

Abstract
A critical overview of the catalytic joining of two different electrophiles, cross-electrophile coupling (XEC), is presented with an emphasis on the central challenge of cross-selectivity. Recent synthetic advances and mechanistic studies have shed light on four possible methods for overcoming this challenge: (1) employing an excess of one reagent; (2) electronic differentiation of starting materials; (3) catalyst–substrate steric matching; and (4) radical chain processes. Each method is described using examples from the recent literature.
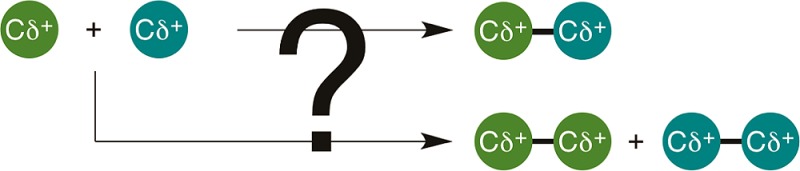
Driven by an aspiration to avoid the challenges of preformed carbon nucleophiles, catalytic methods to selectively cross-couple two different carbon electrophiles, referred to herein as cross-electrophile coupling (XEC), have recently seen rapid development. The central challenge facing XEC methods is selectivity (Figure 1). The two electrophilic starting materials are chemically similar; therefore, both tend to react with a transition-metal catalyst through oxidative addition. This is in contrast to conventional cross-coupling reactions where selectivity is largely engendered by the different reactivity of nucleophiles and electrophiles: nucleophiles react with the catalyst by transmetalation, and electrophiles react by oxidative addition. The purpose of this Synopsis is to introduce the variety of approaches known for the selective joining of two different electrophiles (R1-X and R2-X, Figure 1) using a transition-metal catalyst. The advancement of XEC as a field hinges upon understanding why such reactions are cross-selective. First, a strategy for obtaining high yields without selectivity will be discussed, followed by a discussion of three selectivity models: electronic differentiation of starting materials, catalyst–substrate steric matching, and radical-chain processes.
Figure 1.
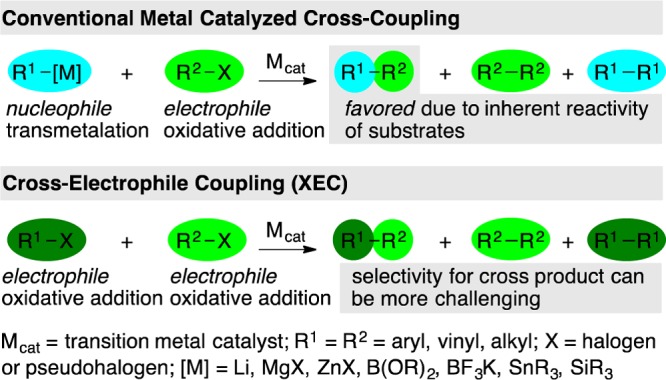
Comparisons of the selectivity challenges of cross-coupling and XEC.
The impetus for developing XEC methods arises from the challenges presented by nucleophilic reagents.1 For example, the most widely used nucleophilic carbon reagents, organoboron compounds, have limited commercial availability compared to halocarbon electrophiles. Additionally, some classes of organoboron reagents are unstable and require special procedures for their use, which ultimately adds steps and time to syntheses.2 Many organometallic reagents or their precursors require special care to exclude water and dioxygen. Similarly, the inherent reactivity of the reagents (RMgX and RZnX) or additives required to facilitate transmetalation (RB(OR′)2 and RSiR′3) place limitations on the use of substrates that have electrophilic functional groups or have acidic protons. Three strategies have been used to alleviate these limitations: (1) use of protected carbon nucleophiles to add stability to these reagents (Figure 2A);3 (2) one-pot, two-step procedures (Figure 2B);4 or (3) selective in situ organozinc or Grignard reagent synthesis from one organic halide concurrent with cross-coupling to a different organic halide (Figure 2C).4a,5 This last approach uses the same substrates—two electrophiles—as XEC but is mechanistically distinct because the reducing agent acts directly upon the substrate rather than the transition-metal catalyst.
Figure 2.
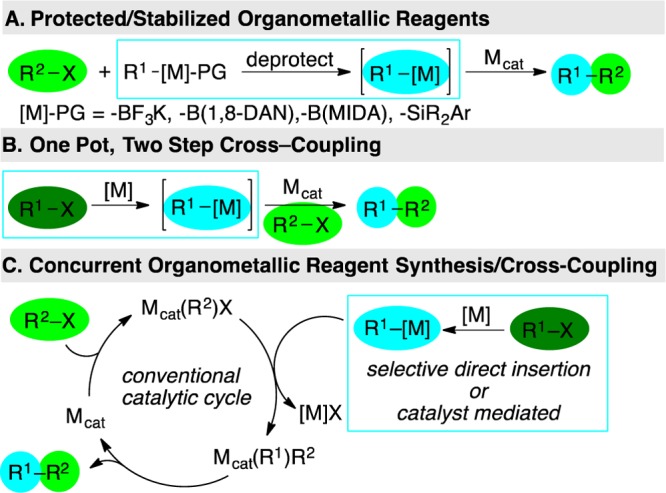
Transition-metal-catalyzed cross-coupling strategies that alleviate limitations imposed by nucleophilic coupling partners.
The dimerization of electrophiles has been known for over 100 years,6−9 yet selective cross-coupling of two different electrophiles has only recently been demonstrated with generality, highlighting the difficulty of suppressing symmetric dimer byproducts. Efforts toward overcoming this challenge began soon after the dimerization reactions were discovered. The Wurtz–Fittig reaction couples alkyl halides with aryl halides through the action of sodium metal and is cross-selective in some cases.10 Major advancements since the 1960s in XEC have come from transition-metal catalysis, wherein most have been electrochemical work.11 Recent efforts have resulted in the development of new cross-selective methods that use familiar chemical reducing agents. A particular advantage of XEC reactions is excellent functional-group compatibility (e.g., acidic protons, protected and unprotected amines and alcohols, carbonyls, high-valent sulfur, and β-leaving groups, but not easily reduced groups like nitro or azido). The development of a fundamental understanding of how and why XEC reactions are cross-selective is critical to advancing the field; thus, this Synopsis will focus on the different strategies currently used to achieve high yields of product (Figure 3).
Figure 3.
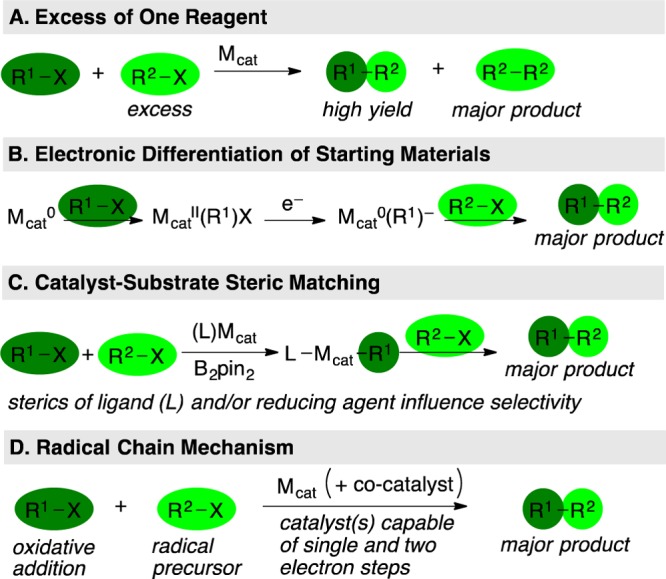
XEC strategies that will be discussed in this Synopsis.
Strategies for Achieving Useful Yields of Cross-Coupled Product in XEC
Equal Substrate Reactivity: Employ an Excess of One Reagent
In cases where the starting materials have nearly identical chemical reactivity, developing selective reactions represents a remarkable challenge. Similar reactivity can be used to an advantage: an excess of one reagent can deliver synthetically useful yields of cross-coupled product without any inherent selectivity. For a fully reversible reaction, the maximum yield obtainable can be high (66% for 2:1 ratio, 75% for 3:1 ratio of reactants).12 For an irreversible reaction, selectivity is higher at first (80% for 2:1, 86% for 3:1) but decreases as the ratio of remaining starting materials changes during the reaction.13 This strategy can be useful if one electrophile is low-cost or the symmetric dimers can be easily separated, as demonstrated in a recent synthesis of flurbiprofen.14
A classic example of this challenge is the cross-Ullmann coupling of two different aryl halides to form nonsymmetrical biaryls (Figure 4A). Reports with cobalt,15 nickel,16 and palladium17 have been published over the years. In each case, using one starting material in excess resulted in higher yields of cross-coupled product at the expense of larger amounts of symmetric dimer. All possible products were not reported, but the symmetric dimer of the aryl halide used in excess was the major product of these reactions, as expected. Better selectivity and higher yields of the cross-coupled product could be obtained when the substrates were better differentiated, such as the coupling of 2-halopyridines with aryl halides, suggesting a different selectivity mechanism is operative (Figure 4A).18
Figure 4.
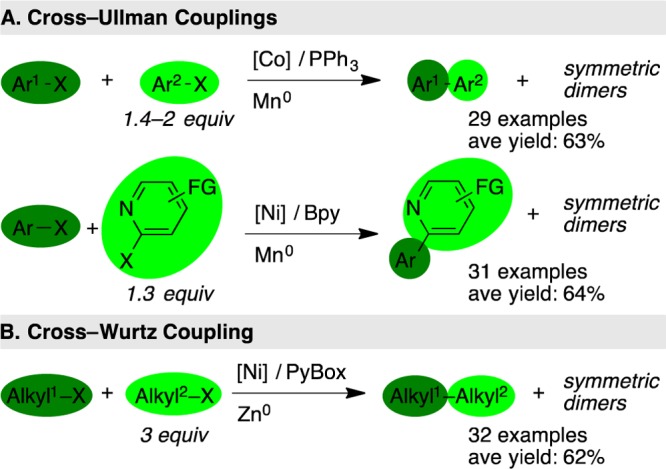
High yields can be obtained with electrophiles of nearly equal reactivity by using an excess of one coupling partner at the expense of large amounts of symmetric dimer byproducts. PyBox = pyridyl bis(oxazoline).
More recently, Gong published a nickel-catalyzed cross-Wurtz coupling of two different alkyl halides (Figure 4B).19 As noted previously for the dimerization of alkyl halides,20 the use of tridentate ligands was found to avoid hydrodehalogenation and β-hydride elimination, two major challenges in the cross-coupling of alkyl halides. Cross-selectivity proved to be more challenging, and the initial report used a 3-fold excess of one alkyl halide to achieve high yields of cross-coupled product. Studies on the byproducts formed and the time course of the reaction revealed that dimerization of the more reactive alkyl halide was usually the major side reaction and this uneven consumption of starting materials was observed early in the reaction. Selectivity appeared to depend upon subtle differences in the reactivity of the substrates. Furthermore, the role of the excess alkyl halide was also complex. For example, 1-bromo-3-butene provided higher yields and selectivities than other primary halides (up to 92% yield and 0.6:1 ratio of cross product:symmetric dimers). The use of only a 2-fold excess of 1-bromo-3-butene was reported to diminish yields by only ∼5% (selectivity not reported), but the use of a single equivalent was reported to give only 30% yield (selectivity not reported). These results suggest that a more general differentiation of the two electrophiles would provide higher yields.
Sequential Oxidative Addition: Electronic Differentiation
Different states of a catalyst (i.e., oxidation states and associated ligands) can differentiate two unlike electrophiles through sequential oxidative additions. In order for this mechanistic model to selectively produce cross-coupled products, the two different active, low-valent catalytic species must each selectively react with one of the starting materials. Key, early electrochemical21 and chemical studies22 had shown that an oxidative addition–reduction–oxidative addition–reductive elimination mechanism was operative in some catalytic Ullmann dimerization reactions. Later, this type of mechanism was proposed by Amatore, Jutand, and Périchon to explain the cross-selective coupling of benzyl bromide (1) with phenylacetyl chloride (2) (Figure 5).23 Selectivity for ketone 3 over bibenzyl (4) or diketone 5 is proposed to arise from the fast oxidative addition of BnBr (1) to Ni(0)Bpy (7) and the selective reaction of anionic complex (Bpy)Ni(0)(Bn)− (9) with phenylacetyl chloride (2) (Figure 5A).24 The anionic nickel complex may be stabilized by the ability of Bpy to accept an electron25 and it is notable that many nickel-catalyzed XEC reactions work best with ligands that are capable of participating in valence tautomerism.26
Figure 5.

Three mechanisms proposed for the XEC of alkyl halides with acid chlorides: (A) sequential oxidative addition, alkyl-first, L = Bpy;23 (B) disproportionation, L = 4,4′-di-tert-butyl-2,2′-bipyridine;29 (C) sequential oxidative addition, acyl first, L = chiral bis(oxazoline) for enantioconvergent reactions, bathophenanthroline for achiral reactions.19a,31
Related XEC methods for the coupling of alkyl iodides and benzyl chlorides with acid chlorides or anhydrides have been developed by several groups and may progress by a similar mechanism. With earlier studies by Mukaiyama27 and Yamamoto28 in mind, we proposed a mechanism that did not involve an intermediate reduction, but also relied upon an alkyl iodide reacting before an acid chloride (Figure 5B).29 Based upon stoichiometric reactivity studies of (L)Ni(0) and a proposed (L)Ni(II)(C(O)R)(O2CR) complex, Gong suggested a (L)Ni(I)(C(O)R) intermediate (14) for the coupling of aryl anhydrides with alkyl bromides and the intermediacy of an alkyl radical in the process (Figure 5C).30 Building off of these results, Reisman realized the enantioconvergent acylation of racemic secondary benzyl chlorides to form enantioenriched ketones with α-stereocenters, simultaneously demonstrating the potential for enantioselective reductive coupling and addressing a long-standing challenge in organic chemistry (Figure 5C).31 Detailed selectivity data were not reported for any of these three reactions, but bialkyl and bibenzyl (4) were noted to be major byproducts by several authors. We did not observe diketone (5), but we did observe decomposition of the acid chloride and small amounts (<10%) of dialkyl ketone (6), presumably formed from decarbonylation of the acid chloride.29
In each of the proposed mechanisms, initial oxidative addition of exclusively one electrophile to a nickel(0) complex is essential, but this selectivity is not always easy to predict. The selectivity observed by Amatore, Jutand, and Périchon23 matches the reported bond strengths: Et-I < Et-Br ∼ Bn-Cl < MeC(O)Cl ∼ PhC(O)Cl < (MeCO)2O.32 However, bond strength is not the only factor, especially for polar bonds, as Gong observed that benzoic anhydride reacted faster than an alkyl bromide with nickel(0).30b It remains unclear whether a unified mechanism exists for these related reactions or if the operating mechanism is dependent upon subtle differences in substrates, ligand, solvent, and additives.
Sequential Oxidative Addition: Steric Differentiation
In cases where substrates are more closely related in reactivity than acid chlorides and alkyl halides, it is possible for steric factors to influence relative electrophile reactivity. We observed steric matching between ligand and substrate in XEC reactions of α,β-unsaturated ketones with aryl halides.33 While high yields were observed in reactions of unhindered aryl halides catalyzed by the nickel complex of a sterically hindered ligand (2,9-dimethylphenanthroline), lower yields were observed with 2-substituted aryl halides and with acyclic E-enones. Switching to a less sterically demanding catalyst derived from 2,2′-bipyridine provided better selectivity and yield in both cases by minimizing enone dimerization (Figure 6A). Presumably, intermediate 17 is too hindered to react with 2-iodotoluene and instead reacts to form enone dimer.
Figure 6.

Steric matching as a selectivity model in XEC and key proposed catalytic intermediates for conjugate addition (A and B) and cross-Wurtz coupling (C). L for 23 = 2-(2-pyridyl)imidazoline.
An even more impressive example was disclosed by Gong (Figure 6B), where the cross-Wurtz coupling of two electronically similar but sterically differentiated alkyl halides proceeded in high yield with bis(pinacolato)diboron (B2pin2) as the reducing agent. The conditions are rather general, coupling not only primary with secondary, but also hindered primary with less hindered primary alkyl halides. The authors propose that different steric requirements of the two proposed intermediates—(L)Ni(I)(Bpin) (23) and (L)Ni(I)(Alkyl) (24)—control selectivity.19b
Heterolytic vs Homolytic Reactivity: Radical-Chain Process
A recent result from our own laboratories has demonstrated a new, general strategy for the coupling of two electrophiles that differentiates the substrates based upon heterolytic and homolytic reactivity trends. We had previously reported the nickel-catalyzed XEC of aryl halides with alkyl halides (Figure 7), which can be run on large scale (>25 mmol),13,34 and the extension of this concept to reactions of alkyl halides with 2-chloropyridines35 and allylic acetates.36 In all of these examples, we reported detailed selectivity data to demonstrate that the reactions were selective for the cross-coupled products. Studies by Gong,37 Peng,38 and Gosmini39 have demonstrated the generality of this approach and, in some cases, improved yields. Selectivity for cross product over symmetric dimers for the work of Gong and Peng are assumed to be similar to those reported by our laboratory because of the similar reaction conditions, but the Gosmini conditions use a cobalt catalyst and may proceed with a different mechanism and with different selectivity.
Figure 7.
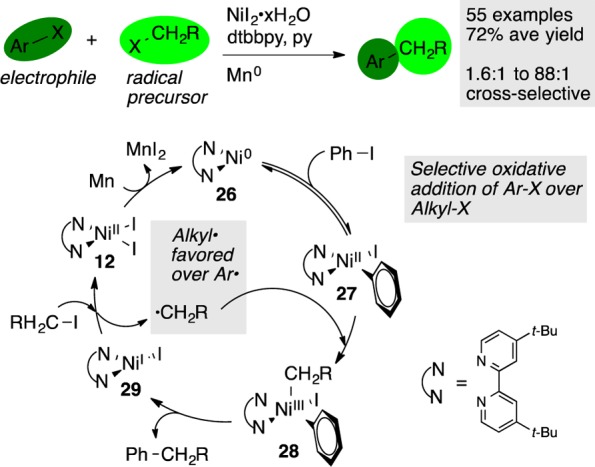
Radical chain cross-coupling of aryl iodides with alkyl iodides. Selectivity derives from the different reactivity of the aryl halides and alkyl halides.
Detailed mechanistic studies in our group recently revealed that selectivity arises from two points: aryl halides react with LNi(0) (26) faster than alkyl halides, but the alkyl halides form radicals more easily than aryl halides.40 Metal-based chain reactions were first proposed by Kochi,41 but an organic radical chain was first proposed by Hegedus.42 Although Durandetti and Périchon had suggested it may play a role in XEC, there was no evidence to differentiate it from two sequential oxidative additions at a single nickel center.43 This would result in a penultimate alkyl(aryl)Ni(IV)X2 intermediate, followed by rapid reductive elimination of the cross-coupled product.44 Our recent report40 showed that the degree of rearrangement of 5-hexenyl iodide, a radical clock, depended on the concentration of nickel in the reaction mixture, consistent with a radical chain mechanism.45 Independently, Hu reported that an isolated organonickel(II) complex reacted with an alkyl radical to form cross-coupled product.46
The key selectivity-determining principle is that one electrophile reacts by a normal, net-two-electron oxidative addition and the other electrophile serves as a radical precursor. For this strategy to be successful, the catalyst must support both single and two-electron steps (Figure 7), which suggests why first-row catalysts have proven so versatile in XEC reactions.
Importantly, selective reactions can be rationally designed using this mechanistic model. For example, epoxides are not reactive under standard reaction conditions because they are slow to form an alkyl radical by reaction with intermediate 29. We were able to develop the first XEC of epoxides with aryl halides by adding a cocatalyst that would assist in the conversion of epoxide to alkyl radical.47 Added NaI and Et3N·HCl converts epoxide 30 into iodohydrin 33 that nickel catalyst 29 can then convert into radical 34. This leads to normal48 (or anti-Markovnikov) epoxide opening product 31. Added titanocene dichloride, converted in situ to Cp2Ti(III)Cl, reacts with epoxide 30 to form the more substituted alkyl radical 35.49 This leads to abnormal48 (or Markovnikov) epoxide opening product 32 (Figure 8).
Figure 8.
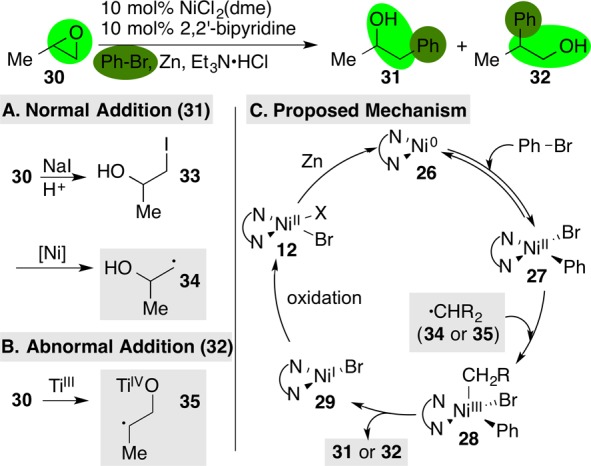
XEC of epoxides with aryl iodides. Iodide (A) or titanium (B) cocatalysis enables selective formation of normal (31) or abnormal (32) addition products.
Conclusion and Outlook
While considerable room for improvement remains, a number of XEC reactions are primed for widespread use in academia and industry. Strengths of the methods currently reported are broad functional-group compatibility, readily available substrates, simple experimental setup, scalability (>25 mmol scale),34b and high yields for some otherwise challenging reactions, like cross-couplings of alkyl halides. In the case of the coupling of alkyl halides with aryl halides, a general mechanism has been elucidated that allows for rational optimization and even de novo reaction design.
While advancement has been rapid, XEC is not yet as well developed as the cross-coupling of carbon nucleophiles with carbon electrophiles. Key challenges to be addressed in the near future are the expansion of the pool of electrophiles (e.g., aryl chlorides, aryl sulfonate esters, alkyl chlorides, other Michael receptors), development of ligands to further improve cross-selectivity, and the conception of new cocatalyst systems. The results of Reisman demonstrate that enantioselective or enantioconvergent reactions are possible and should constitute a major area of interest.
Finally, an improved understanding of the selectivity principles discussed here, as well as others not yet envisioned, should be an emphasis of future studies. For example, selective transmetalation50 or reductive elimination51 both hold promise. Currently, the radical chain mechanism has proven to be the most robust and translatable to other substrates because it is among the most understood. An improved understanding of other mechanisms would enable similarly rapid advancements.
Acknowledgments
This material is based upon work supported by the NSF Graduate Research Fellowship Program under Grant No. (DGE0935947 to D.A.E.) and the NIH (R01 GM097243 to D.J.W.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF or the NIH. D.J.W. is an Alfred P. Sloan Research Fellow. We thank our reviewers for many thoughtful suggestions and Kierra Huihui (University of Rochester) and Jason Brethorst (University of Minnesota) for editorial assistance.
Biographies

Prof. Daniel J. Weix is interested in the development and mechanistic study of transition-metal-catalyzed reactions that involve base metal catalysis and new modes of selectivity. His current research is focused on XEC and the utility of high-spin cobalt complexes.

Dr. Daniel A. Everson is pursuing postdoctoral studies at the University of Minnesota, Twin Cities. His current research is focused on development of new carbon–carbon bond activation and functionalization in the context of natural product synthesis.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Negishi E.-i.; Meijere A. d.. Handbook of Organopalladium Chemistry for Organic Synthesis, 1st ed.; Wiley-VCH Verlag: Weinheim, 2002; Vols. 1 and 2, pp 1–3424. [Google Scholar]; b Meijere A. d.; Diederich F.. Metal-Catalyzed Cross-Coupling Reactions, 2nded.;Wiley-VCH: Weinheim, 2004; pp 1–938. [Google Scholar]
- Knapp D. M.; Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Molander G. A.; Jean-Gérard L. Org. React. 2013, 79, 1–316. [Google Scholar]; b Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. [DOI] [PubMed] [Google Scholar]; c Noguchi H.; Hojo K.; Suginome M. J. Am. Chem. Soc. 2007, 129, 758–759. [DOI] [PubMed] [Google Scholar]; d Nakao Y.; Chen J.; Tanaka M.; Hiyama T. J. Am. Chem. Soc. 2007, 129, 11694–11695. [DOI] [PubMed] [Google Scholar]
- a Amatore M.; Gosmini C. Chem. Commun. 2008, 5019–5021. [DOI] [PubMed] [Google Scholar]; b Sase S.; Jaric M.; Metzger A.; Malakhov V.; Knochel P. J. Org. Chem. 2008, 73, 7380–7382. [DOI] [PubMed] [Google Scholar]; c Alessi M.; Larkin A. L.; Ogilvie K. A.; Green L. A.; Lai S.; Lopez S.; Snieckus V. J. Org. Chem. 2007, 72, 1588–1594. [DOI] [PubMed] [Google Scholar]
- a Krasovskiy A.; Duplais C.; Lipshutz B. H. J. Am. Chem. Soc. 2009, 131, 15592–15593. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Czaplik W. M.; Mayer M.; Jacobi von Wangelin A. Angew. Chem., Int. Ed. 2009, 48, 607–610. [DOI] [PubMed] [Google Scholar]
- Chatterjee A.; Joshi N. N. Tetrahedron 2006, 62, 12137–12158. [Google Scholar]
- Takeda T.; Tsubouchi A. Sci. Synth. 2010, 47a, 247–325. [Google Scholar]
- Hassan J.; Sevignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1469. [DOI] [PubMed] [Google Scholar]
- a Wurtz A. Ann. Chem. Pharm. 1855, 96, 364–375. [Google Scholar]; b Lewis H. F.; Hendricks R.; Yohe G. R. J. Am. Chem. Soc. 1928, 50, 1993–1998. [Google Scholar]
- a Tollens B.; Fittig R. Liebigs Ann. Chem. 1864, 131, 303–323. [Google Scholar]; b Fittig R.; König J. Justus Liebigs Ann. Chem. 1867, 144, 277–294. [Google Scholar]; c Kwa T. L.; Boelhouwer C. Tetrahedron 1969, 25, 5771–5776. [Google Scholar]
- a Jutand A. Chem. Rev. 2008, 108, 2300–2347. [DOI] [PubMed] [Google Scholar]; b Nédélec J.-Y.; Périchon J.; Troupel M. Top. Curr. Chem. 1997, 185, 141–173. [Google Scholar]; c Klein A.; Budnikova Y.; Sinyashin O. J. Organomet. Chem. 2007, 692, 3156–3166. [Google Scholar]
- Chatterjee A. K.; Choi T.-L.; Sanders D. P.; Grubbs R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. [DOI] [PubMed] [Google Scholar]
- Everson D. A.; Jones B. A.; Weix D. J. J. Am. Chem. Soc. 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quasdorf K. W.; Riener M.; Petrova K. V.; Garg N. K. J. Am. Chem. Soc. 2009, 131, 17748–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gomes P.; Fillon H.; Gosmini C.; Labbé E.; Périchon J. Tetrahedron 2002, 58, 8417–8424. [Google Scholar]; b Amatore M.; Gosmini C. Angew. Chem., Int. Ed. 2008, 47, 2089–2092. [DOI] [PubMed] [Google Scholar]
- Qian Q.; Zang Z.; Wang S.; Chen Y.; Lin K.; Gong H. Synlett 2013, 24, 619–624. [Google Scholar]
- a Hassan J.; Hathroubi C.; Gozzi C.; Lemaire M. Tetrahedron Lett. 2000, 41, 8791–8794. [Google Scholar]; b Hassan J.; Hathroubi C.; Gozzi C.; Lemaire M. Tetrahedron 2001, 57, 7845–7855. [Google Scholar]; c Wang L.; Zhang Y.; Liu L.; Wang Y. J. Org. Chem. 2006, 71, 1284–1287. [DOI] [PubMed] [Google Scholar]
- Gosmini C.; Bassene-Ernst C.; Durandetti M. Tetrahedron 2009, 65, 6141–6146. [Google Scholar]
- a Yu X.; Yang T.; Wang S.; Xu H.; Gong H. Org. Lett. 2011, 13, 2138–2141. [DOI] [PubMed] [Google Scholar]; b Xu H.; Zhao C.; Qian Q.; Deng W.; Gong H. Chem. Sci. 2013, 4, 4022–4029. [Google Scholar]
- a Prinsell M. R.; Everson D. A.; Weix D. J. Chem. Commun. 2010, 46, 5743–5745. [DOI] [PubMed] [Google Scholar]; b Goldup S. M.; Leigh D. A.; McBurney R. T.; McGonigal P. R.; Plant A. Chem. Sci. 2010, 1, 383–386. [Google Scholar]
- Amatore C.; Jutand A. Organometallics 1988, 7, 2203–2214. [Google Scholar]
- Colon I.; Kelsey D. R. J. Org. Chem. 1986, 51, 2627–2637. [Google Scholar]
- Amatore C.; Jutand A.; Périchon J.; Rollin Y. Monatsh. Chem. 2000, 131, 1293–1304. [Google Scholar]
- a Schiavon G.; Bontempelli G.; Corain B. J. Chem. Soc., Dalton Trans. 1981, 1074–1081. [Google Scholar]; b Troupel M.; Rollin Y.; Sibille S.; Périchon J.; Fauvarque J. F. J. Organomet. Chem. 1980, 202, 435–446. [Google Scholar]
- Scarborough C. C.; Wieghardt K. Inorg. Chem. 2011, 50, 9773–9793. [DOI] [PubMed] [Google Scholar]
- a Ciszewski J. T.; Mikhaylov D. Y.; Holin K. V.; Kadirov M. K.; Budnikova Y. H.; Sinyashin O.; Vicic D. A. Inorg. Chem. 2011, 50, 8630–8635. [DOI] [PubMed] [Google Scholar]; b Luca O. R.; Crabtree R. H. Chem. Soc. Rev. 2013, 42, 1440–1459. [DOI] [PubMed] [Google Scholar]
- Onaka M.; Matsuoka Y.; Mukaiyama T. Chem. Lett. 1981, 10, 531–534. [Google Scholar]
- Yamamoto T.; Kohara T.; Yamamoto A. Bull. Chem. Soc. Jpn. 1981, 54, 2161–2168. [Google Scholar]
- Wotal A. C.; Weix D. J. Org. Lett. 2012, 14, 1476–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu F.; Lu W.; Qian Q.; Ren Q.; Gong H. Org. Lett. 2012, 14, 3044–3047. [DOI] [PubMed] [Google Scholar]; b Yin H.; Zhao C.; You H.; Lin K.; Gong H. Chem. Commun. 2012, 48, 7034–7036. [DOI] [PubMed] [Google Scholar]
- Cherney A. H.; Kadunce N. T.; Reisman S. E. J. Am. Chem. Soc. 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]
- a Blanksby S. J.; Ellison G. B. Acc. Chem. Res. 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]; b Feng Y.; Huang H.; Liu L.; Guo Q.-X. Phys. Chem. Chem. Phys. 2003, 5, 685–690. [Google Scholar]; c Laarhoven L. J. J.; Mulder P.; Wayner D. D. M. Acc. Chem. Res. 1999, 32, 342–349. [Google Scholar]; d Sanderson R. T.Chemical Bonds and Bond Energy, 2nd ed.; Academic Press: New York, 1976; p 218. [Google Scholar]
- Shrestha R.; Dorn S. C. M.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Everson D. A.; Shrestha R.; Weix D. J. J. Am. Chem. Soc. 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]; b Everson D. A.; George D. T.; Weix D. J.; Buergler J. F.; Wood J. L. Org. Synth. 2013, 90, 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson D. A.; Buonomo J. A.; Weix D. J. Synlett 2014, 25, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anka-Lufford L. L.; Prinsell M. R.; Weix D. J. J. Org. Chem. 2012, 77, 9989–10000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang S.; Qian Q.; Gong H. Org. Lett. 2012, 14, 3352–3355. [DOI] [PubMed] [Google Scholar]; b Dai Y.; Wu F.; Zang Z.; You H.; Gong H. Chem.–Eur. J. 2012, 18, 808–812. [DOI] [PubMed] [Google Scholar]
- Yan C.-S.; Peng Y.; Xu X.-B.; Wang Y.-W. Chem.–Eur. J. 2012, 18, 6039–6048. [DOI] [PubMed] [Google Scholar]
- a Amatore M.; Gosmini C. Chem.–Eur. J. 2010, 16, 5848–5852. [DOI] [PubMed] [Google Scholar]; b Qian X.; Auffrant A.; Felouat A.; Gosmini C. Angew. Chem., Int. Ed. 2011, 50, 10402–10405. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kochi J. K. Pure Appl. Chem. 1980, 52, 571–605. [Google Scholar]; b Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 7547–7560. [Google Scholar]; c Kochi J. Acc. Chem. Res. 1974, 7, 351–360. [Google Scholar]
- Hegedus L. S.; Miller L. L. J. Am. Chem. Soc. 1975, 97, 459–460. [Google Scholar]
- Durandetti M.; Nédélec J.-Y.; Périchon J. J. Org. Chem. 1996, 61, 1748–1755. [DOI] [PubMed] [Google Scholar]
- Ananikov V. P.; Musaev D. G.; Morokuma K. J. Am. Chem. Soc. 2002, 124, 2839–2852. [DOI] [PubMed] [Google Scholar]
- Kinney R. J.; Jones W. D.; Bergman R. G. J. Am. Chem. Soc. 1978, 100, 7902–7915. [Google Scholar]
- Breitenfeld J.; Ruiz J.; Wodrich M. D.; Hu X. J. Am. Chem. Soc. 2013, 135, 12004–12012. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Weix D. J. J. Am. Chem. Soc. 2014, 136, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R. E.; Isaacs N. S. Chem. Rev. 1959, 59, 737–799. [Google Scholar]
- a RajanBabu T. V.; Nugent W. A. J. Am. Chem. Soc. 1994, 116, 986–997. [Google Scholar]; b Gansäuer A.; Fan C.-A.; Keller F.; Karbaum P. Chem.–Eur. J. 2007, 13, 8084–8090. [DOI] [PubMed] [Google Scholar]
- Osakada K.; Yamamoto T. Coord. Chem. Rev. 2000, 198, 379–399. [Google Scholar]
- Ananikov V. P.; Musaev D. G.; Morokuma K. Organometallics 2005, 24, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]