substrate into the PCR tubes. If you are using inoculum and substrate from the same species, the starting dilution of each inoculum over NBH should be not less than 100-fold (i.e., for the first dilution (100-fold), add 1 μl of inoculum into 99 μl of substrate). However, if inoculum and substrate are from different species, a smaller dilution is appropriate; we recommend a 1:2 dilution as the starting point. Use at least triplicates for each sample to evaluate the reproducibility of amplification. The final reaction volume should be between 80 and 100 μl; however, when making up dilutions, consider that you may need to take some volume of the mixture for nonamplified controls (see Step 12) and/or to make up the serial solutions (see Step 13).
-
12|
Take an aliquot from the sample before starting PMCA and freeze it for later use as a nonamplification control (frozen). For example, for a tenfold-diluted inoculum and a final volume of 100 μl, add 12 μl of inoculum into 108 μl of 10% (wt/vol) substrate. Mix well by pipetting. Take 20 μl of the mixture, freeze it and keep it as a nonamplification control. Submit the remaining sample to PMCA.
-
13|
If desired, make serial tenfold dilutions of the inoculum-NBH substrate mixture in NBH substrate in order to assess the amplification rate by comparing it with the signal of nonamplified samples (Fig. 3). We recommend testing at least eight tenfold dilutions of the sample. The use of clean forceps is recommended when manipulating the tubes.
-
14|
Place tubes in a rack and then into the clean water-filled sonicator horn (which is inside a dry incubator set at 37 °C). Start sonication/incubation cycles (PMCA procedure). Each cycle should consist of 29 min 40 s of incubation, followed by a 20-s sonication. Repeat the cycles for 24–72 h, depending on the extent of amplification required.
▲ CRITICAL STEP The final volume in each tube should be between 80 and 120 μl.
▲ CRITICAL STEP We strongly recommend spacing the tubes in the rack. Tubes should be placed either as single tubes or as pairs.
▲ CRITICAL STEP The settings of the sonicator should be specifically modified depending on the prion strains/species used. We recommend using the optimal settings for amplification of hamster 263K prions (250–260 W potency) as a starting point.
▲ CRITICAL STEP Among sonication cycles, the sonicator horn releases sediments that could affect PrPSc amplification (Supplementary Fig. 3). For that reason, the water in the sonication horn should be periodically changed (once every PMCA round is recommended).
▲ CRITICAL STEP Whenever possible, we recommend using a thermal probe to check the temperature inside the tube during sonication. The temperature should be between 37 and 45 °C (but not higher than 50 °C).
? TROUBLESHOOTING
-
15|
After PMCA completion, either freeze the sample for later analysis of PrPSc content (Step 17) or proceed with the use of the PMCA product obtained as inoculum in a new PMCA round (i.e., proceed with Step 16). Several consecutive rounds of PMCA are necessary when very high levels of amplification are required, such as for the detection of very small quantities of PrPSc (for example, for prion detection in blood)17,32.
? TROUBLE SHOOTING
Figure 3.

Schematic representation of the serial PMCA (sPMCA) assay. In a typical sPMCA assay, the resulting product of a PMCA round is used as an inoculum in a new PMCA reaction (usually by diluting the PMCA product tenfold in fresh brain homogenate substrate). (a,b) By using this procedure, it is possible to increase the detection levels in order to detect up to a single infectious prion. In the left side of the figure, schematic representations of the dilution setup is shown both for the experimental samples (a) and negative controls (b). (a) Western blots show how the detection of PrP27–30 increases among PMCA rounds (right), detecting the equivalent of a 10−10 263K brain dilution in a second PMCA round. Numbers on top of each lane correspond to the dilution of the brain homogenate used to trigger amplification. Gaps in PrP27–30 detection as shown for the second PMCA round (10−9 dilution) are infrequently seen and are usually normalized in the following PMCA rounds. (b) Negative controls consisting only of NBH and submitted to the same procedures do not show any PrPSc signal if all proper precautions are followed (for details, please refer to the TROUBLESHOOTING section and references). In this panel, the numbers on top of the gel refer to multiple replicates. Results showed in this figure are obtained from PMCA assays performed as explained in the protocol using four silica beads per tube. MW, molecular weight ladder.
Serial PMCA (sPMCA) ● TIMING variable; ~73 h per round of standard cycles
-
16|
Dilute PMCA products tenfold into fresh NBH substrate and proceed with a new round of PMCA by repeating Steps 10–14 (Fig. 3a). The rounds of sPMCA can be repeated as many times as needed to reach the required detection threshold. However, we do not recommend performing more than seven rounds of sPMCA, as this increases the chance of de novo prion formation24,30.
? TROUBLESHOOTING
■PAUSE POINT Samples after amplification can be stored frozen, ideally for not longer than 1 month, at − 20 °C until PrPSc detection as described below.
Visualizing samples by western blotting ● TIMING 10 h
-
17|
Take frozen control and amplified samples and treat them with PK. The PK concentration and the protease incubation conditions depend on the prion strain/species used. For 263K prions, the final PK concentration recommended is 50 μg ml −1. Place the samples in an Eppendorf thermomixer at 37 °C for 1 h with 450 r.p.m. agitation.
▲ CRITICAL STEP As different prion strains show different extents of resistance to proteolytic degradation44, the optimal PK treatment condition should be determined in advance. It is crucial to ensure that no PrPC remains undigested after PK treatment, because this can affect the quantification of the PrPSc product. One of the advantages of western blot analysis is that it is easy to differentiate incomplete PrPC digestion from the signal corresponding to the protease-resistant core of PrPSc, as the latter shows a faster electrophoretic mobility owing to the removal of ~90 residues from the N-terminal part of the protein (Fig. 4).
? TROUBLESHOOTING
-
18|
Stop the PK digestion by adding LDS-PAGE loading buffer at a final ratio of 1:2.
-
19|
Heat the samples at 95–100 °C for 10 min. Samples can be frozen at this stage for later analysis.
■PAUSE POINT Freeze the samples at − 20 °C for later analysis.
-
20|
Load the same volume of the samples (typically 20 μl) into 4–12% (wt/vol) NuPAGE Bis-Tris gels.
▲ CRITICAL STEP We recommend loading PrPC (non-PK-digested brain homogenate from healthy animals of the same species) as an electrophoretic mobility control (Figs. 3 and 4).
-
21|
Run the samples for 20 min at 70 V and 1 h and 40 min at 135 V.
-
22|
Transfer onto Hybond ECL membranes for 1 h at 800 mA at room temperature using a wet Trans-blot chamber from Bio-Rad.
-
23|
Block the membranes using 5% (wt/vol) dry nonfat milk in 0.05% (vol/vol) Tween-20 (prepared in PBS) for 1 h at room temperature with shaking.
-
24|
Incubate the membranes with gentle shaking using an appropriate PrP-specific antibody diluted in 0.05% (vol/vol) Tween-20 (prepared in PBS) for 1 h at room temperature. For example, for hamster and human PrP detection, we use the monoclonal 3F4 antibody at a dilution of 1:5,000 and for mouse PrP we use the monoclonal 6D11 antibody at a 1:5,000 dilution.
-
25|
Wash the membranes using 0.05% (vol/vol) Tween-20 (prepared in PBS; 1 × 10 min and 2 × 5 min).
-
26|
Incubate the membranes with gentle shaking using horseradish peroxidase–linked polyclonal antibody mouse IgG (diluted 3,000 times in 0.05% (vol/vol) Tween-20 (prepared in PBS)) for 1 h at room temperature.
-
27|
Wash the membranes using 0.05% (vol/vol) Tween-20 (prepared in PBS; 1 × 10 min and 2 × 5 min).
-
28|
Develop the membranes with the ECL system and EpiChemi Darkroom as recommended by the manufacturer.
? TROUBLESHOOTING
-
29|
To calculate the amplification rate by comparing the dilutions of the nonamplified and amplified materials with similar intensities, estimate the signal intensity by using an image analysis program.
Figure 4.
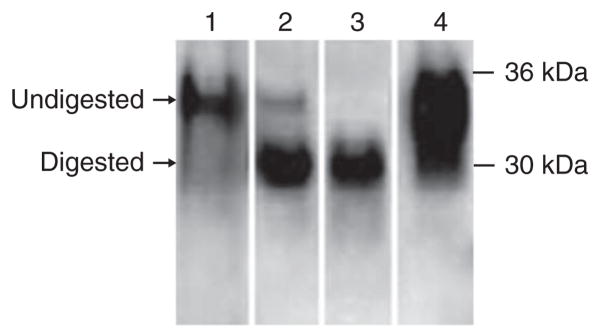
Distinguishing complete and incomplete proteolytic digestion of PrPSc. A frequent problem associated with the detection of PrPSc after PMCA amplification is ensuring that the product being detected is bona fide PrPSc and not some kind of amorphous aggregate that prevents access to the protease used to eliminate unconverted PrPC. Fortunately, gel electrophoresis and western blotting enable these possibilities to be discriminated. Incomplete digestion (lane 1) leads to a signal with the molecular weight similar to PrPC not treated with PK (lane 4), whereas the typical signal for PrP27–30 has a clearly lower molecular weight (around 29 kDa; lane 3). Lanes 1–3 show the results of experiments containing a similar amount of hamster PrPSc, in which PK digestion did not digest (lane 1), digested partially (lane 2) and digested completely (lane 3) the N-terminal fragment of the protein. To depict the migration of full-length PrPC, lane 4 corresponds to a hamster NBH not treated with PK.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Solution |
|---|---|---|
| 2 | Brain is not well perfused | It is very important that the brain used as substrate is as free of blood as possible. This can be monitored easily by the color of the tissue (see Supplementary Fig. 2 to compare a well-perfused and a poorly perfused brain). To achieve good perfusion, it is important to apply high pressure to obtain a properly perfused tissue. Buffer released from the nose and mouth during the process is a good indicator of a high-quality brain perfusion |
| 10 and Box 2 | Tubes are cracked or open after PMCA | We strongly recommend using the tubes described here. When other types of tubes are used, they can break or open during PMCA cycling. If this happens during an experiment, discard the entire experiment and thoroughly clean the sonicator to prevent cross-contamination |
| 14 and Box 2 | Some portion of the sample gets trapped in the tube cap | Owing to temperature and sonication, part of the sample may get stuck in the tube cap. In addition, some evaporation may occur. It is important to check at least once a day the status of the samples in order to make sure that temperature/sonication strength are adequate and that the sample is actually at the bottom of the tube. Shaking the rack carefully to homogenate the samples is recommended. Do not spin the samples as potential PrPSc seeds may decrease with subsequent loss in amplification efficiency. This problem may be more frequent when generating spontaneous prions (Box 2) because of the large number of rounds of PMCA performed |
| 14 | The performance of the sonicator decreases over time | The sonicator that is currently being used for PMCA was not designed for continuous use. It is not an uncommon problem to observe that after a period of heavy use, the ultrasound output decreases. We suggest constantly monitoring the performance of the sonicator by testing the amplification efficiency with a standard sample (substrate and inoculum) for which the amplification rate is known. The power to reach optimal amplification should be adjusted accordingly to recover the amplification efficiency. If the decrease in sonication efficiency is too large, the sonicator can be repaired (contact the company) or replaced |
| 15, 16 and Box 1 | PrPSc amplification in negative control samples | There are two main reasons for signal in negative controls: cross-contamination or de novo formation of prions. Under the conditions described in this protocol, no de novo generation of PrPSc should be observed in less than seven rounds of sPMCA. So, the most likely interpretation of false positives is contamination. The extremely high-amplification power of PMCA, and especially of sPMCA, makes very important to take many precautions to avoid contamination. We recommend the following measures to minimize cross-contamination: All infected materials and reagents should be carefully handled. Make sure to work on a prion-free surface (i.e., by using new disposable pads between experiments) Never use equipment or devices (e.g., sonicators, homogeneizers, glassware) used with prion-infected materials to prepare PMCA substrates of noninfected samples. As much as possible, have dedicated devices for each set of samples As recommended by a recent article from Cosseddu et al.48, using 0.5-ml screw-cap Multiply Safecup tubes may help to decrease cross-contamination Work under conditions used to manipulate sterile material (double gloves, biosafety cabinet, change filter tips everytime, avoid spills, keep neatly clean bench, clean frequently bench, pipettes and other materials with 2 N NaOH) After each round of PMCA, the gently spin down samples before opening the tube to remove material present in the lid, which arises during sonication or from condensation of the liquid. Avoid opening tubes with your hands, as PrPSc from one tube can be spread by gloves. Use NaOH-disinfected forceps instead Change the water and clean the sonicator cup with 2N NaOH after each experiment Do not mix prions from different sources in one sonicator If you are attempting to detect PrPSc with very high sensitivity (e.g., in blood samples) do not simultaneously manipulate samples with large quantities of infectious material. When serial PMCA rounds are performed, always manipulate tubes containing or suspected to contain higher amounts of PrPSc at the end Always include a large number of controls to assess false positives in each experiment. For this purpose, tubes containing only NBH substrate should be run simultaneously |
| 17 | Incomplete digestion after PK treatment | It is not uncommon to observe incomplete digestion with PK when the efficiency of amplification is low. In the absence of a good amplification, PrPC gets aggregated into amorphous structures resistant to PK. The efficiency of PK digestion can be improved by increasing the protease concentration, raising the temperature (up to 45 °C) during PK treatment or adding 0.1% (wt/vol) SDS to the PK digestion buffer. These aggregates are easily differentiated from PrPSc by their electrophoretical migration and their inconsistent signal in subsequent PMCA rounds |
| 28 | No, little or highly variable amplification | There could be many reasons for low or variable amplification efficiency. To start implementing PMCA, we recommend working with the 263K prion strain, using healthy Syrian golden hamster brain homogenate as substrate, as in our experience this is the material in which it is easiest to obtain a high rate of PrPSc amplification. Once the amplification rate is adequate for this prion strain, then the technology can be adapted for other species/strains. The main variables to check when improving the PMCA efficiency and reproducibility are as follows: the quality of the normal brain homogenate used as substrate; the accurate preparation of the conversion buffer (changes in salt and/or detergent concentration can substantially modify the rate of PrPSc conversion); the quantity of PrPSc present in the inoculum (brain homogenate from infected animals can carry significantly different amounts of PrPSc); the position of the tubes in the sonicator’s horn; and the correct performance of the sonicator |
● TIMING
Steps 1–6, substrate preparation: 4 h Steps 7–9, inoculum preparation: 2 h
Steps 10–15, PMCA procedure: variable; ~75 h for one round of standard 144 PMCA cycles
Step 16, serial PMCA (sPMCA): variable; ~73 h for each additional round of standard 144 PMCA cycles Steps 17–29, visualizing samples by western blotting: 10 h
Box 1, quantitative PMCA: ~323 h for four rounds of qPMCA
Box 2, de novo formation of infectious prions: 1,200–1,250 h for ten rounds of PMCA
ANTICIPATED RESULTS
As shown in Figure 3a, the presence of PrP27-30 can be assessed by western blotting. In addition to its PK resistance, PrP27-30 will be distinguished from PrPC by its different electrophoretical mobility (Fig. 4). A good PMCA procedure should be able to detect 263K brain dilutions of 10 −6 and 10 −9, representing amplification rates of around 1,000- and 1,000,000-fold in a first and second PMCA round, respectively32. Similar levels of detection have been described for the amplification of prions from other species, including mouse and cervid21,45–47. Although infrequent, and owing to the nature of a tissue homogenate (heterogeneous distribution of aggregates), gaps in PrP27–30 detection are sometimes observed between different dilutions of the inoculum among PMCA rounds (for example, see the second PMCA round in Fig. 3a). For that reason, we strongly recommend running at least triplicates of each PMCA reaction in order to assess this type of variability. These gaps are usually normalized in the subsequent PMCA round. Negative controls should not show any PrP signal, as shown in Figure 3b.
Acknowledgments
We thank the many former laboratory members whose input has been important for reaching the optimal PMCA procedure, especially G. Saborio, L. Anderes, C. Adessi, K. Maundrell, J. Castilla, P. Saa, K. Abid, M. Barria, D. Gonzalez-Romero and B. Chen. We also thank M.-J. Liberona for critical reading of the manuscript. This work was partially supported by US National Institutes of Health grants R01 NS049173, P01 AI077774 and P01 AG014359 to C.S.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS: R.M. designed the experiments, carried out the work, analyzed the results and wrote part of the manuscript. C.D.-A. performed various PMCA assays, wrote part of the manuscript and drafted the figures. M.V.C. performed various PMCA optimizations and wrote part of the manuscript. R.D.-E. performed experiments with recombinant prion protein and wrote part of the manuscript, and C.S. is the principal investigator on the project and was responsible for coordinating research activity, funding and producing the final version of the article.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 3.Collee JG, Bradley R. BSE: a decade on—Part I. Lancet. 1997;349:636–641. doi: 10.1016/S0140-6736(96)01310-4. [DOI] [PubMed] [Google Scholar]
- 4.Collee JG, Bradley R. BSE: a decade on—Part 2. Lancet. 1997;349:715–721. doi: 10.1016/S0140-6736(96)08496-6. [DOI] [PubMed] [Google Scholar]
- 5.Llewelyn CA, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 6.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–529. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 7.Wroe SJ, et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet. 2006;368:2061–2067. doi: 10.1016/S0140-6736(06)69835-8. [DOI] [PubMed] [Google Scholar]
- 8.Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci. 2011;36:151–158. doi: 10.1016/j.tibs.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soto C, Satani N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol Med. 2011;36:151–158. doi: 10.1016/j.molmed.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 11.Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002;25:390–394. doi: 10.1016/s0166-2236(02)02195-1. [DOI] [PubMed] [Google Scholar]
- 12.Caughey B, Kocisko DA, Raymond GJ, Lansbury PT., Jr Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem Biol. 1995;2:807–817. doi: 10.1016/1074-5521(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 13.Ghetti B, et al. Prion protein amyloidosis. Brain Pathol. 1996;6:127–145. doi: 10.1111/j.1750-3639.1996.tb00796.x. [DOI] [PubMed] [Google Scholar]
- 14.Jarrett JT, Lansbury PT., Jr Seeding ‘one-dimensional crystallization’ of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 15.Soto C, Estrada L, Castilla J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci. 2006;31:150–155. doi: 10.1016/j.tibs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem. 2006;281:35245–35252. doi: 10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Montalban N, et al. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 2011;7:e1001277. doi: 10.1371/journal.ppat.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Borges N, Castilla J. PMCA. A decade of in vitro prion replication. Curr Chem Biol. 2010;4:200–207. [Google Scholar]
- 20.Pritzkow S, et al. Quantitative detection and biological propagation of scrapie seeding activity in vitro facilitate use of prions as model pathogens for disinfection. PLoS ONE. 2011;6:e20384. doi: 10.1371/journal.pone.0020384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castilla J, et al. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green KM, et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008;4:e1000139. doi: 10.1371/journal.ppat.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyerett C, et al. In vitro strain adaptation of CWD prions by serial protein misfolding cyclic amplification. Virology. 2008;382:267–276. doi: 10.1016/j.virol.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abid K, Morales R, Soto C. Cellular factors implicated in prion replication. FEBS Lett. 2010;584:2409–2414. doi: 10.1016/j.febslet.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Supattapone S. Biochemistry. What makes a prion infectious? Science. 2010;327:1091–1092. doi: 10.1126/science.1187790. [DOI] [PubMed] [Google Scholar]
- 28.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry. 2010;49:3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castilla J, et al. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134:757–768. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 2009;5:e1000421. doi: 10.1371/journal.ppat.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soto C. Diagnosing prion diseases: needs, challenges and hopes. Nat Rev Microbiol. 2004;2:809–819. doi: 10.1038/nrmicro1003. [DOI] [PubMed] [Google Scholar]
- 32.Castilla J, Saa P, Soto C. Detection of prions in blood. Nat Med. 2005;11:982–985. doi: 10.1038/nm1286. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Romero D, Barria MA, Leon P, Morales R, Soto C. Detection of infectious prions in urine. FEBS Lett. 2008;582:3161–3166. doi: 10.1016/j.febslet.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murayama Y, et al. Urinary excretion and blood level of prions in scrapie-infected hamsters. J Gen Virol. 2007;88:2890–2898. doi: 10.1099/vir.0.82786-0. [DOI] [PubMed] [Google Scholar]
- 35.Tattum MH, Jones S, Pal S, Collinge J, Jackson GS. Discrimination between prion-infected and normal blood samples by protein misfolding cyclic amplification. Transfusion. 2010;50:996–1002. doi: 10.1111/j.1537-2995.2010.02595.x. [DOI] [PubMed] [Google Scholar]
- 36.Saa P, Castilla J, Soto C. Presymptomatic detection of prions in blood. Science. 2006;313:92–94. doi: 10.1126/science.1129051. [DOI] [PubMed] [Google Scholar]
- 37.Chen B, Morales R, Barria MA, Soto C. Estimating prion concentration in fluids and tissues by quantitative PMCA. Nat Methods. 2010;7:519–520. doi: 10.1038/nmeth.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barret A, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77:8462–8469. doi: 10.1128/JVI.77.15.8462-8469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morales R, et al. Reduction of prion infectivity in packed red blood cells. Biochem Biophys Res Commun. 2008;377:373–378. doi: 10.1016/j.bbrc.2008.09.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE. 2009;4:e4848. doi: 10.1371/journal.pone.0004848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haley NJ, Mathiason CK, Zabel MD, Telling GC, Hoover EA. Detection of sub-clinical CWD infection in conventional test-negative deer long after oral exposure to urine and feces from CWD+ deer. PLoS ONE. 2009;4:e7990. doi: 10.1371/journal.pone.0007990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nichols TA, et al. Detection of protease-resistant cervid prion protein in water from a CWD-endemic area. Prion. 2009;3:171–183. doi: 10.4161/pri.3.3.9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagaoka K, et al. Sensitive detection of scrapie prion protein in soil. Biochem Biophys Res Commun. 2010;397:626–630. doi: 10.1016/j.bbrc.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 44.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murayama Y, et al. Efficient in vitro amplification of a mouse-adapted scrapie prion protein. Neurosci Lett. 2006;413:270–273. doi: 10.1016/j.neulet.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 46.Barria MA, Telling GC, Gambetti P, Mastrianni JA, Soto C. Generation of a new form of human PrPSc in vitro by interspecies transmission from cervid prions. J Biol Chem. 2011;286:7490–7495. doi: 10.1074/jbc.M110.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurt TD, et al. Efficient in vitro amplification of chronic wasting disease PrPRES. J Virol. 2007;81:9605–9608. doi: 10.1128/JVI.00635-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cosseddu GM, et al. Ultraefficient PrPSc amplification highlights potentialities and pitfalls of the PMCA technology. PLoS Pathog. 2011;7:e1002370. doi: 10.1371/journal.ppat.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorne L, Terry LA. In vitro amplification of PrPSc derived from the brain and blood of sheep infected with scrapie. J Gen Virol. 2008;89:3177–3184. doi: 10.1099/vir.0.2008/004226-0. [DOI] [PubMed] [Google Scholar]