

Abstract
Oligonucleotides modified with conformationally restricted nucleotides such as locked nucleic acid (LNA) monomers are used extensively in molecular biology and medicinal chemistry to modulate gene expression at the RNA level. Major efforts have been devoted to the design of LNA derivatives that induce even higher binding affinity and specificity, greater enzymatic stability, and more desirable pharmacokinetic profiles. Most of this work has focused on modifications of LNA’s oxymethylene bridge. Here, we describe an alternative approach for modulation of the properties of LNA: i.e., through functionalization of LNA nucleobases. Twelve structurally diverse C5-functionalized LNA uridine (U) phosphoramidites were synthesized and incorporated into oligodeoxyribonucleotides (ONs), which were then characterized with respect to thermal denaturation, enzymatic stability, and fluorescence properties. ONs modified with monomers that are conjugated to small alkynes display significantly improved target affinity, binding specificity, and protection against 3′-exonucleases relative to regular LNA. In contrast, ONs modified with monomers that are conjugated to bulky hydrophobic alkynes display lower target affinity yet much greater 3′-exonuclease resistance. ONs modified with C5-fluorophore-functionalized LNA-U monomers enable fluorescent discrimination of targets with single nucleotide polymorphisms (SNPs). In concert, these properties render C5-functionalized LNA as a promising class of building blocks for RNA-targeting applications and nucleic acid diagnostics.
Introduction
The development of novel conformationally restricted nucleotides is a vibrant area of research.1,2 Efforts are driven by the interesting properties of oligodeoxyribonucleotides (ONs) modified with classic examples of conformationally restricted nucleotides such as homo-DNA,3 hexitol nucleic acid (HNA),4 cyclohexane nucleic acid (CeNA),5 bicyclo DNA,6 tricyclo DNA,7 or locked nucleic acid (LNA),8,9 which is also known as bridged nucleic acid (BNA).10 ONs comprising these building blocks display high affinity toward complementary DNA/RNA often due to reduced entropic binding penalties11 and are accordingly in high demand for a wide range of nucleic acid targeting applications in molecular biology, biotechnology, and pharmaceutical science.12 Their use as RNA-targeting antisense oligonucleotides to decrease gene expression is a particularly prominent example.12b
LNA is an especially interesting member of this compound class because it induces some of the greatest duplex stabilizations observed to date (Figure 1).8−10 Modulation of gene expression through LNA-mediated targeting of mRNA, pre-mRNA, or miRNA has accelerated gene function studies and led to the development of LNA-based drug candidates against diseases of genetic origin.13,14 Other applications of LNA include its use as an in situ hybridization probe to monitor spatiotemporal expression patterns of miRNAs.15
Figure 1.

Structure of LNA-T and C5-functionalized analogues thereof studied herein.
Many analogues of LNA have been synthesized with the aim of further improving the binding affinity/specificity, enzymatic stability and pharmokinetic characteristics of LNA.1,2,16 The vast majority of these efforts have focused on modifying the oxymethylene bridge spanning the C2′/C4′-positions and/or introducing minor-groove-oriented substituents on the bridge. These structural perturbations have resulted in improved enzymatic stability and altered biodistribution and/or toxicity profiles but have generally not resulted in major improvements in binding affinity and specificity.
C5-functionalized pyrimidine DNA monomers have also attracted considerable attention,17,18 as they enable predictable positioning of functional entities in the major groove of nucleic acid duplexes.19 Small C5-alkynyl substituents such as propyn-1-yl and 3-aminopropyn-1-yl induce considerable duplex thermostabilization relative to unmodified duplexes, while large hydrophobic substituents typically decrease duplex thermostability. Attachment of polarity-sensitive fluorophores to the C5 position of DNA pyrimidine monomers has produced several interesting oligonucleotide probes for structural studies of nucleic acids and applications in nucleic acid diagnostics.12c,20
In light of the above and our ongoing interest in LNA chemistry,12c,21 we recently set out to study C5-alkynyl-functionalized LNA uridine (U) monomers, on the basis of the hypothesis that these monomers will exhibit beneficial properties from both compound classes, i.e., high affinity toward RNA complements and good mismatch discrimination (LNA), along with the ability to position blocking groups in the major groove to confer protection against enzymatic degradation (C5 substituent). The results from our preliminary studies have been very encouraging.22 ONs modified with small C5-alkynyl-functionalized LNA-U monomers display high affinity toward complementary RNA and moderate protection against 3′-exonucleases, while ONs modified with large C5-alkynyl-functionalized LNA-U monomers display greatly increased enzymatic stability but decreased RNA affinity.
In the present article, we describe full experimental details concerning the synthesis of 12 different C5-functionalized LNA-U phosphoramidites, their incorporation into ONs, and the characterization of these modified ONs by means of thermal denaturation experiments, analysis of thermodynamic parameters, nuclease stability experiments, and fluorescence spectroscopy. The monomers in question were selected to ensure a representation of substituents with different sizes, polarities, linker chemistries, and fluorescence characteristics (Figure 1).
Results and Discussion
Synthesis of Phosphoramidites
Our route to target phosphoramidites 5b–l initiates from LNA uridine diol 1, which is obtained from commercially available diacetone-α-d-allose in ∼52% yield (Scheme 1).23 C5-iodination of 1 was accomplished through treatment with iodine and ceric ammonium nitrate (CAN) in acetic acid at 80 °C for ∼40 min to afford nucleoside 2 in 87% yield. Prolonged heating and/or higher reaction temperatures result in the formation of nonpolar impurities, which complicate purification and reduce product yield. Subsequent O5′-dimethoxytritylation using standard conditions afforded the key intermediate 3 in 84% yield. Terminal alkynes24 were then coupled with 3 under typical Sonogashira conditions25 to provide C5-alkynyl-functionalized LNA uridines 4a–j in 53–87% yield. Careful deoxygenation is critical to the outcome of these reactions, as they otherwise do not proceed to completion. Finally, O3′-phosphitylation using 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite afforded target phosphoramidites 5b–j in 43–83% yield.
Scheme 1. Synthesis of C5-Alkynyl-Functionalized LNA Uridine Phosphoramidites.

Abbreviations: CAN, ceric ammonium nitrate; DMTrCl, 4,4′-dimethoxytrityl chloride; PCl, 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite; DIPEA, N,N′-diisopropylethylamine.
In order to obtain C5-triazolyl-functionalized LNA uridine phosphoramidites 5k and 5l, C5-ethynyl-functionalized LNA uridine 4b (obtained via standard TBAF-mediated desilylation of 4a) was reacted with 1-azidopyrene26 or 1-azidomethylpyrene27 in a Cu(I)-catalyzed azide alkyne Huisgen 1,3-dipolar cycloaddition,28 followed by standard O3′-phosphitylation (Scheme 2).
Scheme 2. Synthesis of C5-Triazolyl-Functionalized LNA Uridine Phosphoramidites.

ON Synthesis
Phosphoramidites 5b–l were used in machine-assisted solid-phase DNA synthesis (0.2 μmol scale) to incorporate monomers K–Z into ONs. Standard conditions were used except for extended hand-coupling (generally 15 min with 4,5-dicyanoimidazole or 5-[3,5-bis(trifluoromethyl)phenyl]-1H-tetrazole as activator) when using 5b–l, which typically resulted in stepwise coupling yields of >95%. The composition and purity of all modified ONs was ascertained by MALDI MS analysis (Table S1, Supporting Information) and ion-pair reversed-phase HPLC, respectively. ONs containing a single incorporation in the 5′-GTGABATGC context are denoted K1–M1 and so on. Similar conventions apply for ONs in the B2–B4 series (Table 1). Reference DNA and RNA strands are denoted D1/D2 and R1/R2, respectively.
Table 1. ΔTm Values of Duplexes between ONs Modified with C5-Functionalized LNA Monomers and Complementary DNA/RNA Measured Relative to Unmodified Duplexesa.
| ΔTm/mod (°C) |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ON | duplex | B = | L | K | M | N | O | P | Q | S | V | W | X | Y | Z |
| B1 | 5′-GTG ABA TGC | +5.0 | +7.0 | +7.0 | +8.0 | +4.5 | +4.0 | +1.0 | –5.5 | –6.5 | –8.5 | –12.5 | –10.5 | –5.5 | |
| D2 | 3′-CAC TAT ACG | ||||||||||||||
| D1 | 5′-GTG ATA TGC | +4.0 | +5.5 | +5.5 | +6.5 | +3.0 | +1.0 | +0.5 | –5.0 | –7.5 | –9.5 | –12.0 | –13.5 | –6.5 | |
| B2 | 3′-CAC BAT ACG | ||||||||||||||
| D1 | 5′-GTG ATA TGC | +6.5 | +5.5 | +7.0 | +9.5 | +4.5 | +3.5 | +1.0 | –3.5 | –4.0 | –10.5 | –11.5 | –12.5 | –5.5 | |
| B3 | 3′-CAC TAB ACG | ||||||||||||||
| D1 | 5′-GTG ATA TGC | +5.5 | +5.5 | +5.5 | +8.0 | nd | +3.0 | <−10.0 | <−10.0 | +0.5 | –6.5 | <−10.0 | –4.0 | –2.0 | |
| B4 | 3′-CAC BAB ACG | ||||||||||||||
| B1 | 5′-GTG ABA TGC | +9.5 | +11.0 | +9.5 | +13.0 | +6.0 | +5.5 | +4.0 | –2.0 | –4.0 | –2.0 | –12.0 | –2.0 | –1.5 | |
| R2 | 3′-CAC UAU ACG | ||||||||||||||
| R1 | 5′-GUG AUA UGC | +6.5 | +8.5 | +8.0 | +10.0 | –0.5 | +2.0 | +3.5 | ±0 | –6.0 | –1.5 | –12.0 | –10.0 | –5.0 | |
| B2 | 3′-CAC BAT ACG | ||||||||||||||
| R1 | 5′-GUG AUA UGC | +9.5 | +8.5 | +10.0 | +12.5 | +2.5 | +2.0 | +2.5 | –1.0 | ±0 | –5.5 | –11.0 | –9.0 | –1.0 | |
| B3 | 3′-CAC TAB ACG | ||||||||||||||
| R1 | 5′-GUG AUA UGC | +8.0 | +8.5 | +8.0 | +11.0 | nd | +4.5 | <−8.5 | <−8.5 | +2.0 | –5.5 | <−8.5 | –2.0 | –0.5 | |
| B4 | 3′-CAC BAB ACG | ||||||||||||||
ΔTm = change in Tm values relative to unmodified reference duplexes D1:D2 (Tm ≡ 29.5 °C), D1:R2 (Tm ≡ 27.0 °C), and D2:R1 (Tm ≡ 27.0 °C); Tm values were determined as the first-derivative maximum of denaturation curves (A260 vs T) recorded in medium salt phosphate buffer ([Na+] = 110 mM, [Cl–] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4)), using 1.0 μM of each strand. Tm values are averages of at least two measurements within 1.0 °C; see Figure 1 for structures of monomers. nd = not determined. Data for duplexes between L/K/N/Q/S-modified ONs and complementary RNA has been previously reported in ref (22).
Thermal Denaturation Experiments: Binding Affinity
The thermostabilities of duplexes between modified ONs and complementary DNA/RNA were evaluated by determining their thermal denaturation temperature (Tm) in medium salt phosphate buffer ([Na+] = 110 mM, pH 7.0). Tm’s of modified duplexes are discussed relative to Tm’s of unmodified reference duplexes (Table 1).
As anticipated, ONs modified with one or two conventional LNA-T monomers form very thermostable duplexes with RNA targets in particular (see ΔTm values for L1–L4, Table 1). Interestingly, ONs modified with LNA monomers featuring small C5-alkynyl moieties generally result in the formation of even more thermostable duplexes (compare ΔTm values of K/M/N series with those of the L series, Table 1). The effect is most pronounced for ONs modified with aminopropynyl-functionalized monomer N, which display increases in Tm values of up to +13 °C per modification. The greater thermostability of duplexes modified with K/M/N monomers is most likely the result of enhanced stacking interactions18a and, in the case of monomer N, favorable electrostatic interactions and/or hydration in the major groove, in a manner similar to that previously suggested for C5-aminopropynyl-modified DNA.18e,18h
In contrast, duplexes modified with LNA monomers that are conjugated to medium-sized hydrophobic C5-alkynyl substituents are less thermostable than the corresponding LNA-modified duplexes (compare ΔTm values of the O/P-series with those of the L series, Table 1). The trend is particularly prominent in DNA:RNA duplexes, presumably due to a suboptimal fit of the C5-alkynyl substituent in the narrow major groove of A/B-type duplexes. However, other factors, such as different influences on hydration,18c cannot be ruled out. The resulting duplexes are, nevertheless, still significantly more stable that the unmodified reference duplexes.
ONs modified with LNA monomers that are conjugated to long hydrophobic C5-alkynyl substituents display even lower affinity toward their targets (see ΔTm values of the Q/S series, Table 1). It is particularly noteworthy that duplexes involving the doubly modified Q4 or S4 do not display transitions above 10 °C. Similar observations have been made with doubly cholesterol-modified 2′-amino-LNA.29 We hypothesize that interactions between the hydrophobic groups in single-stranded Q4 or S4 interfere with duplex formation. The fact that DNA duplexes with interstrand zipper arrangements of two Q monomers are rather thermostable supports this hypothesis (see Table S2, Supporting Information).
Similarly, ONs modified with LNA monomers that are conjugated to large hydrophobic fluorophores generally form very thermolabile duplexes, regardless of whether the fluorophore is attached via an alkynyl or triazoyl linker (see ΔTm values of the V–Z series, Table 1). The use of monomers in which the fluorophore is attached to the nucleobase via a short rigid linker, such as in monomers W–Y, results in particularly unstable duplexes. Once again, we speculate that these trends reflect a poor fit of the fluorophore in the major groove; short rigid linkers between the fluorophore and nucleobase moieties may prevent the fluorophore from sampling more suitable conformational space. Interestingly, with the exception of pyrene- and perylene-functionalized W4 and X4, duplexes entailing the doubly modified B4 ONs are considerably more stable than those entailing their singly modified counterparts (e.g., compare ΔTm/mod of B4:D1 relative to B2:D1 and B3:D1, Table 1). Similar stabilizing trends have been reported for other densely fluorophore modified duplexes and were attributed to the formation of chromophore arrays in the major groove.19 The presence of pyrene excimer signals in the steady-state fluorescence emission spectra of duplexes between V4/Y4/Z4 and DNA/RNA complements supports this hypothesis (Figure S3, Supporting Information).
Thermodynamic Analysis of Duplexes Modified with C5-Functionalized LNA-U Monomers
The Tm-based conclusions are largely corroborated through analysis of the thermodynamic parameters for duplex formation, which were derived from thermal denaturation curves through curve fitting.30 Thus, the formation of duplexes between conventional LNA L1–L3 and complementary DNA or RNA is 4–7 and 8–13 kJ/mol more favorable, respectively, in comparison to unmodified reference duplexes (see ΔΔG298 values for L1–L3, Table 2). The greater stability of LNA-modified duplexes is generally a result of lower enthalpy (ΔΔH < 0 kJ/mol for L2 and L3, Table 2), but entropic stabilization is also observed (Δ(T298ΔS) < 0 kJ/mol for L1, Table 2).
Table 2. Thermodynamic Parameters for Formation of Duplexes Modified with C5-Functionalized LNA Monomersa.
| +complementary
DNA |
+complementary
RNA |
||||||
|---|---|---|---|---|---|---|---|
| ON | sequence | ΔG298 [ΔΔG298] (kJ/mol) | ΔH [ΔΔH] (kJ/mol) | –T298ΔS [Δ(T298ΔS)] (kJ/mol) | ΔG298 [ΔΔG298] (kJ/mol) | ΔH [ΔΔH] (kJ/mol) | –T298ΔS [Δ(T298ΔS)] (kJ/mol) |
| D1 | 5′-GTG ATA TGC | –42 | –314 | 271 | –36 | –278 | 241 |
| D2 | 3′-CAC TAT ACG | –42 | –314 | 271 | –39 | –293 | 254 |
| L1 | 5′-GTG ALA TGC | –47 [−5] | –297 [+17] | 250 [−21] | –49 [−13] | –309 [−31] | 260 [+19] |
| L2 | 3′-CAC LAT ACG | –46 [−4] | –332 [−18] | 286 [+15] | –47 [−8] | –331 [−38] | 283 [+29] |
| L3 | 3′-CAC TAL ACG | –49 [−7] | –332 [−18] | 283 [+12] | –50 [−11] | –340 [−47] | 290 [+36] |
| K1 | 5′-GTG AKA TGC | –49 [−7] | –350 [−36] | 301 [+30] | –53 [−17] | –424 [−146] | 371 [+130] |
| K2 | 3′-CAC KAT ACG | –49 [−7] | –349 [−35] | 300 [+29] | –49 [−10] | –367 [−74] | 317 [+63] |
| K3 | 3′-CAC TAK ACG | –52 [−10] | –372 [−58] | 319 [+48] | –57 [−18] | –414 [−121] | 357 [+103] |
| M1 | 5′-GTG AMA TGC | –51 [−9] | –390 [−76] | 339 [+68] | –52 [−16] | –386 [−108] | 334 [+93] |
| M2 | 3′-CAC MAT ACG | –50 [−8] | –394 [−80] | 344 [+73] | –51 [−12] | –398 [−105] | 347 [+93] |
| M3 | 3′-CAC TAM ACG | –51 [−9] | –360 [−46] | 309 [+38] | –51 [−12] | –367 [−74] | 316 [+62] |
| N1 | 5′-GTG ANA TGC | –51 [−9] | –353 [−39] | 302 [+31] | –51 [−15] | –324 [−46] | 272 [+31] |
| N2 | 3′-CAC NAT ACG | –49 [−7] | –362 [−48] | 313 [+42] | –52 [−13] | –364 [−71] | 312 [+58] |
| N3 | 3′-CAC TAN ACG | –52 [−10] | –361 [−47] | 309 [+38] | –52 [−13] | –325 [−32] | 272 [+18] |
| O1 | 5′-GTG AOA TGC | –47 [−5] | –337 [−23] | 290 [+19] | –46 [−10] | –337 [−59] | 291 [+50] |
| O2 | 3′-CAC OAT ACG | –44 [−2] | –322 [−8] | 278 [+7] | –43 [−4] | –366 [−73] | 322 [+68] |
| O3 | 3′-CAC TAO ACG | –46 [−4] | –324 [−10] | 278 [+7] | –44 [−5] | –340 [−47] | 296 [+42] |
| P1 | 5′-GTG APA TGC | –45 [−3] | –334 [−20] | 289 [+18] | –45 [−9] | –327 [−49] | 282 [+41] |
| P2 | 3′-CAC PAT ACG | –43 [−1] | –324 [−10] | 281 [+10] | –43 [−4] | –351 [−58] | 308 [+54] |
| P3 | 3′-CAC TAP ACG | –44 [−2] | –339 [−25] | 294 [+23] | –43 [−4] | –365 [−72] | 321 [+67] |
| Q1 | 5′-GTG AQA TGC | –45 [−3] | –346 [−32] | 301 [+30] | –45 [−9] | –347 [−69] | 302 [+61] |
| Q2 | 3′-CAC QAT ACG | –45 [−3] | –411 [−97] | 371 [+100] | –46 [−7] | –377 [−84] | 331 [+77] |
| Q3 | 3′-CAC TAQ ACG | –43 [−1] | –287 [+27] | 243 [−28] | –43 [−4] | –360 [−67] | 317 [+63] |
| S1 | 5′-GTG ASA TGC | –37 [+5] | –317 [−3] | 280 [+9] | –39 [−3] | –333 [−55] | 294 [+53] |
| S2 | 3′-CAC SAT ACG | –39 [+3] | –380 [−66] | 342 [+71] | –42 [−3] | –359 [−66] | 316 [+62] |
| S3 | 3′-CAC TAS ACG | –40 [+2] | –380 [−66] | 339 [+68] | –40 [−1] | –355 [−62] | 315 [+61] |
Parameters were determined from thermal denaturation curves, which were recorded as described in Table 1. ΔΔG298, ΔΔH, and Δ(T298ΔS) are calculated relative to reference duplexes D1:D2, D1:R2, and D2:R1.
Formation of duplexes entailing ONs modified with K/M/N monomers, which are conjugated to small and/or relatively polar alkynes, is 1–7 kJ/mol more favorable than formation of the corresponding LNA-modified duplexes (compare ΔΔG298 values for the K/M/N series vs L series, Table 2). The additional duplex stabilization is generally enthalpic in origin, which is consistent with improved base stacking due to the extended π surface of the C5-alkynyl-functionalized LNA monomers (compare ΔΔH values for the K/M/N series vs L series, Table 2); similar trends have been previously reported for C5-propynyl-functionalized DNA monomers.31
Duplexes involving ONs modified with monomers O/P/Q, which are conjugated to moderately large hydrophobic alkynyl substituents, are 0–7 kJ/mol less favorable than the corresponding LNA-modified duplexes (compare ΔΔG298 values for the O/P/Q series vs the L series, Table 2). Comparison with K-modified duplexes suggests that the hydrophobic substituents counteract the favorable enthalpy of the extended π surfaces (compare ΔΔH values for the O/P/Q series vs the K series, Table 2). One possible interpretation of this is that the hydrophobic substituents disrupt hydration in the major groove.
DNA duplexes modified with C5-cholesterol-functionalized LNA monomer S are less stable than the control duplex, whereas duplexes with RNA are slightly more stable (see ΔΔG298 values for S1–S3, Table 2). The favorable enthalpic contribution of the alkyne functionality is fully counteracted by low entropy in DNA duplexes but only partially counteracted in DNA:RNA duplexes (compare ΔΔH vs Δ(T298ΔS) for S1–S3, Table 2).
Thermal Denaturation Studies: Binding Specificity
The binding specificities of centrally modified ONs (B1 series) were determined by using DNA/RNA targets with mismatched nucleotides opposite of the modified monomer. As expected,8−10 LNA-modified ON L1 displays improved binding specificity relative to unmodified reference strand D1, as evidenced by the more pronounced decreases in Tm values of mismatched duplexes (compare ΔTm values for L1 and D1, Table 3). Interestingly, many of the C5-functionalized LNA monomers induce additional improvements in binding specificity (note ΔTm values of K1/M1/N1/O1/P1/Q1, Table 3). It is recognized that nucleotide modifications, which improve target affinity as well as binding specificity, are desirable for nucleic acid targeting applications.32 Cholesterol-functionalized LNA S1 and fluorophore-functionalized LNAs V1/W1/X1/Y1/Z1 display poor discrimination of mismatched DNA targets but maintain reasonable specificity against RNA targets (Table 3). These trends are indicative of different binding modes of the pyrene and perylene moieties in DNA:DNA vs DNA:RNA duplexes. Intercalation of aromatic units, which is known to stabilize mismatched base pairs,33 is more favorable in DNA:DNA than in DNA:RNA duplexes.34 For a discussion of the binding specificities of double-modified ONs (B4-series), see the Supporting Information (Table S3).
Table 3. Discrimination of Mismatched DNA/RNA Targets by Singly Modified LNAs and Reference ONsa.
| DNA:
3′-CAC TBT ACG |
RNA:
3′-CAC UBU ACG |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm | ΔTm |
Tm | ΔTm |
||||||
| ON | sequence | A | C | G | T | A | C | G | U |
| D1 | 5′-GTG ATA TGC | 29.5 | –16.5 | –8.0 | –15.5 | 27.0 | <−17.0 | –4.5 | <−17.0 |
| L1 | 5′-GTG ALA TGC | 34.5 | –18.0 | –11.0 | –16.0 | 36.5 | –19.0 | –8.0 | –18.5 |
| K1 | 5′-GTG AKA TGC | 36.5 | –20.0 | –15.5 | –18.5 | 38.0 | –20.5 | –13.5 | –22.0 |
| M1 | 5′-GTG AMA TGC | 36.5 | –20.0 | –11.5 | –18.5 | 36.5 | –18.5 | –9.5 | –20.0 |
| N1 | 5′-GTG ANA TGC | 37.5 | –19.0 | –12.0 | –17.5 | 40.0 | –18.5 | –11.5 | –22.5 |
| O1 | 5′-GTG AOA TGC | 34.0 | –20.5 | –16.5 | –18.0 | 33.0 | –20.0 | –9.5 | –20.0 |
| P1 | 5′-GTG APA TGC | 33.5 | –21.5 | –17.0 | –20.5 | 32.5 | –20.5 | –11.5 | –19.5 |
| Q1 | 5′-GTG AQA TGC | 30.5 | –18.0 | –13.0 | –16.5 | 31.0 | –19.5 | –10.0 | –20.0 |
| S1 | 5′-GTG ASA TGC | 24.0 | –11.5 | –10.0 | –11.0 | 25.0 | –15.0 | –9.0 | <−15.0 |
| V1 | 5′-GTG AVA TGC | 23.0 | –7.5 | –10.0 | –7.5 | 23.0 | <−13.0 | –10.5 | <−13.0 |
| W1 | 5′-GTG AWA TGC | 21.0 | +6.0 | –7.0 | +3.0 | 25.0 | <−15.0 | <−15.0 | <−15.0 |
| X1 | 5′-GTG AXA TGC | 17.0 | +4.5 | ±0.0 | +3.5 | 15.0 | <−5.0 | <−5.0 | <−5.0 |
| Y1 | 5′-GTG AYA TGC | 19.0 | –1.0 | –4.0 | –2.5 | 25.0 | <−15.0 | –8.0 | <−15.0 |
| Z1 | 5′-GTG AZA TGC | 24.0 | –10.0 | <−14.0 | –9.5 | 25.5 | –1.5 | <−15.5 | <−15.5 |
3′-Exonuclease Stability of C5-Functionalized LNA
Next, we examined the enzymatic stability of select C5-functionalized LNAs and reference strands in the presence of snake venom phosphodiesterase (SVPDE), a 3′-exonuclease. As expected, unmodified D2 is quickly degraded (>95% cleavage after 15 min), while the singly modified LNA L2 offers moderate protection against SVPDE (>95% cleaved after 50 min) (Figure 2). ONs modified with a single C5-ethynyl- or C5-aminopropynyl-functionalized LNA monomer are markedly more resistant toward SVPDE degradation (∼55% and 35% cleavage of K2 and N2, respectively, after 2 h). Interestingly, ONs that are modified with LNA monomers conjugated to large hydrophobic substituents are completely inert against SVPDE-mediated degradation, following a brief period of cleavage (see degradation profiles for Q2 and S2; Figure 2). M2/O2/P2 also display markedly increased 3′-exonuclease resistance (Figure S1, Supporting Information). As expected, these trends are even more pronounced with the doubly modified B4 series (Figure 2). Thus, the data strongly suggest that large hydrophobic C5-alkynyl substituents offer effective protection from enzymatic degradation.
Figure 2.
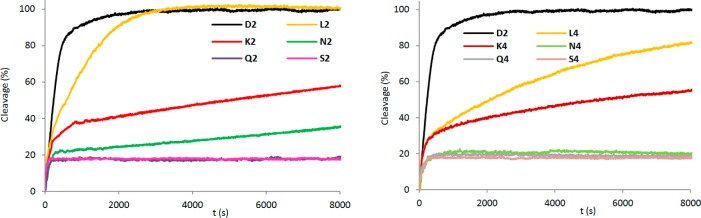
3′-Exonuclease (SVPDE) degradation of singly (left, 3′-CAC BAT ACG) and doubly modified (right, 3′-CAC BAB ACG) C5-functionalized LNA and reference strands. Nuclease degradation studies were performed in magnesium buffer (50 mM Tris-HCl, 10 mM Mg2+, pH 9.0) by using 3.3 μM ONs and 0.03 U of SVPDE. Data depicted in the left panel have been previously reported in ref (22).
Fluorescence Properties of C5-Functionalized LNA
Steady-state fluorescence emission spectra of ONs modified with C5-fluorophore-functionalized LNA monomers and the corresponding duplexes with complementary or mismatched DNA targets were recorded to gain further insight into the binding modes of the fluorophores. In addition to studying the fluorescence properties of B1 and B4 probes in the presence or absence of matched/mismatched DNA/RNA (Figures S2 and S3, Supporting Information), we also studied centrally modified 13-mer ONs (V5–Z5 series) and their duplexes with matched/mismatched DNA targets (Figure 3). The thermal denaturation characteristics of these ONs (Table S4, Supporting Information) closely follow those of the singly modified 9-mer ONs: i.e. (i) the corresponding duplexes with DNA/RNA targets are less stable than unmodified reference duplexes (only Z5-modified duplexes are slightly more stable) and (ii) W5–Y5 display very poor thermal discrimination of mismatched DNA targets, while V5 and Z5 display similar binding specificity as the unmodified reference strands.
Figure 3.

Steady-state fluorescence emission spectra of single-stranded B5 ONs (5′-CG CAA CBC AAC GC) and the corresponding duplexes with fully complementary or singly mismatched DNA strands (mismatched nucleotide opposite of modification is specified). Conditions: λex 344 nm (V5/Y5/Z5), λex 375 nm (W5), λex 448 nm (X5); T = 5 °C. Note that different axis scales are used.
V/Y/Z-modified duplexes exhibit emission peaks of varying broadness at ∼390/402 nm (V), ∼381/398 nm (Y), and ∼376/396/416 nm (Z), respectively, which are typical emission maxima for electronically isolated pyrene units (Figure 3). As expected for duplexes modified with the 1-ethynylpyrene fluorophore,35 the duplex between W5 and complementary DNA exhibits broad red-shifted emission centered around ∼465 nm, which is indicative of strong electronic coupling between the pyrene and nucleobase moiety. Interestingly, the emission intensities of pyrene-functionalized ONs V5/W5/Y5/Z5 increase upon binding to complementary DNA (∼3.8-, ∼3.9-, ∼3.1-, and ∼51-fold increases for V5, W5, Y5 and Z5, respectively, Figure 3). In contrast, much smaller increases are observed upon hybridization with mismatched DNA targets. The intensity differences are most likely due to different positioning of the pyrene moieties in matched vs mismatched duplexes, in a manner similar to that proposed for the corresponding DNA analogues of monomers V/W/Y/Z.18g,18j,35b Thus, the pyrene moieties likely point into the nonquenching environment of the major groove in matched duplexes (nucleobase in anti conformation), while they are intercalating into mismatched duplexes leading to nucleobase-mediated quenching36 of pyrene fluorescence (nucleobase in syn conformation). Regardless of the mechanism, the results strongly suggest that V/W/Y/Z-modified ONs are promising probes for the detection of nucleic acid targets and fluorescent discrimination of single-nucleotide polymorphisms (SNPs).
Duplexes between perylene-functionalized X5 and complementary DNA display broad emission maxima at ∼487 and ∼517 nm, whereas the emission maxima are red-shifted by ∼10 nm in mismatched DNA duplexes (Figure 3). The emission intensity of X5 does not change significantly upon binding with complementary DNA but is reduced by 30–60% upon binding to mismatched targets, presumably due to nucleobase-mediated quenching of intercalating perylene units.
Recently, we examined the SNP-discriminating properties of V-modified ONs and compared them to probes modified with the corresponding DNA analogue of monomer V.37 We found that there are distinct advantages to conjugating the 1-pyrenecarboxamido fluorophore to the C5-position of LNA-U, including (i) greater increases in fluorescence intensity upon target binding, (ii) formation of more brightly fluorescent duplexes, and (iii) stricter fluorescent discrimination of DNA targets with SNP sites. Force field calculations suggested that the extreme pucker of the LNA skeleton influences the rotational freedom around the N1–C1′ glycosyl bond due to steric hindrance between H6 and H3′, leading to different positioning and modulated photophysical properties of the C5-fluorophore relative to the analogous DNA monomer.37
Direct comparison of Y5 and Z5 with the corresponding DNA-based probes Y5d and Z5d(18j) (for structures of the DNA analogues of V/Y/Z monomers, see Figure S4 in the Supporting Information) reveals similar advantages (Figure 4). Thus, the results suggest that conjugation of fluorophores to the C5 position of LNA monomers is an effective strategy toward the generation of building blocks with interesting photophysical properties.
Figure 4.
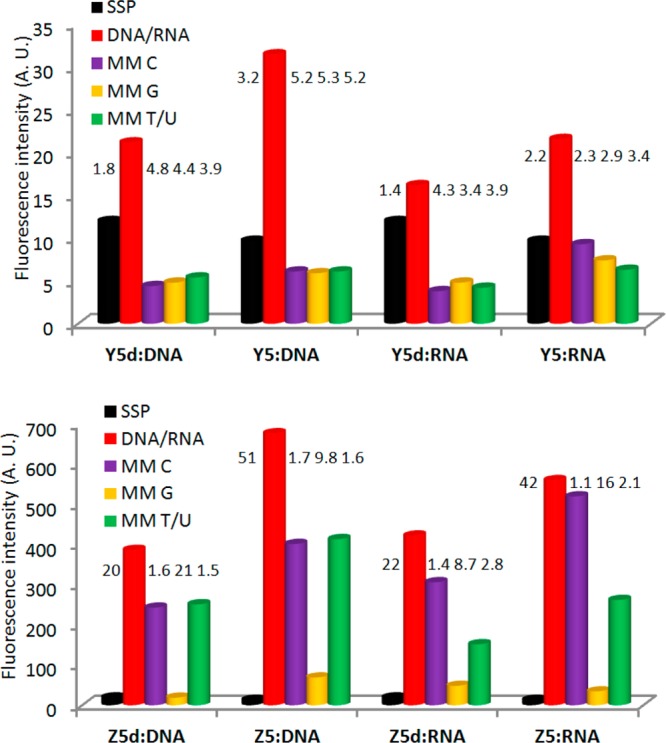
Fluorescence intensity of single-stranded probes (SSPs) in the presence or absence of complementary or singly mismatched DNA/RNA strands. Mismatched nucleotide opposite of modification is specified. Hybridization-induced increases and discrimination factors (defined as the fluorescence intensity of duplexes with complementary DNA/RNA divided by the intensity of SSPs or duplexes with mismatched DNA/RNA, respectively) are given above the corresponding histograms. Intensity recorded at λem 382 nm for Y5/Y5d and λem 377 nm for Z5/Z5d at T = 5 °C. Note that different y-axis scales are used.
The large increases in fluorescence intensity upon hybridization of Z5 with complementary targets prompted us to examine the potential of Z-modified ONs as hybridization probes38 in greater detail. Three additional 13-mer ONs were therefore prepared in which the nucleotides flanking monomer Z were systematically varied (Table S4, Supporting Information). Although the increases in fluorescence intensities upon hybridization with DNA targets are less pronounced (4–17-fold, Figure 5) and the resulting duplexes are significantly less fluorescent than with Z5,39 moderate to excellent fluorescent discrimination of mismatched DNA targets is observed with all Z-modified probes (discrimination factors from 1.5 to 48, Figure 5). Accordingly, Z-modified ONs constitute an interesting addition to the existing pool of pyrene-based hybridization probes.18j,40
Figure 5.
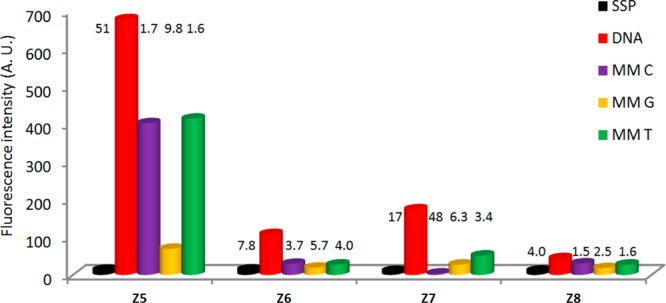
Fluorescence intensity of single-stranded probes (SSPs) in the presence or absence of complementary or singly mismatched DNA strands. Mismatched nucleotide opposite of modification is specified. Hybridization-induced increases and discrimination factors are given above the corresponding histograms. Target: 5′-CG CAA BZB AAC GC, where B = C/A/G/T for ON5–8, respectively. Intensity recorded at λem 377 nm at T = 5 °C.
Conclusion
The hybridization characteristics and enzymatic stabilities of ONs modified with LNA uridines can be extensively modulated through conjugation of different entities to the C5 position of the nucleobase. Only two extra steps, relative to conventional LNA synthesis, are needed. Monomers that are conjugated to small alkynyl substituents result in significantly greater target affinity and specificity than regular LNA monomers. Conjugation of bulky moieties confers complete protection against 3′-exonucleases but also decreases target affinity. ONs modified with C5-fluorophore-functionalized LNA uridines display improved photophysical characteristics relative to the corresponding DNA-based probes, including greater hybridization-induced increases in fluorescence intensity, formation of more brightly fluorescent duplexes, and strict fluorescent discrimination of single-nucleotide polymorphisms.20b These properties render C5-functionalized LNA as promising building blocks for RNA-targeting applications and nucleic acid diagnostics, although concerns regarding the potential toxicity of C5-alkynyl entities41 must be alleviated prior to biological evaluation.
The present study suggests that it is possible to combine desirable properties from LNA (target affinity/specificity) and C5-functionalized DNA monomers (positioning of functional entities in the major groove) into one compound class. The subsequent article in this issue demonstrates that the properties of ONs modified with α-l-LNA uridines also can be modulated through functionalization of the nucleobase.42 We therefore anticipate that C5 functionalization of pyrimidines will serve as a general and synthetically straightforward approach for modulation of pharmocodynamic and pharmacokinetic properties of oligonucleotides modified with LNA8−10,13 or other conformationally restricted monomers.1−7,16 Efforts aiming at delineating whether the biophysical properties of LNA purines also can be improved through functionalization of the nucleobase are ongoing, and the results from these studies will be reported shortly.
Experimental Section
Representative Protocol for EDC-Mediated Coupling of Carboxylic Acids with Propargylamine to Furnish Alkynes Ae–Ag43 Used in Sonogashira Couplings
The appropriate carboxylic acid and 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride (EDC·HCl) were added to propargylamine in anhydrous CH2Cl2, and the reaction mixture was stirred under an argon atmosphere until analytical TLC indicated full conversion (quantities, volumes, reaction time, and temperature are specified below). At this point, CH2Cl2 (40 mL) was added and the organic phase was washed with 5% aqueous citric acid (2 × 20 mL) and H2O (20 mL). The aqueous phase was back-extracted with CH2Cl2 (25 mL), the combined organic layers were concentrated to dryness, and the resulting residue was purified by column chromatography (0–4% MeOH in CH2Cl2) to afford the desired product (quantities and yields specified below).
N-(Prop-2-ynyl)-2-(adamant-1-yl)ethanamide (Ae)
1-Adamantaneacetic acid (1.60 g, 8.24 mmol), EDC·HCl (1.80 g, 9.42 mmol), and propargylamine (0.60 mL, 9.38 mmol) in anhydrous CH2Cl2 (30 mL) were set up, reacted (14 h at room temperature), and worked up, and the product was purified as described above to afford alkyne Ae(43) (1.40 g, 76%) as a white solid material: Rf = 0.8 (10% MeOH/CH2Cl2, v/v); MALDI-HRMS m/z 254.1527 ([M + Na]+, C15H21NO·Na+, calcd 254.1515); 1H NMR (CDCl3) δ 5.56 (br s, 1H, NH), 4.01 (dd, 2H, J = 5.2 Hz, 2.5 Hz, CH2NH), 2.19 (t, 1H, J = 2.5 Hz, HC≡C), 1.93–1.97 (m, 3H, 3 × CH), 1.92 (s, 2H, CH2CONH), 1.58–1.70 (12H, 6 × CH2-ada); 13C NMR (CDCl3) δ 170.8, 80.0, 71.6 (HC≡C), 51.6 (CH2CONH), 42.8 (CH2-ada), 37.0 (CH2-ada), 33.1, 29.2 (CH2NH), 28.9 (CH-ada).
N-(Prop-2-ynyl)dodecanamide (Af)
Lauric acid (dodecanoic acid, 1.60 g, 8.00 mmol), EDC·HCl (1.80 g, 9.42 mmol), and propargylamine (0.60 mL, 9.38 mmol) in anhydrous CH2Cl2 (30 mL) were set up, reacted (12 h at room temperature), and worked up, and the product was purified as described above to afford alkyne Af(43) (1.40 g, 76%) as a white solid material: Rf = 0.8; MALDI-HRMS m/z 260.1978 ([M + Na]+, C15H27NO·Na+, calcd 260.1985); 1H NMR (CDCl3) δ 5.66 (bs, ex, 1H, NH), 4.03 (dd, 2H, J = 5.0 Hz, 2.5 Hz, CH2C≡CH), 2.19 (t, 1H, J = 2.5 Hz, HC≡C), 2.16 (2d, 2H, J = 7.7 Hz, CH2CO), 1.61 (quintet, 2H, J = 7.7 Hz, CH2CH2CO), 1.20–1.30 (m, 16H, 8 × CH2), 0.85 (t, 3H, J = 6.5 Hz, CH3); 13C NMR (CDCl3) δ 172.9, 79.9, 71.7, 36.7 (CH2CO), 32.1 (CH2), 29.8 (CH2), 29.7 (CH2), 29.53 (CH2), 29.47 (CH2), 29.4 (CH2C≡CH), 25.8 (CH2CH2CO), 22.9, 14.3 (CH3). 1H NMR data are in agreement with literature reports.44
N-(Prop-2-ynyl)octadecanamide (Ag)
Stearic acid (octadecanoic acid, 1.42 g, 5.00 mmol), EDC·HCl (1.15 g, 6.00 mmol), and propargylamine (0.40 mL, 6.25 mmol) in anhydrous CH2Cl2 (30 mL) were set up, reacted (12 h at room temperature), and worked up, and the product was purified as described above to afford alkyne Ag(43) (1.40 g, 87%) as a white solid material: Rf = 0.1 (CH2Cl2); FAB-HRMS m/z 321.3020 ([M]+, C21H39NO+, calcd 321.3032); 1H NMR (CDCl3) δ 5.54 (br s, 1H, ex, NH), 4.03 (dd, 2H, J = 5.5 Hz, 2.5 Hz, CH2NH), 2.20 (t, 1H, J = 2.5 Hz, HC≡C), 2.17 (t, 2H, J = 7.5 Hz, CH2CO), 1.61 (quintet, 2H, J = 7.5 Hz, CH2CH2CO), 1.23–1.27 (m, 28H, 14 × CH2), 0.87 (t, 1H, J = 7.0 Hz, CH3); 13C NMR (CDCl3) δ 172.7, 79.7, 71.5 (HC≡C), 36.5 (CH2CO), 31.9 (CH2), 29.69 (CH2), 29.684 (CH2), 29.677 (CH2), 29.66 (CH2), 29.65 (CH2), 29.64 (CH2), 29.59 (CH2), 29.46 (CH2), 29.35 (CH2), 29.31 (CH2), 29.2 (CH2), 29.1 (CH2NH), 25.5 (CH2CH2CONH), 22.7 (CH2), 14.1 (CH3). The NMR data are in excellent agreement with literature reports.45
(1S,3R,4R,7S)-7-Hydroxy-1-hydroxymethyl-3-(5-iodoracil-1-yl)-2,5-dioxabicyclo[2.2.1]heptane (2)
To a solution of nucleoside 1(23) (4.00 g, 15.62 mmol) in glacial AcOH (150 mL) were added iodine (2.40 g, 9.44 mmol) and ceric ammonium nitrate (4.26 g, 7.77 mmol), and the reaction mixture was stirred at 80 °C for ∼40 min. After it was cooled to room temperature, the mixture was evaporated to dryness and the resulting residue was suspended in MeOH (150 mL). The mixture was concentrated and the resulting residue adsorbed on silica gel and purified by column chromatography (0–15% MeOH/CH2Cl2, v/v) to afford nucleoside 2 (5.21 g, 87%) as a white solid material: Rf = 0.4 (10% MeOH/CH2Cl2, v/v); FAB-HRMS m/z 382.9732 ([M + H]+, C10H11IN2O6H+, calcd 382.9735); 1H NMR (DMSO-d6) δ 11.69 (s, 1H, ex, NH), 8.13 (s, 1H, H6), 5.65 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.40 (s, 1H, H1′), 5.27 (t, 1H, ex, J = 5.4 Hz, 5′–OH), 4.14 (s, 1H, H2′), 3.91 (d, 1H, J = 4.5 Hz, H3′), 3.79–3.82 (d, 1H, J = 8.5 Hz, H5″), 3.68–3.75 (m, 2H, H5′), 3.58–3.62 (d, 1H, J = 8.5 Hz, H5″); 13C NMR (DMSO-d6) δ 160.5, 149.6, 143.5 (C6), 88.8, 86.4 (C1′), 78.5 (C2′), 70.8 (C5″), 68.4, 68.2 (C3′), 55.3 (C5′).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-(5-iodoracil-1-yl)-2,5-dioxabicyclo[2.2.1]heptane (3)
Diol 2 (5.00 g, 13.0 mmol) was coevaporated with anhydrous pyridine (100 mL) and redissolved in anhydrous pyridine (100 mL). To this was added 4,4′-dimethoxytrityl chloride (DMTr-Cl, 5.75 g, 16.9 mmol), and the reaction mixture was stirred at room temperature for 16 h, whereupon solvent was evaporated off. The residue was dissolved in CH2Cl2 (300 mL) and washed with saturated aqueous NaHCO3 (300 mL). The aqueous layer was back-extracted with CH2Cl2 (2 × 100 mL), and the combined organic layer was washed with saturated aqueous NaHCO3 (100 mL), dried (Na2SO4), evaporated to near dryness, and coevaporated with toluene/absolute EtOH (100 mL, 1/2, v/v). The resulting residue was purified by column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford key intermediate 3 (7.52 g, 82%) as a slightly yellow solid material: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); FAB-HRMS m/z 684.0977 ([M]+, C31H29IN2O8+, calcd 684.0969); 1H NMR (DMSO-d6) δ 11.74 (s, 1H, ex, NH), 7.96 (s, 1H, H6), 7.23–7.45 (m, 9H, Ar), 6.91 (d, 4H, J = 8.5 Hz, Ar), 5.70 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.44 (s, 1H, H1′), 4.24 (s, 1H, H2′), 4.07 (d, 1H, J = 4.5 Hz, H3′), 3.74–3.76 (m, 8H, 2 × OCH3, 2 × H5″), 3.39–3.42 (d, 1H, J = 11.0 Hz, H5′), 3.28–3.31 (d, 1H, J = 11.0 Hz, H5′, overlap with H2O); 13C NMR (DMSO-d6) δ 160.5, 158.1, 158.0, 149.7, 144.6, 142.7 (C6), 135.3, 135.2, 129.7 (Ar), 129.6 (Ar), 127.9 (Ar), 127.5 (Ar), 126.6 (Ar), 113.3 (Ar), 87.5, 86.9 (C1′), 85.6, 78.8 (C2′), 71.3 (C5″), 69.4 (C3′), 68.9, 58.9 (C5′), 55.0 (CH3O).
Representative Protocol for Sonogashira Couplings (4a–j)
Key intermediate 3, Pd(PPh3)4, CuI, and alkyne were added to anhydrous DMF (quantities and volumes specified below), and the reaction chamber was degassed and placed under an argon atmosphere. To this was added Et3N, and the reaction mixture was stirred in the dark until analytical TLC indicated full conversion of the starting material (reaction time and temperature specified below), whereupon solvents were evaporated off. The resulting residue was taken in up in EtOAc (100 mL) and washed with brine (2 × 50 mL) and saturated aqueous NaHCO3 (50 mL). The combined aqueous phase was back-extracted with EtOAc (100 mL), the combined organic phase dried (Na2SO4) and evaporated to dryness, and the resulting residue purified by column chromatography (0–5% MeOH in CH2Cl2 (v/v) to afford the desired product.
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(trimethylsilylethynyl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4a)
Nucleoside 3 (0.68 g, 1.00 mmol), Pd(PPh3)4 (120 mg, 0.10 mmol), CuI (40 mg, 0.20 mmol), trimethylsilylacetylene (294 mg, 0.42 mL, 3.00 mmol), and Et3N (0.60 mL, 4.27 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred stirred at room temperature for 12 h. After workup and purification, nucleoside 4a (0.56 g, 85%) was obtained as a brown solid material. Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 677.2297 ([M + Na]+, C36H38N2O8Si·Na+, calcd 677.2290); 1H NMR (DMSO-d6) δ 11.69 (s, 1H, ex, NH), 7.86 (s, 1H, H6), 7.20–7.45 (m, 9H, Ar), 6.89 (d, 4H, J = 8.5 Hz, Ar), 5.72 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.39 (s, 1H, H1′), 4.28 (s, 1H, H2′), 4.08 (d, 1H, J = 4.5 Hz, H3′), 3.75–3.80 (m, 2H, 2 × H5″), 3.73 (s, 6H, 2 × OCH3), 3.40–3.43 (d, 1H, J = 11.0 Hz, H5′), 3.30–3.33 (d, 1H, J = 11.0 Hz, H5′, overlap with H2O signal), −0.04 (s, 9H, Me3Si); 13C NMR (DMSO-d6) δ 161.5, 158.05, 158.03, 148.9, 144.6, 142.5 (C6), 135.4, 135.2, 129.7 (Ar), 129.5 (Ar), 127.8 (Ar), 127.6 (Ar), 126.6 (Ar), 113.2 (Ar), 97.9, 97.5, 96.9, 87.7, 87.1 (C1′), 85.5, 78.6 (C2′), 71.3 (C5″), 69.5 (C3′), 58.9 (C5′), 55.0 (CH3O), −0.47 (Me3Si).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-3-(5-ethynyluracil-1-yl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (4b)
To a solution of nucleoside 4a (0.53 g, 0.81 mmol) in THF (20 mL) was added tetrabutylammonium fluoride in THF (TBAF, 1 M, 1.2 mL, 1.2 mmol), and the reaction mixture was stirred at room temperature for 2 h. EtOAc (50 mL) was added and the organic phase washed with brine (2 × 30 mL) and H2O (30 mL). The aqueous phase was back-extracted with EtOAc (30 mL). The combined organic phase was dried (Na2SO4) and evaporated to dryness and the resulting residue purified by column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford nucleoside 4b (0.37 g, 78%) as a lightly brown solid material: Rf = 0.4 (5% MeOH/CH2Cl2, v/v); ESI-HRMS m/z 621.1666 ([M + K]+, C33H30N2O8·K+, calcd 621.1634); 1H NMR (DMSO-d6) δ 11.70 (s, 1H, ex, NH), 7.88 (s, 1H, H6), 7.21–7.45 (m, 9H, Ar), 6.88–6.92 (m, 4H, Ar), 5.70 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.45 (s, 1H, H1′), 4.27 (s, 1H, H2′), 4.05 (d, 1H, J = 4.5 Hz, H3′), 3.95 (s, 1H, CH), 3.77 (s, 2H, 2 × H5″), 3.75 (s, 6H, 2 × CH3O), 3.43–3.45 (d, 1H, J = 11.0 Hz, H5′), 3.28–3.31 (d, 1H, J = 11.0 Hz, H5′, overlap with H2O signal); 13C NMR (DMSO-d6) δ 161.7, 158.1, 149.0, 144.6, 142.2 (C6), 135.3, 135.2, 129.7 (Ar), 129.6 (Ar), 127.9 (Ar), 127.5 (Ar), 126.7 (Ar), 113.3 (Ar), 97.2, 87.5, 86.9 (C1′), 85.7, 83.6, 78.9 (C2′), 76.2, 71.4 (C5″), 69.4 (C3′), 59.0 (C5′), 55.0 (CH3O).
(1R,3R,4R,7S)-3-[5-(3-Benzoyloxypropyn-1-yl)uracil-1-yl]-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (4c)
Nucleoside 3 (0.50 g, 0.73 mmol), Pd(PPh3)4 (90 mg, 0.07 mmol), CuI (30 mg, 0.14 mmol), prop-2-ynyl benzoate46 (180 mg, 1.12 mmol), and Et3N (0.40 mL, 2.84 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the reaction mixture was stirred at room temperature for 12 h. After workup and purification, nucleoside 4c (0.37 g, 70%) was obtained as a light brown solid material: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 739.2289 ([M + Na]+, C41H36N2O10·Na, calcd 739.2262); 1H NMR (DMSO-d6) δ 11.72 (s, 1H, ex, NH), 7.91–7.93 (d, 2H, J = 7.7 Hz, Bzortho), 7.89 (s, 1H, H6), 7.65–7.70 (t, 1H, J = 7.7 Hz, Bzpara), 7.49–7.54 (t, 2H, J = 7.7 Hz, Bzmeta), 7.42–7.46 (d, 2H, J = 8.5 Hz, DMTr), 7.28–7.34 (m, 6H, DMTr), 7.18–7.22 (t, 1H, J = 7.5 Hz, DMTr), 6.87–6.91 (2d, 4H, J = 9.0 Hz, DMTr), 5.74 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.42 (s, 1H, H1′), 4.93–4.97 (d, 1H, J = 16.0 Hz, CH2OBz), 4.86–4.90 (d, 1H, J = 16.0 Hz, CH2OBz), 4.25 (s, 1H, H2′), 4.10 (d, 1H, J = 4.5 Hz, H3′), 3.78–3.82 (d, 1H, J = 8.0 Hz, H5″), 3.75–3.76 (d, 1H, J = 8.0 Hz, H5″), 3.71 (s, 6H, 2 × CH3O), 3.53–3.56 (d, 1H, J = 11.0 Hz, H5′), 3.27–3.30 (d, 1H, J = 11.0 Hz, H5′, overlap with H2O); 13C NMR (DMSO-d6) δ 164.9, 161.6, 158.1, 158.0, 149.0, 144.6, 142.7 (C6), 135.4, 135.0, 133.5 (CBzpara), 129.7 (DMTr), 129.6 (DMTr), 129.2 (CBzortho), 129.0, 128.7 (CBzmeta), 127.8 (DMTr), 127.5 (DMTr), 126.6 (DMTr), 113.20 (DMTr), 113.17 (DMTr), 96.8, 87.6, 86.9 (C1′), 86.3, 85.6, 79.2, 78.7 (C2′), 71.3 (C5″), 69.4 (C3′), 58.7 (C5′), 54.9 (CH3O), 53.1 (CH2OBz).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(3-trifluoroacetylaminopropyn-1-yl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4d)
Nucleoside 3 (0.50 g, 0.73 mmol), Pd(PPh3)4 (90 mg, 0.07 mmol), CuI (30 mg, 0.14 mmol), 2,2,2-trifluoro-N-(prop-2-ynyl)acetamide47 (180 mg, 1.46 mmol), and Et3N (0.40 mL, 2.84 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 12 h. After workup and purification, nucleoside 4d (0.41 g, 80%) was obtained as a brown solid material: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 714.2247 ([M+Li]+, C36H32F3N3O9·Li+, calcd 714.2245); 1H NMR (DMSO-d6) δ 11.69 (s, 1H, ex, NH(U)), 9.95 (t, 1H, ex, J = 5.5 Hz, NHCH2), 7.78 (s, 1H, H6), 7.22–7.46 (m, 9H, Ar), 6.90 (dd, 4H, J = 9.0 Hz, 3.5 Hz, Ar), 5.73 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.41 (s, 1H, H1′), 4.25 (s, 1H, H2′), 3.97–4.10 (m, 3H, H3′, CH2NH), 3.79–3.83 (2d, 2H, J = 8.0 Hz, 2 × H5″), 3.74 (s, 6H, 2 × CH3O), 3.56–3.58 (d, 1H, J = 11.0 Hz, H5′), 3.26–3.28 (d, 1H, J = 11.0 Hz, H5′); 13C NMR (DMSO-d6) δ 161.6, 158.11, 158.06, 155.9 (q, J = 36.1 Hz, COCF3), 149.0, 144.6, 142.1 (C6), 135.4, 134.9, 129.8 (Ar), 129.6 (Ar), 127.8 (Ar), 127.5 (Ar), 126.6 (Ar), 115.6 (q, J = 287 Hz, CF3), 113.22 (Ar), 113.20 (Ar), 97.2, 87.6, 87.2, 86.9 (C1′), 85.6, 78.7 (C2′), 75.4, 71.3 (C5″), 69.6 (C3′), 59.1 (C5′), 55.0 (CH3O), 29.4 (CH2NH); 19F (DMSO-d6, 470 MHz) δ −74.7.
(1R,3R,4R,7S)-3-[5-(3-(1-Adamantylmethylcarbonyl)aminopropyn-1-yl)uracil-1-yl]-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (4e)
Nucleoside 3 (0.50 g, 0.73 mmol), Pd(PPh3)4 (90 mg, 0.05 mmol), CuI (30 mg, 0.10 mmol), N-(prop-2-ynyl)-1-adamantaneacetamide (220 mg, 1.00 mmol), and Et3N (0.40 mL, 2.84 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 12 h. After workup and purification, nucleoside 4e (0.43 g, 76%) was obtained as a white solid material: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 810.3330 ([M + Na]+, C46H49N3O9·Na+, calcd 810.3361); 1H NMR (DMSO-d6) δ 11.66 (s, 1H, ex, NH(U)), 8.00 (t, 1H, ex, J = 5.5 Hz, NHCO), 7.75 (s, 1H, H6), 7.42–7.45 (m, 2H, Ar), 7.23–7.34 (m, 7H, Ar), 6.89–6.92 (2d, 4H, J = 9.0 Hz, Ar), 5.72 (d, 1H, ex, J = 4.5 Hz, 3′–OH), 5.42 (s, 1H, H1′), 4.24 (s, 1H, H2′), 4.01 (d, 1H, J = 4.5 Hz, H3′), 3.87–3.93 (dd, 1H, J = 17.5 Hz, 5.5 Hz, CH2NHCO), 3.82–3.87 (dd, 1H, J = 17.5 Hz, 5.5 Hz, CH2NHCO), 3.78–3.82 (2d, 2H, J = 8.2 Hz, H5″), 3.75 (s, 3H, CH3O), 3.74 (s, 3H, CH3O), 3.54–3.56 (d, 1H, J = 11.0 Hz, H5′), 3.27–3.30 (d, 1H, J = 11.0 Hz, H5′, overlap with H2O), 1.82–1.87 (m, 5H, 3× ada-CH/CH2CONH), 1.53–1.63 (m, 12H, 6 × ada-CH2); 13C NMR (DMSO-d6) δ 169.6, 161.7, 158.12, 158.07, 149.0, 144.7, 141.5 (C6), 135.4, 134.9, 129.8 (Ar), 129.6 (Ar), 127.9 (Ar), 127.5 (Ar), 126.7 (Ar), 113.24 (Ar), 113.23 (Ar), 97.7, 89.6, 87.5, 86.9 (C1′), 85.6, 78.8 (C2′), 74.2, 71.4 (C5″), 69.6 (C3′), 59.1 (C5′), 54.9 (CH3O), 49.5 (CH2CONH), 42.0 (ada-CH2), 36.4 (ada-CH2), 32.3, 28.4 (CH2NHCO), 28.0 (ada-CH).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-3-[5-(3-dodecanoylaminopropyn-1-yl)uracil-1-yl]-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (4f)
Nucleoside 3 (200 mg, 0.29 mmol), Pd(PPh3)4 (34 mg, 0.03 mmol), CuI (11 mg, 0.06 mmol), N-(prop-2-ynyl)lauroylamide (110 mg, 0.44 mmol), and Et3N (0.18 mL, 1.29 mmol) in DMF (3 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 15 h. After workup and purification, nucleoside 4f (202 mg, 87%) was obtained as a white solid material: Rf = 0.2 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 816.3835 ([M + Na]+, C46H55N3O9·Na+, calcd 816.3831); 1H NMR (DMSO-d6) δ 11.67 (s, 1H, ex, NH(U)), 8.08 (t, 1H, J = 5.4 Hz, NHCH2), 7.76 (s, 1H, H6), 7.22–7.46 (m, 9H, Ar), 6.87–6.96 (m, 4H, Ar), 5.72 (d, 1H, ex, J = 4.7 Hz, 3′–OH), 5.42 (s, 1H, H1′), 4.25 (s, 1H, H2′), 4.03 (d, 1H, J = 4.7 Hz, H3′), 3.88–3.94 (dd, 1H, J = 12.4 Hz, 5.4 Hz, CH2NHCO), 3.81–3.88 (dd, 1H, J = 12.4 Hz, 5.4 Hz, CH2NHCO), 3.78–3.82 (2d, 2H, J = 8.0 Hz, H5″), 3.75 (s, 6H, 2 × CH3O), 3.55–3.57 (d, 1H, J = 10.9 Hz, H5′), 3.27–3.30 (d, 1H, J = 10.9 Hz, H5′, overlap with H2O), 2.05 (t, 2H, J = 7.4 Hz,CH2CONH), 1.43–1.48 (m, 2H, CH2CH2CONH), 1.19–1.28 (m, 16H, 8 × CH2), 0.85 (t, 3H, J = 7.0 Hz, CH3); 13C NMR (DMSO) δ 171.7, 161.7, 158.11, 158.06, 149.0, 144.6, 141.6 (C6), 135.4, 134.9, 129.8 (Ar), 129.6 (Ar), 127.9 (Ar), 127.5 (Ar), 126.6 (Ar), 113.22 (Ar), 113.21 (Ar), 97.7, 89.5, 87.5, 86.9 (C1′), 85.6, 78.8 (C2′), 74.3, 71.4 (C5″), 69.6 (C3′), 59.0 (C5′), 55.0 (CH3O), 35.0 (CH2CONH), 31.2 (CH2), 28.94 (CH2), 28.92 (CH2), 28.8 (CH2), 28.7 (CH2), 28.63 (CH2), 28.60 (CH2), 28.52 (CH2NHCO), 25.0 (CH2CH2CONH), 22.0 (CH2CH3), 13.9 (CH3).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(3-octadecanoylaminopropyn-1-yl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4g)
Nucleoside 3 (0.34 g, 0.50 mmol), Pd(PPh3)4 (60 mg, 0.05 mmol), CuI (20 mg, 0.10 mmol), N-(prop-2-ynyl)stearamide (0.28 g, 1.00 mmol), and Et3N (0.30 mL, 2.13 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at 40 °C for 6 h. After workup and purification, nucleoside 4g (0.29 g, 68%) was obtained as a brown solid material, which was used in the next step without further purification: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); FAB-HRMS m/z 877.4844 ([M]+, C52H67N3O9+, calcd 877.4877); 1H NMR (CDCl3) δ 9.45 (br s, 1H, ex, NH(U)), 8.05 (s, 1H, H6), 7.22–7.50 (m, 9H, Ar), 6.85–6.89 (dd, 4H, J = 9.0 Hz, 1.5 Hz, Ar), 5.56–5.59 (m, 2H, 1 ex, H1′, NHCH2), 4.53 (s, 1H, H2′), 4.29 (s, 1H, H3′), 3.78–4.01 (m, 10H, 2 × H5″, CH2NH, 2 × CH3O), 3.53–3.57 (d, 1H, J = 11.0 Hz, H5′), 3.49–3.52 (d, 1H, J = 11.0 Hz, H5′), 3.35 (br s, 1H, ex, 3′–OH), 1.85–1.89 (m, 2H, CH2CONH), 1.44–1.51 (m, 2H, CH2CH2CONH), 1.23–1.28 (m, 28H, 14 × CH2), 0.89 (t, 3H, J = 6.5 Hz, CH3); 13C NMR (CDCl3) δ 172.7, 162.1, 158.69, 158.67, 148.6, 144.6, 141.9 (C6), 135.5, 135.4, 130.02 (Ar), 130.01 (Ar), 128.1 (Ar), 128.0 (Ar), 127.0 (Ar), 113.45 (Ar), 113.43 (Ar), 99.1, 89.9, 88.4, 87.4 (C1′), 86.6, 79.1 (C2′), 74.2, 71.8 (C5″), 70.5 (C3′), 58.5 (C5′), 55.3 (CH3O), 36.1 (CH2CONH), 31.9 (CH2), 29.9 (CH2NH), 29.69 (CH2), 29.68 (CH2), 29.66 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.34 (CH2), 29.33 (CH2), 25.4 (CH2CH2CONH), 22.7 (CH2), 14.1 (CH3). A small impurity of silicon grease was observed at ∼1 ppm in the 13C NMR.48
(1R,3R,4R,7S)-3-[5-(3-Cholesterylcarbonylaminopropyn-1-yl)uracil-1-yl]-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (4h)
Nucleoside 3 (0.34 g, 0.50 mmol), Pd(PPh3)4 (60 mg, 0.05 mmol), CuI (20 mg, 0.10 mmol), cholesterylprop-2-ynylcarbamate49 (0.47 g, 1.00 mmol), and Et3N (0.30 mL, 2.13 mmol) in DMF (8 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 12 h. After workup and purification, nucleoside 4h (0.27 g, 53%) was obtained as a brown solid material, which was used in the next step without further purification: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); FAB-HRMS m/z 1046.5560 ([M + Na]+, C62H77N3O10·Na+, calcd 1046.5507); 1H NMR (CDCl3) δ 9.40 (br s, 1H, ex, NH(U)), 7.97 (s, 1H, H6), 7.22–7.49 (m, 9H, Ar), 6.87 (d, 4H, J = 9.0 Hz, Ar), 5.58 (s, 1H, H1′), 5.34 (d, 1H, J = 5.0 Hz, HC=C-chol), 4.96 (bs, 1H, ex, NHCH2), 4.53 (s, 1H, H2′), 4.40–4.47 (m, 1H, HC-O-chol) 4.23 (bs, 1H, H3′), 3.83–3.98 (m, 4H, 2 × H5″, CH2NH), 3.80 (s, 3H, CH3O), 3.79 (s, 6H, 2 × CH3O), 3.58–3.61 (d, 1H, J = 11.0 Hz, H5′), 3.51–3.53 (d, 1H, J = 11.0 Hz, H5′), 3.27 (bs, 1H, ex, 3′-OH), 0.87–2.29 (m, 40 H, chol), 0.69 (s, 3H, CH3-chol); 13C NMR (CDCl3) δ 162.1, 158.63, 158.60, 155.5, 148.7, 144.5, 141.8 (C6), 139.8, 135.44, 135.39, 130.0 (Ar), 128.1 (Ar), 128.0 (Ar), 127.0 (Ar), 122.5 (=CH, chol), 113.4 (Ar), 99.2, 90.1, 88.4, 87.4 (C1′), 86.6, 79.1 (C2′), 74.7 (OCH-chol), 74.1, 71.9 (C5″), 70.6 (C3′), 58.6 (C5′), 56.7 (CH-chol), 56.2 (CH-chol), 55.2 (CH3O), 50.0 (CH-chol), 42.3, 39.8 (CH2-chol), 39.5 (CH2-chol), 38.5 (CH2-chol), 37.0 (CH2-chol), 36.5, 36.2 (CH2-chol), 35.8 (CH-chol), 31.9 (CH-chol/CH2NH), 28.2 (CH2-chol), 28.1 (CH2-chol), 28.0 (CH-chol), 24.3 (CH2-chol), 23.8 (CH2-chol), 22.8 (CH3-chol), 22.5 (CH3-chol), 21.0 (CH2-chol), 19.3 (CH3-chol), 18.7 (CH3-chol), 11.8 (CH3-chol). Signals at 41.4, 29.0, 22.6, 20.4, 19.4, 14.3, and 11.4 ppm, presumably arising from a small contamination of unreacted cholesterylprop-2-ynylcarbamate, were also observed in the 13C NMR spectrum.
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(2-(1-pyrenyl)ethynyl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4i)
Nucleoside 3 (0.34 g, 0.50 mmol), Pd(PPh3)4 (60 mg, 0.05 mmol), CuI (20 mg, 0.10 mmol), 1-ethynylpyrene50 (0.28 g, 1.00 mmol), and Et3N (0.30 mL, 2.84 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 12 h. Following workup and purification, nucleoside 4i (0.31 g, 80%) was obtained as a slightly yellow solid, which was used in the next step without further purification: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 805.2554 ([M + Na]+, C49H38N2O8·Na+, calcd 805.2520); 1H NMR (DMSO-d6) δ 11.89 (s, 1H, ex, NH), 8.35 (d, 1H, J = 8.5 Hz, Ar), 8.31–8.34 (d, 1H, J = 8.5 Hz, Ar), 8.25–8.28 (d, 1H, J = 8.0 Hz, Ar), 8.14–8.23 (m, 4H, H6, Ar), 8.09–8.12 (ap t, 1H, J = 8.0 Hz, Ar), 7.93 (d, 1H, J = 8.5 Hz, Ar), 7.67 (d, 1H, J = 8.0 Hz, Ar), 7.49–7.50 (m, 2H, Ar), 7.33–7.38 (m, 4H, Ar), 7.26–7.30 (ap t, 2H, J = 7.5 Hz, Ar), 7.03–7.06 (ap t, 1H, J = 7.5 Hz, Ar), 6.78–6.85 (m, 4H, Ar), 5.78 (d, 1H, ex, J = 4.5 Hz, 3′-OH), 5.53 (s, 1H, H1′), 4.34 (s, 1H, H2′), 4.24 (d, 1H, J = 4.5 Hz, H3′), 3.78–3.82 (2d, 2H, J = 7.5 Hz, H5″), 3.43–3.52 (m, 7H, OCH3, H5′), 3.38–3.42 (d, 1H, J = 11.0 Hz, H5′); 13C NMR (DMSO-d6) δ 161.7, 158.02, 157.95, 149.1, 144.4, 141.4 (C6), 135.4, 130.8, 130.7, 130.6, 130.4, 129.6 (Ar), 129.5 (Ar), 128.8 (Ar), 128.2 (Ar), 128.1 (Ar), 127.8 (Ar), 127.7 (Ar), 127.1 (Ar), 126.63 (Ar), 126.58 (Ar), 125.7 (Ar), 125.6 (Ar), 124.8 (Ar), 124.5 (Ar), 123.5, 123.3, 116.9, 113.17 (Ar), 113.16 (Ar), 98.2, 91.3, 88.2, 87.8, 87.2 (C1′), 85.6, 78.8 (C2′), 71.4 (C5″), 69.4 (C3′), 58.7 (C5′), 54.7 (OCH3), 54.6 (OCH3). A trace of pyridine was observed in the 13C NMR.48
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(2-(3-perylenyl)ethynyl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4j)
Nucleoside 3 (0.50 g, 0.73 mmol), Pd(PPh3)4 (90 mg, 0.07 mmol), CuI (30 mg, 0.14 mmol), 3-ethynylperylene51 (0.27 g, 1.00 mmol), and Et3N (0.40 mmol, 2.84 mmol) in DMF (10 mL) were reacted as described in the representative Sonogashira protocol, and the mixture was stirred at room temperature for 12 h. After workup and purification, nucleoside 4j (0.49 g, 80%) was obtained as a brown solid material: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 855.2675 ([M + Na]+, C53H40N2O8·Na+, calcd 855.2677); 1H NMR (DMSO-d6) δ 11.86 (s, 1H, ex, NH), 8.34–8.40 (m, 3H, Ar), 8.22 (d, 1H, J = 8.0 Hz, Ar), 8.13 (s, 1H, H6), 8.02 (d, 1H, J = 8.5 Hz, Ar), 7.79–7.85 (m, 2H, Ar), 7.55 (dt, 2H, J = 8.0 Hz, 2.0 Hz, Ar), 7.48–7.51 (m, 2H, Ar), 7.28–7.37 (m, 7H, Ar), 7.22 (d, 1H, J = 8.0 Hz, Ar), 7.09–7.12 (t, 1H, J = 7.5 Hz, Ar), 6.83–6.88 (m, 4H, Ar), 5.77 (d, 1H, ex, J = 4.5 Hz, 3′-OH), 5.51 (s, 1H, H1′), 4.33 (s, 1H, H2′), 4.22 (d, 1H, J = 4.5 Hz, H3′), 3.77–3.83 (m, 2H, 2 × H5″), 3.59 (s, 3H, OCH3), 3.56 (s, 3H, OCH3), 3.48–3.51 (d, 1H, J = 11.0 Hz, H5′), 3.35–3.39 (d, 1H, J = 11.0 Hz, H5′); 13C NMR (DMSO-d6) δ 161.7, 158.05, 158.00, 149.0, 144.4, 141.3 (C6), 135.43, 135.37, 134.2, 133.6, 130.9, 130.7, 130.4 (Ar), 130.1, 129.9, 129.6 (Ar), 129.5 (Ar), 128.5 (Ar), 128.2 (Ar), 127.9 (Ar), 127.7 (Ar), 127.60, 127.57, 127.47 (Ar), 127.0 (Ar), 126.9 (Ar), 126.7 (Ar), 125.7 (Ar), 121.5 (Ar), 121.2, 121.1 (Ar), 120.0 (Ar), 119.4, 113.2 (Ar), 98.1, 90.9, 88.5, 87.7, 87.1 (C1′), 85.6, 78.8 (C2′), 71.4 (C5″), 69.4 (C3′), 58.7 (C5′), 54.80 (CH3O), 54.75 (CH3O).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(1-(1-pyrenyl)-1H-1,2,3-triazol-4-yl)-uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4k)
To a solution of nucleoside 4b (0.25 g, 0.36 mmol) and 1-pyrenyl azide26 (110 mg, 0.45 mmol) in THF/H2O/t-BuOH (10 mL, 3/1/1, v/v/v) were added aqueous sodium ascorbate (1 M, 0.70 mL, 0.70 mmol) and aqueous CuSO4 (7.5%, w/v, 0.65 mL, 0.19 mmol). The solution was stirred at room temperature for 2 h, whereupon it was taken up in EtOAc (50 mL) and brine (50 mL). The layers were separated, and the organic phase was washed with saturated aqueous NaHCO3 (50 mL). The combined aqueous phase was back-extracted with EtOAc (50 mL). The combined organic phase was then dried (Na2SO4) and evaporated to dryness and the resulting residue purified by column chromatography (0–75% EtOAc in petroleum ether, v/v) to afford nucleoside 4k (230 mg, 76%) as a slightly yellow solid material: Rf = 0.4 (70% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 848.2712 ([M + Na]+, C49H39N5O8·Na+, calcd 848.2691); 1H NMR (DMSO-d6) δ 11.85 (s, 1H, ex, NH), 8.84 (s, 1H, H-Tz), 8.58 (s, 1H, H6), 8.48 (d, 1H, J = 8.0 Hz, Ar), 8.44–8.47 (d, 1H, J = 7.5 Hz, Ar), 8.40–8.43 (d, 1H, J = 8.0 Hz, Ar), 8.35–8.38 (d, 1H, J = 9.0 Hz, Ar), 8.32–8.35 (d, 1H, J = 9.0 Hz, Ar), 8.30 (d, 1H, J = 9.0 Hz, Ar), 8.16–8.20 (overlapping d and t, 2H, Ar), 7.79 (d, 1H, J = 8.0 Hz, Ar), 7.45–7.48 (d, 2H, J = 7.5 Hz, Ar), 7.28–7.39 (m, 6H, Ar), 7.17–7.20 (t, 1H, J = 7.5 Hz, Ar), 6.88–6.93 (2d, 4H, J = 9.0 Hz, Ar), 5.79 (d, 1H, ex, J = 4.5 Hz, 3′-OH), 5.64 (s, 1H, H1′), 4.43 (s, 1H, H2′), 4.13 (d, 1H, J = 4.5 Hz, H3′), 3.94–3.97 (d, 1H, J = 9.0 Hz, H5″), 3.86–3.90 (d, 1H, J = 9.0 Hz, H5″), 3.681 (s, 3H, CH3O), 3.675 (s, 3H, CH3O), 3.57–3.60 (d, 1H, J = 11.0 Hz, H5′), 3.34–3.37 (m, 1H, J = 11.0 Hz, H5′); 13C NMR (DMSO-d6) δ 161.3, 158.05, 158.02, 149.4, 144.6, 139.4, 135.3, 135.2, 135.0 (C6), 131.6, 130.6, 130.14, 130.09, 129.7 (Ar), 129.6 (Ar), 129.5 (Ar), 128.7 (Ar), 127.8 (Ar), 127.6 (Ar), 127.0 (Ar), 126.6 (Ar), 126.5 (Ar), 126.1 (Ar), 125.3, 125.1 (Ar), 124.8 (CH-Tz), 124.0, 123.7 (Ar), 123.3, 120.9 (Ar), 113.24 (Ar), 113.19 (Ar), 104.3, 87.6, 87.2 (C1′), 85.6, 79.0 (C2′), 71.5 (C5″), 70.0 (C3′), 59.5 (C5′), 54.9 (CH3O).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-7-hydroxy-3-[5-(1-(pyren-1-ylmethyl)-1H-1,2,3-triazol-4-yl)-uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (4l)
To a solution of nucleoside 4b (0.33 g, 0.56 mmol) and 1-azidomethylpyrene27 (200 mg, 0.78 mmol) in THF/H2O/t-BuOH (10 mL, 3/1/1, v/v/v) were added aqueous sodium ascorbate (1 M, 1.00 mL, 1.00 mmol) and aqueous CuSO4 (7.5%, w/v, 1.00 mL, 0.30 mmol). The reaction mixture was stirred at room temperature for 2 h, whereupon it was taken up in EtOAc (50 mL) and brine (50 mL). The layers were separated, and the organic phase was washed with saturated aqueous NaHCO3 (50 mL). The combined aqueous phase was back-extracted with EtOAc (50 mL). The combined organic phase was dried (Na2SO4) and evaporated to dryness and the resulting residue purified by column chromatography (0–75% EtOAc in petroleum ether, v/v) to afford nucleoside 4l (0.43 g, 91%) as a slightly yellow solid material: Rf = 0.4 (70% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 862.2869 ([M + Na]+, C50H41N5O8·Na+, calcd 862.2847); 1H NMR (DMSO-d6) δ 11.70 (s, 1H, ex, NH), 8.56 (d, 1H, J = 9.0 Hz, Ar), 8.44 (s, 1H, H-Tz), 8.29–8.36 (m, 5H, H6, Ar), 8.20–8.23 (d, 1H, J = 9.0 Hz, Ar), 8.17–8.20 (d, 1H, J = 9.0 Hz, Ar), 8.09–8.12 (t, 1H, J = 8.0 Hz, Ar), 8.07 (d, 1H, J = 8.0 Hz, Ar), 7.39–7.42 (d, 2H, J = 7.5 Hz, Ar), 7.22–7.33 (m, 6H, Ar), 7.09–7.12 (t, 1H, J = 7.5 Hz, Ar), 6.88 (d, 4H, J = 8.0 Hz, Ar), 6.40 (s, 2H, CH2Py), 5.69 (d, 1H, ex, J = 4.5 Hz, 3′-OH), 5.55 (s, 1H, H1′), 4.32 (s, 1H, H2′), 3.91–3.96 (m, 2H, H3′, H5″), 3.81–3.84 (d, 1H, J = 8.0 Hz, H5″), 3.70 (s, 3H, CH3O), 3.68 (s, 3H, CH3O), 3.50–3.54 (d, 1H, J = 11.0 Hz, H5′), 3.25–3.29 (d, 1H, J = 11.0 Hz, H5′); 13C NMR (DMSO-d6) δ 161.2, 158.1, 158.0, 149.2, 144.7, 138.9, 135.1, 134.2 (C6), 131.0, 130.7, 130.1, 129.7 (Ar), 129.6 (Ar), 129.1, 128.4, 128.2 (Ar), 127.8 (Ar), 127.6 (Ar), 127.5 (Ar), 127.2 (Ar), 126.52 (Ar), 126.45 (Ar), 125.7 (Ar), 125.5 (Ar), 125.0 (Ar), 124.0, 123.7, 122.7 (Ar), 122.4 (CH-Tz), 113.3 (Ar), 113.2 (Ar), 104.5, 87.5, 87.1 (C1′), 85.6, 79.0 (C2′), 71.6 (C5″), 70.0 (C3′), 59.8 (C5′), 54.9 (CH3O), 54.8 (CH3O), 50.7 (CH2Py).
Representative Procedure for O3′-Phosphitylation
Alcohols 5b–l were dried by coevaporation with anhydrous 1,2-dichloroethane and dissolved in anhydrous CH2Cl2. To this were added anhydrous N,N′-diisopropylethylamine (DIPEA) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (PCl-reagent) (quantities and volumes specified below), and the reaction mixture was stirred at room temperature until analytical TLC indicated complete conversion (2 h unless otherwise mentioned). Unless otherwise mentioned, the reaction mixture was diluted with CH2Cl2 (25 mL) and washed with aqueous NaHCO3 (2 × 10 mL), the combined aqueous phases were back-extracted with CH2Cl2 (2 × 10 mL), and the combined organic phases were dried (Na2SO4) and evaporated to dryness. Regardless of the workup procedure, the resulting residue was purified by silica gel column chromatography (typically 0–4% MeOH/CH2Cl2, v/v) and subsequent trituration from CH2Cl2 and petroleum ether to provide the target phosphoramidites.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-(5-ethynyluracil-1-yl)-2,5-dioxabicyclo[2.2.1]heptane (5b)
Nucleoside 4b (0.34 g 0.58 mmol), DIPEA (0.50 mL, 2.90 mmol), PCl-reagent (0.20 mL, 0.87 mmol), and anhydrous CH2Cl2 (10 mL) were mixed, reacted, worked up, and purified as described in the representative protocol to provide nucleoside 5b (0.38 g, 83%) as a white foam: Rf = 0.5 (2% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 805.2973 ([M + Na]+, C42H47N4O9P·Na+, calcd 805.2958); 31P NMR (CDCl3) δ 149.8, 149.3.
(1R,3R,4R,7S)-3-[5-(3-Benzoyloxypropyn-1-yl)uracil-1-yl]-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-2,5-dioxabicyclo[2.2.1]heptane (5c)
Nucleoside 4c (0.30 g, 0.42 mmol), DIPEA (300 μL, 1.67 mmol), PCl-reagent (121 μL, 0.54 mmol), and anhydrous CH2Cl2 (5 mL) were mixed and reacted (5 h) as described above. At this point the reaction mixture was concentrated to one-third volume and diluted with diethyl ether (100 mL) and the organic phase sequentially washed with H2O (35 mL), H2O/DMF (70 mL, 1/1, v/v), H2O (35 mL), and brine (35 mL). The organic phase was evaporated to dryness and the resulting residue purified as described in the representative protocol to provide nucleoside 5c (150 mg, 43%) as a white foam: Rf = 0.6 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 939.3356 ([M + Na]+, C50H53N4O11P·Na+, calcd 939.3341); 31P NMR (CDCl3) δ 149.8, 149.3.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(3-trifluoroacetylaminopropyn-1-yl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5d)
Nucleoside 4d (0.37 g 0.52 mmol), DIPEA (0.44 mL, 2.52 mmol), PCl-reagent (0.18 mL, 0.78 mmol), and anhydrous CH2Cl2 (10 mL) were mixed, reacted, worked up, and purified as described in the representative protocol to provide nucleoside 5d (0.39 g, 80%) as a white foam: Rf = 0.5 (2% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 930.3068 ([M + Na]+, C45H49F3N5O10P·Na+, calcd 930.3061); 31P NMR (CDCl3) δ 149.7, 149.1.
(1R,3R,4R,7S)-3-[5-(3-(1-Adamantylmethylcarbonyl)aminopropyn-1-yl)uracil-1-yl]-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-2,5-dioxabicyclo[2.2.1]heptane (5e)
Nucleoside 4e (204 mg, 0.26 mmol), DIPEA (184 μL, 1.06 mmol), and PCl-reagent (106 μL, 0.48 mmol) in anhydrous CH2Cl2 (4 mL) were mixed and reacted as described in the representative protocol. At this point, ice-cold EtOH (1 mL) was added and the solvents were evaporated off. Purification as described in the representative protocol provided nucleoside 4e (190 mg, 74%) as a slightly yellow foam: Rf = 0.4 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 1010.4408 ([M + Na]+, C55H66N5O10P·Na+, calcd 1010.4440); 31P NMR (CDCl3) δ 149.8, 149.2. A minor impurity at ∼14 ppm was observed.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(3-dodecanoylaminopropyn-1-yl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5f)
Nucleoside 4f (175 mg, 0.22 mmol), DIPEA (154 μL, 0.88 mmol), N-methylimidazole (14 μL, 0.18 mmol), PCl-reagent (75 μL, 0.33 mmol), and anhydrous CH2Cl2 (3 mL) were mixed and reacted (2.5 h). The solvents were evaporated off, and the resulting residue was purified as described in the representative protocol to provide nucleoside 5f (183 mg, 83%) as a white foam: Rf = 0.4 (4% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 1016.4983 ([M + Na]+, C55H72N5O10P·Na+, calcd 1016.4909); 31P NMR (CDCl3) δ 149.8, 149.2.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(3-octadecanoylaminopropyn-1-yl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5g)
Nucleoside 4g (0.25 g, 0.28 mmol), DIPEA (0.24 mL, 1.37 mmol), PCl-reagent 0.10 mL, 0.42 mmol), and anhydrous CH2Cl2 (10 mL) were mixed, reacted, worked up, and purified as described in the representative protocol to provide nucleoside 5g (180 mg, 60%) as a white foam: Rf = 0.5 (2% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1100.5836 ([M + Na]+, C61H84N5O10P·Na+, calcd 1100.5848); 31P NMR (CDCl3) δ 149.8, 149.2.
(1R,3R,4R,7S)-3-[5-(3-Cholesterylcarbonylaminopropyn-1-yl)uracil-1-yl]-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-2,5-dioxabicyclo[2.2.1]heptane (5h)
Nucleoside 4h (240 mg, 0.23 mmol), DIPEA (0.19 mL, 1.08 mmol), PCl-reagent (0.08 mL, 0.34 mmol), and anhydrous CH2Cl2 (10 mL) were mixed, reacted, worked up, and purified as described in the representative protocol to provide nucleoside 5h (190 mg, 66%) as a white foam: Rf = 0.5 (2% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1246.6571 ([M + Na]+, C71H94N5O11P·Na+, calcd 1246.6579); 31P NMR (CDCl3) δ 149.8, 149.3.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(2-(1-pyrenyl)ethynyl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5i)
Nucleoside 4i (0.20 g, 0.26 mmol), DIPEA (225 μL, 1.28 mmol), PCl-reagent (114 μL, 0.51 mmol), and anhydrous CH2Cl2 (4 mL) were mixed and reacted (4.5 h) as described in the representative protocol. Solvents were evaporated off, and the resulting residue was purified as described in the representative protocol to provide nucleoside 5i (188 mg, 75%) as a pale yellow foam: Rf = 0.5 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 1005.3661 ([M + Na]+, C58H55N4O9P·Na+, calcd 1005.3606); 31P NMR (CDCl3) δ 149.7, 149.3.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(2-(3-perylenyl)ethynyl)uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5j)
Nucleoside 4j (0.47 g, 0.56 mmol), DIPEA (0.40 mL, 2.26 mmol), PCl-reagent (165 μL, 0.73 mmol), and anhydrous CH2Cl2 (4 mL) were mixed, reacted (3 h), worked up, and purified as described in the representative protocol to provide nucleoside 5j (0.43 g, 78%) as a yellow foam: Rf = 0.4 (4% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1055.3751 ([M + Na]+, C62H57N4O9P·Na+, calcd 1055.3763); 31P NMR (CDCl3) δ 149.7, 149.3
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(1-(1-pyrenyl)-1H-1,2,3-triazol-4-yl)-uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5k)
Nucleoside 4k (180 mg, 0.22 mmol), DIPEA (160 μL, 0.88 mmol), PCl-reagent (63 μL, 0.28 mmol), and anhydrous CH2Cl2 (1.5 mL) were mixed and reacted (2.5 h) as described in the representative protocol. At this point, the reaction mixture was diluted with EtOAc (20 mL) and washed with H2O (2 × 25 mL). The organic phase was dried (Na2SO4) and evaporated to dryness and the resulting residue purified as described in the representative protocol to provide nucleoside 5k (184 mg, 82%) as a white foam: Rf = 0.7 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1048.3789 ([M + Na]+, C58H56N7O9P·Na+, calcd 1048.3769); 31P NMR (CDCl3) δ 149.8, 149.1.
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-3-[5-(1-(pyren-1-ylmethyl)-1H-1,2,3-triazol-4-yl)-uracil-1-yl]-2,5-dioxabicyclo[2.2.1]heptane (5l)
Nucleoside 4l (0.41 g, 0.49 mmol), DIPEA (345 μL, 1.95 mmol), PCl-reagent (165 μL, 0.73 mmol), and anhydrous CH2Cl2 (5 mL) were mixed, reacted (2.5 h), worked up, and purified as described in the representative protocol to provide nucleoside 5l (190 mg, 40%) as a white foam: Rf = 0.5 (3% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 1062.3909 ([M + Na]+, C59H58N7O9P·Na+, calcd 1062.3934); 31P NMR (CDCl3) δ 149.6, 149.1.
Synthesis and Purification of ONs
L1–4, K1–4, N1–4, Q1–4, S1–4, and V5 were prepared and characterized with respect to identity (MALDI-MS) and purity (>80%, ion-pair reverse-phase HPLC) in previous studies.22,37 All of the other ONs were synthesized, worked up, purified, and characterized essentially as previously described.37 Briefly, ONs were synthesized on a 0.2 μmol scale using an automated DNA synthesizer and long chain alkyl amine controlled pore glass columns with a pore size of 500 Å. Standard reagents were used. The following hand-coupling conditions were employed to incorporate monomers K–Z into ONs, which generally resulted in coupling yields in excess of 95% (coupling time; activator; phosphoramidite solvent): monomers K/L/M/N/O/Q/S/W/Z (15 min; 0.25 M 4,5-dicyanoimidazole in CH3CN; CH3CN), monomer P (15 min; 0.25 M 4,5-dicyanoimidazole in CH3CN; CH2Cl2), monomer V (30 min; 0.25 M 4,5-dicyanoimidazole in CH3CN; CH2Cl2), and monomers X/Y/Z (15 min; 0.25 M 5-[3,5-bis(trifluoromethyl)phenyl]-1H-tetrazole52 in CH3CN; CH2Cl2). ONs were cleaved from the solid support and protecting groups removed through treatment with concentrated aqueous ammonia (55 °C, 24 h). ONs were purified by ion-pair reverse-phase HPLC (XTerra MS C18 column) using a gradient of 0.05 M triethylammonium acetate in water and 25% water in CH3CN, followed by detritylation (80% aqueous AcOH) and precipitation (NaOAc/NaClO4/acetone, −18 °C for 12–16 h). The identity of all synthesized ONs was verified by MALDI MS analysis (Table S1, Supporting Information) recorded in positive ion mode on a quadrupole time-of-flight tandem mass spectrometer equipped with a MALDI source. Purity (>80%) was verified by ion-pair reverse-phase HPLC running in analytical mode.
Biophysical Characterization Studies
Thermal denaturation temperatures and steady-state fluorescence emission spectra were determined essentially as previously described.37 Briefly, thermal denaturation temperatures were determined as the maximum of the first derivative of the thermal denaturation curve (A260 vs T) recorded in medium salt buffer (Tm buffer: 110 mM NaCl, 0.1 mM EDTA, pH adjusted with 10 mM Na2HPO4/NaH2PO4). A temperature ramp of 0.5 °C/min was used in all experiments. Reported thermal denaturation temperatures are an average of at least two experiments within ±1.0 °C.
Thermodynamic parameters for duplex formation were determined through baseline fitting of denaturation curves (van’t Hoff analysis) using software provided with the UV/vis spectrometer. Bimolecular reactions, two-state melting behavior, and a heat capacity change of ΔCp = 0 upon hybridization were assumed. A minimum of two experimental denaturation curves were each analyzed to minimize errors arising from baseline choice. Averages are listed.
3′-Exonuclease degradation studies were performed by observing the change in absorbance at 260 nm and 37 °C as a function of time for a solution of ONs (3.3 μM) in magnesium buffer (600 μL, 50 mM Tris-HCl, 10 mM MgCl2, pH 9.0) to which SVPDE (snake venom phosphodiesterase) dissolved in H2O was added (12 μL, 0.52 μg, 0.03 U).
Steady-state fluorescence emission spectra were recorded using the same buffers and ON concentrations (1.0 μM) as in thermal denaturation studies. Fluorescence emission spectra were recorded at 5 °C to ensure maximum hybridization. Deoxygenation was deliberately not applied to the samples, since the scope of the work was to determine fluorescence under aerated conditions prevailing in bioassays. Steady-state fluorescence emission spectra were obtained as an average of five scans using λex 344 nm for V/Y/Z-modified ONs, λex 375 nm for W-modified ONs, λex 448 nm for X-modified ONs, excitation slit 5.0 nm, emission slit 5.0 nm, and a scan speed of 600 nm/min.
Acknowledgments
We appreciate financial support from the Idaho NSF EPSCoR, the BANTech Center at the University of Idaho, and Award No. GM088697 from the National Institute of General Medical Sciences, National Institutes of Health. Scholarships from the College of Graduate Studies (M.E.Ø.) and the National Science Foundation under Award No. 0648202 and the Department of Defense ASSURE (Awards to Stimulate and Support Undergraduate Research Experiences) Program (D.J.R.) are appreciated. We thank Dr. Alexander Blumenfeld (Department of Chemistry), Dr. Gary Knerr (Department of Chemistry) and Dr. Lee Deobald (EBI Murdock Mass Spectrometry Center, University of Idaho) for NMR and mass spectrometric analyses. Preliminary synthetic efforts by T. Santhosh Kumar (University of Southern Denmark) are appreciated.
Supporting Information Available
Text, tables, and figures giving general experimental details, NMR spectra for all new compounds, MS data for all new modified ONs, additional Tm data, 3′-exonuclease degradation and fluorescence data, and structures of modified DNA monomers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For recent reviews on conformationally restricted nucleotides, see e.g.:; a Herdewijn P. Chem. Biodiv. 2010, 7, 1–59. [DOI] [PubMed] [Google Scholar]; b Obika S.; Abdur Rahman S. M.; Fujisaka A.; Kawada Y.; Baba T.; Imanishi T. Heterocycles 2010, 81, 1347–1392. [Google Scholar]; c Prakash T. P. Chem. Biodiv. 2011, 8, 1616–1641. [DOI] [PubMed] [Google Scholar]; d Zhou C.; Chattopadhyaya J. Chem. Rev. 2012, 112, 3808–3832. [DOI] [PubMed] [Google Scholar]
- For recent representative examples, see:; a Seth P. P.; Vasquez G.; Allerson C. A.; Berdeja A.; Gaus H.; Kinberger G. A.; Prakash T. P.; Migawa M. T.; Bhat B.; Swayze E. E. J. Org. Chem. 2010, 75, 1569–1581. [DOI] [PubMed] [Google Scholar]; b Li Q.; Yuan F.; Zhou C.; Plashkevych O.; Chattopadhyaya J. J. Org. Chem. 2010, 75, 6122–6140. [DOI] [PubMed] [Google Scholar]; c Liu Y.; Xu J.; Karimiahmadabadi M.; Zhou C.; Chattopadhyaya J. J. Org. Chem. 2010, 75, 7112–7128. [DOI] [PubMed] [Google Scholar]; d Upadhayaya R.; Deshpande S. A.; Li Q.; Kardile R. A.; Sayyed A. Y.; Kshirsagar E. K.; Salunke R. V.; Dixit S. S.; Zhou C.; Foldesi A.; Chattopadhyaya J. J. Org. Chem. 2011, 76, 4408–4431. [DOI] [PubMed] [Google Scholar]; e Shrestha A. R.; Hari Y.; Yahara A.; Osawa T.; Obika S. J. Org. Chem. 2011, 76, 9891–9899. [DOI] [PubMed] [Google Scholar]; f Hanessian S.; Schroeder B. R.; Giacometti R. D.; Merner B. L.; Østergaard M. E.; Swayze E. E.; Seth P. P. Angew. Chem., Int. Ed. 2012, 51, 11242–11245. [DOI] [PubMed] [Google Scholar]; g Madsen A. S.; Wengel J. J. Org. Chem. 2012, 77, 3878–3886. [DOI] [PubMed] [Google Scholar]; h Haziri A. I.; Leumann C. J. J. Org. Chem. 2012, 77, 5861–5869. [DOI] [PubMed] [Google Scholar]; i Gerber A.-B.; Leumann C. J. Chem. Eur. J. 2013, 19, 6990–7006. [DOI] [PubMed] [Google Scholar]; j Morihiro K.; Kodama T.; Kentefu; Moai Y.; Veedu R. N.; Obika S. Angew. Chem., Int. Ed. 2013, 52, 5074–5078. [DOI] [PubMed] [Google Scholar]; k Hari Y.; Osawa T.; Kotobuki Y.; Yahara A.; Shrestha A. R.; Obika S. Bioorg. Med. Chem. 2013, 21, 4405–4412. [DOI] [PubMed] [Google Scholar]; l Hari Y.; Morikawa T.; Osawa T.; Obika S. Org. Lett. 2013, 15, 3702–3705. [DOI] [PubMed] [Google Scholar]; m Migawa M. T.; Prakash T. P.; Vasquez G.; Seth P. P.; Swayze E. E. Org. Lett. 2013, 15, 4316–4319. [DOI] [PubMed] [Google Scholar]; n Hanessian S.; Schroeder B. R.; Merner B. L.; Chen B.; Swayze E. E.; Seth P. P. J. Org. Chem. 2013, 78, 9051–9063. [DOI] [PubMed] [Google Scholar]; o Hanessian S.; Wagger J.; Merner B. L.; Giacometti R. D.; Østergaard M. E.; Swayze E. E.; Seth P. P. J. Org. Chem. 2013, 78, 9064–9075. [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. Science 1999, 284, 2118–2124. [DOI] [PubMed] [Google Scholar]
- Hendrix C.; Rosemeyer H.; Verheggen I.; Van Aerschot A.; Seela F.; Herdewijn P. Chem. Eur. J. 1997, 3, 110–120. [Google Scholar]
- Wang J.; Verbeure B.; Luyten I.; Lescrinier E.; Froeyen M.; Hendrix C.; Rosemeyer H.; Seela F.; Van Aerschot A.; Herdewijn P. J. Am. Chem. Soc. 2000, 122, 8595–8602. [Google Scholar]
- Bolli M.; Trafelet H. U.; Leumann C. Nucleic Acids Res. 1996, 24, 4660–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renneberg D.; Leumann C. J. J. Am. Chem. Soc. 2002, 124, 5993–6002. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Nielsen P.; Koshkin A. A.; Wengel J. Chem. Commun. 1998, 455–456. [Google Scholar]
- Kaur H.; Babu B. R.; Maiti S. Chem. Rev. 2007, 107, 4672–4697. [DOI] [PubMed] [Google Scholar]
- Obika S.; Nanbu D.; Hari Y.; Andoh J.-I.; Morio K.-I.; Doi T.; Imanishi T. Tetrahedron Lett. 1998, 39, 5401–5404. [Google Scholar]
- Kool E. T. Chem. Rev. 1997, 97, 1473–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Duca M.; Vekhoff P.; Oussedik K.; Halby L.; Arimondo P. B. Nucleic Acids Res. 2008, 36, 5123–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bennett C. F.; Swayze E. E. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [DOI] [PubMed] [Google Scholar]; c Østergaard M. E.; Hrdlicka P. J. Chem. Soc. Rev. 2011, 40, 5771–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Watts J. K.; Corey D. R. J. Pathol. 2012, 226, 365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Matsui M.; Corey D. R. Drug Discuss. Today 2012, 17, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dong H.; Lei J.; Ding L.; Wen Y.; Ju H.; Zhang X. Chem. Rev. 2013, 113, 6207–6233. [DOI] [PubMed] [Google Scholar]
- a Wahlestedt C.; Salmi P.; Good L.; Kela J.; Johnsson T.; Hokfelt T.; Broberger C.; Porreca F.; Lai J.; Ren K. K.; Ossipov M.; Koshkin A.; Jacobsen N.; Skouv J.; Oerum H.; Jacobsen M. H.; Wengel J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Graziewicz M. A.; Tarrant T. K.; Buckley B.; Roberts J.; Fulton L.; Hansen H.; Ørum H.; Kole R.; Sazani P. Mol. Ther. 2008, 16, 1316–1322. [DOI] [PubMed] [Google Scholar]; c Straarup E. M.; Fisker N.; Hedtjarn M.; Lindholm M. W.; Rosenbohm C.; Aarup V.; Hansen H. F.; Ørum H.; Hansen J. B.; Koch T. Nucleic Acids Res. 2010, 38, 7100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lanford R. E.; Hildebrandt-Eriksen E. S.; Petri A.; Persson R.; Lindow M.; Munk M. E.; Kauppinen S.; Ørum H. Science 2010, 327, 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Obad S.; Dos Santos C. O.; Petri A.; Heidenblad M.; Broom O.; Ruse C.; Fu C.; Lindow M.; Stenvang J.; Straarup E. M.; Hansen H. F.; Koch T.; Pappin D.; Hannon G. J.; Kauppinen S. Nat. Genet. 2011, 43, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an updated clinical status of LNA, see: http://www.santaris.com/product-pipeline.
- Wienholds E.; Kloostermann W. P.; Misk W. P.; Alvarez-Saavedra E.; Berezikov E.; Bruijn E.; Horvitz H. R.; Kauppinen S.; Plasterk R. H. A. Nature 2005, 309, 310–311. [DOI] [PubMed] [Google Scholar]
- For particularly interesting earlier examples see:; a Sørensen M. D.; Kværnø L.; Bryld T.; Håkansson A. E.; Verbeure B.; Gaubert G.; Herdewijn P.; Wengel J. J. Am. Chem. Soc. 2002, 124, 2164–2176. [DOI] [PubMed] [Google Scholar]; b Sørensen M. D.; Petersen M.; Wengel J. Chem. Commun. 2003, 2130–2131. [DOI] [PubMed] [Google Scholar]; c Morita K.; Takagi M.; Hasegawa C.; Kaneko M.; Tsutsumi S.; Sone J.; Ishikawa T.; Imanishi T.; Koizumi M. Bioorg. Med. Chem. 2003, 11, 2211–2226. [DOI] [PubMed] [Google Scholar]; d Fluiter K.; Frieden M.; Vreijling J.; Rosenbohm C.; De Wissel M. B.; Christensen S. M.; Koch T.; Ørum H.; Baas F. ChemBioChem 2005, 6, 1104–1109. [DOI] [PubMed] [Google Scholar]; e Albæk N.; Petersen M.; Nielsen P. J. Org. Chem. 2006, 71, 7731–7740. [DOI] [PubMed] [Google Scholar]; f Varghese O. P.; Barman J.; Pathmasiri W.; Plashkevych O.; Honcharenko D.; Chattopadhyaya J. J. Am. Chem. Soc. 2006, 128, 15173–15187. [DOI] [PubMed] [Google Scholar]; g Abdur Rahman S. M.; Seki S.; Obika S.; Yoshikawa H.; Miyashita K.; Imanishi T. J. Am. Chem. Soc. 2008, 130, 4886–4896. [DOI] [PubMed] [Google Scholar]; h Mitsuoka Y.; Kodama T.; Ohnishi R.; Hari Y.; Imanishi T.; Obika S. Nucleic Acids Res. 2009, 37, 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Zhou C.; Liu Y.; Andaloussi M.; Badgujar N.; Plashkevych O.; Chattopadyaya J. J. Org. Chem. 2009, 74, 118–134. [DOI] [PubMed] [Google Scholar]; j Seth P. P.; Siwkowski A.; Allerson C. R.; Vasquez G.; Lee S.; Prakash T. P.; Wancewicz E. V.; Witchell D.; Swayze E. E. J. Med. Chem. 2009, 52, 10–13. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Luyten I.; Herdewijn P. Eur. J. Med. Chem. 1998, 33, 515–576. [Google Scholar]; b Ahmadian M.; Bergstrom D. E. In Modified Nucleotides in Biochemistry, Biotechnology and Medicine, 1st ed .;Herdewijn P., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 251–276. [Google Scholar]
- For particularly interesting examples from the original research literature, see:; a Wagner R. W.; Matteucci M. D.; Lewis J. G.; Gutierrez A. J.; Moulds C.; Froehler B. C. Science 1993, 260, 1510–1513. [DOI] [PubMed] [Google Scholar]; b Hashimoto H.; Nelson M. G.; Switzer C. J. Am. Chem. Soc. 1993, 115, 7128–7134. [Google Scholar]; c Sagi J.; Szemzo A.; Ebinger K.; Szabolcs A.; Sagi G.; Ruff E.; Otvos L. Tetrahedron Lett. 1993, 34, 2191–2194. [Google Scholar]; d Ahmadian M.; Zhang P. M.; Bergstrom D. E. Nucleic Acids Res. 1998, 26, 3127–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Heystek L. E.; Zhou H. Q.; Dande P.; Gold B. J. Am. Chem. Soc. 1998, 120, 12165–12166. [Google Scholar]; f Kottysch T.; Ahlborn C.; Brotzel F.; Richert C. Chem. Eur. J. 2004, 10, 4017–4028. [DOI] [PubMed] [Google Scholar]; g Okamoto A.; Kanatani K.; Saito I. J. Am. Chem. Soc. 2004, 126, 4820–4827. [DOI] [PubMed] [Google Scholar]; h Booth J.; Brown T.; Vadhia S. J.; Lack O.; Cummins W. J.; Trent J. O.; Lane A. N. Biochemistry 2005, 44, 4710–4719. [DOI] [PubMed] [Google Scholar]; i Skorobogatyi M. V.; Malakhov A. D.; Pchelintseva A. A.; Turban A. A.; Bondarev S. L.; Korshun V. A. ChemBioChem 2006, 7, 810–816. [DOI] [PubMed] [Google Scholar]; j Østergaard M. E.; Guenther D. C.; Kumar P.; Baral B.; Deobald L.; Paszczynski A. J.; Sharma P. K.; Hrdlicka P. J. Chem. Commun. 2010, 4929–4931. [DOI] [PubMed] [Google Scholar]
- a Barbaric J.; Wagenknecht H. A. Org. Biomol. Chem. 2006, 4, 2088–2090. [DOI] [PubMed] [Google Scholar]; b Kocalka P.; Andersen N. K.; Jensen F.; Nielsen P. ChemBioChem 2007, 8, 2106–2116. [DOI] [PubMed] [Google Scholar]; c Nguyen T. N.; Brewer A.; Stulz E. Angew. Chem., Int. Ed. 2009, 48, 1974–1977. [DOI] [PubMed] [Google Scholar]; d Kaura M.; Kumar P.; Hrdlicka P. J. Org. Biomol. Chem. 2012, 10, 8575–8578. [DOI] [PubMed] [Google Scholar]
- Sinkeldam R. W.; Greco N. J.; Tor Y. Chem. Rev. 2010, 110, 2579–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent example, see:Andersen N. K.; Anderson B. A.; Wengel J.; Hrdlicka P. J. J. Org. Chem. 2013, 78, 12690–12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard M. E.; Kumar P.; Baral B.; Raible D. J.; Kumar T. S.; Anderson B. A.; Guenther D. C.; Deobald L.; Paszczynski A. J.; Sharma P. K.; Hrdlicka P. J. ChemBioChem 2009, 10, 2740–2743. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Østerggard M. E.; Hrdlicka P. J. Curr. Protocols Nucleic Acid Chem. 2011, 44, 4.43.1–4.43.22. [DOI] [PubMed] [Google Scholar]
- Terminal alkynes were obtained from commercial vendors, synthesized via EDC-mediated coupling between functionalized carboxylic acids and propargylamine (see Scheme S1, Supporting Information) or prepared according to known literature protocols (see the Experimental Section).
- a Sonogashira K.; Tohda Y.; Hagihara N. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar]; b Agrofoglio L. A.; Gillaizeau I.; Saito Y. Chem. Rev. 2003, 103, 1875–1916. [DOI] [PubMed] [Google Scholar]
- Schrock A. K.; Schuster G. B. J. Am. Chem. Soc. 1984, 106, 5234–5240. [Google Scholar]
- Park S. Y.; Yoon J. H.; Hong C. S.; Souane R.; Kim J. S.; Mathews S. E.; Vicens J. J. Org. Chem. 2008, 73, 8212–8218. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Bryld T.; Lomholt C. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1645–1647. [DOI] [PubMed] [Google Scholar]
- Mergny J. L.; Lacroix L. Oligonucleotides 2003, 13, 515–537. [DOI] [PubMed] [Google Scholar]
- Gyi J. I.; Gao D.; Conn G. L.; Trent J. O.; Brown T.; Lane A. N. Nucleic Acids Res. 2003, 31, 2683–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidov V. V.; Frank-Kamenetskii M. D. Trends Biochem. Sci. 2004, 29, 62–71. [DOI] [PubMed] [Google Scholar]
- a Korshun V. A.; Stetsenko D. A.; Gait M. J. J. Chem. Soc., Perkin Trans. 1 2002, 1092–1104. [Google Scholar]; b Dohno C.; Saito I. ChemBioChem 2005, 6, 1075–1081. [DOI] [PubMed] [Google Scholar]; c Kumar T. S.; Madsen A. S.; Østergaard M. E.; Sau S. P.; Wengel J.; Hrdlicka P. J. J. Org. Chem. 2009, 74, 1070–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin V.; Hansen H. F.; Koch T. R.; Armitage B. A. J. Biomol. Struct. Dyn. 2004, 21, 841–850. [DOI] [PubMed] [Google Scholar]
- a Mayer E.; Valis L.; Wagner C.; Rist M.; Amann N.; Wagenknecht H.-A. ChemBioChem 2004, 5, 865–868. [DOI] [PubMed] [Google Scholar]; b Hwang G. T.; Seo Y. J.; Kim S. J.; Kim B. H. Tetrahedron Lett. 2004, 45, 3543–3546. [Google Scholar]
- a Dougherty G.; Pilbrow J. R. Int. J. Biochem. 1984, 16, 1179–1192. [DOI] [PubMed] [Google Scholar]; b Manoharan M.; Tivel K. L.; Zhao M.; Nafisi K.; Netzel T. L. J. Phys. Chem. 1995, 99, 17461–17472. [Google Scholar]; c Seo Y. J.; Ryu J. H.; Kim B. H. Org. Lett. 2005, 7, 4931–4933. [DOI] [PubMed] [Google Scholar]
- Østergaard M. E.; Kumar P.; Baral B.; Guenther D. C.; Anderson B. A.; Ytreberg F. M.; Deobald L.; Paszczynski A. J.; Sharma P. K.; Hrdlicka P. J. Chem. Eur. J. 2011, 17, 3157–3165. [DOI] [PubMed] [Google Scholar]
- Seitz O.; Schmuck E. C.; Wennemers H.. Homogeneous DNA Detection. In Highlights in Bioorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004;pp 311–328. [Google Scholar]
- Quantum yields were not determined for Z-modified duplexes. However, direct comparison with Z5d:DNA, which has a relative fluorescence quantum yield of ∼16%,18j suggests that Z5:DNA has a quantum yield of 20–30%.
- a Yamana K.; Iwase R.; Furutani S.; Tsuchida H.; Zako H.; Yamaoka T.; Murakami A. Nucleic Acids Res. 1999, 27, 2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yamana K.; Zako H.; Asazuma K.; Iwase R.; Nakano H.; Murakami A. Angew. Chem., Int. Ed. 2001, 40, 1104–1106. [DOI] [PubMed] [Google Scholar]; c Mahara A.; Iwase R.; Sakamoto T.; Yamana K.; Yamaoka T.; Murakami A. Angew. Chem., Int. Ed. 2002, 41, 3648–3650. [DOI] [PubMed] [Google Scholar]; d Hrdlicka P. J.; Babu B. R.; Sørensen M. D.; Harrit N.; Wengel J. J. Am. Chem. Soc. 2005, 127, 13293–13299. [DOI] [PubMed] [Google Scholar]; e Astakhova I. V.; Korshun V. A.; Wengel J. Chem. Eur. J. 2008, 14, 11010–11026. [DOI] [PubMed] [Google Scholar]; f Wang G.; Bobkov G. V.; Mikhailov S. N.; Schepers G.; van Aerschot A.; Rozenski J.; van der Auweraer M.; Herdewijn P.; Feyter S. D. ChemBioChem 2009, 10, 1175–1185. [DOI] [PubMed] [Google Scholar]; g Østergaard M. E.; Cheguru P.; Papasani M. R.; Hill R. A.; Hrdlicka P. J. J. Am. Chem. Soc. 2010, 132, 14221–14228. [DOI] [PubMed] [Google Scholar]; h Astakhova I. V.; Lindegaard D.; Korshun V. A.; Wengel J. Chem. Commun. 2010, 46, 8362–8364. [DOI] [PubMed] [Google Scholar]; i Förster U.; Lommel K.; Sauter D.; Grünewald C.; Engels J. W.; Wachtveitl J. ChemBioChem 2010, 11, 664–672. [DOI] [PubMed] [Google Scholar]; j Mansawat W.; Boonlua C.; Siriwong K.; Vilaivan T. Tetrahedron 2012, 68, 3988–3995. [Google Scholar]
- Shen L.; Siwkowski A.; Wancewicz E. V.; Lesnik E.; Butler M.; Witchell D.; Vasquez G.; Ross B.; Acevedo O.; Inamati G.; Sasmor H.; Manoharan M.; Monia B. P. Antisense Nucleic Acid Drug Dev. 2003, 13, 129–142. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Baral B.; Anderson B. A.; Guenther D. C.; Østergaard M. E.; Sharma P. K.; Hrdlicka P. J. J. Org. Chem. 2014, 10.1021/jo5006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For structures of alkynes Ae–g, see Scheme S1 (Supporting Information).
- Suenaga M.; Kaneko Y.; Kadokawa J.; Nishikawa T.; Mori H.; Tabata M. Macromol. Biosci. 2006, 6, 1009–1018. [DOI] [PubMed] [Google Scholar]
- Godeau G.; Barthélémy P. Langmuir 2009, 25, 8447–8450. [DOI] [PubMed] [Google Scholar]
- Ott K.; Schmidt K.; Kircher B.; Schumacher P.; Wiglenda T.; Gust R. J. Med. Chem. 2005, 48, 622–629. [DOI] [PubMed] [Google Scholar]
- Trybulski E.; Zhang J.; Kramss R. H.; Mangano R. M. J. Med. Chem. 1993, 36, 3533–3541. [DOI] [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. J. Org. Chem. 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- Qu J.; Shiotsuki M.; Sanda F.; Masuda T. Macromol. Chem. Phys. 2007, 208, 823–832. [Google Scholar]
- Wenting W.; Wanhua W.; Shaomin J.; Hunin G.; Jiazhang Z. Eur. J. Inorg. Chem. 2010, 4470–4482. [Google Scholar]
- Andronova V. L.; Skorobogtyi M. V.; Manasova E. V.; Berlin Y. A.; Korshun V. A.; Galegov G. A. Russ. J. Bioorg. Chem. 2003, 29, 262–266. [DOI] [PubMed] [Google Scholar]
- Initial screening revealed that this activator results in higher stepwise coupling yields than the normal 4,5-dicyanoimidazole activator.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.