

Abstract
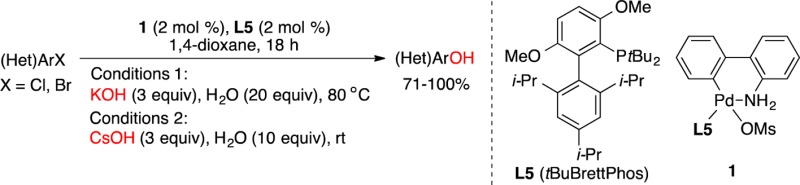
A method for the hydroxylation of aryl and heteroaryl halides, promoted by a catalyst based on a biarylphosphine ligand tBuBrettPhos (L5) and its corresponding palladium precatalyst (1), is described. The reactions allow the cross-coupling of both potassium and cesium hydroxides with (hetero)aryl halides to afford a variety of phenols and hydroxylated heteroarenes in high to excellent yield.
Phenols, hydroxylated heteroarenes, and phenol derivatives [e.g., alkyl (hetero)aryl ethers, bi(hetero)aryl ethers] represent important structural motifs found in natural products,1 materials,2 pharmaceuticals,3 and agrochemicals4 (Figure 1). Furthermore, phenol subunits are common building blocks and versatile synthetic intermediates. Phenols are traditionally prepared by nucleophilic aromatic substitution,5 hydrolysis of arenediazonium salts (Sandmeyer reactions),6 and deprotection of phenol precursors (e.g., ethers, esters, carbamates),7 but these methods generally exhibit limited substrate scope and/or may require harsh reaction conditions. Recently, methods to generate phenols from the corresponding boronic acids have been developed, including the oxidation of (hetero)arylboronic acids,8 and the copper-catalyzed oxidative cross-coupling between (hetero)arylboronic acids and hydroxide ion.9 Unfortunately, these strategies may require multiple synthetic operations, depending on the availability of the organoboron starting materials.10 Alternatively, functionalized phenols may be synthesized via the iridium-catalyzed C–H activation/borylation/oxidation of arenes,11 as well as the transition-metal-catalyzed C–H activation/oxidation of arenes.12 However, these methods are restricted by the C–H activation step, which can suffer from a lack of regioselectivity12g,12i,12j or require neighboring group assistance.12a−12i
Figure 1.
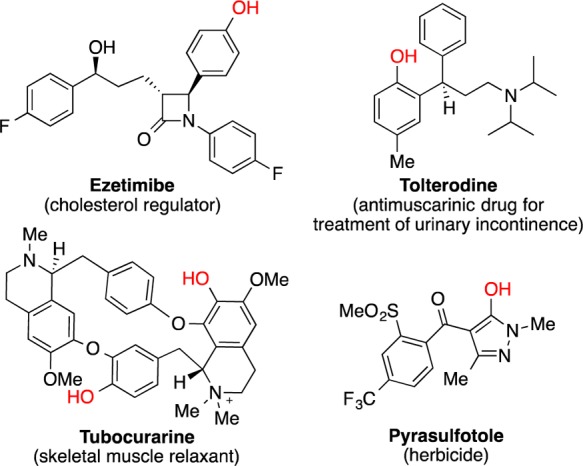
Selected pharmaceutical and pesticide molecules containing the phenol and hydroxylated heteroarene motifs.
The transition-metal-catalyzed cross-coupling of (hetero)aryl halides with hydroxide salts represents a direct, convenient, regioselective, and relatively atom-economical process to afford phenols and hydroxylated heteroarenes.13−15 The Cu-catalyzed hydroxylation of aryl bromides and iodides has been applied to prepare a wide range of functionalized phenols.14 These Cu-catalyzed protocols are generally limited to the use of electron-deficient aryl chloride coupling partners;14c,14g,14i however, the analogous Pd-catalyzed process is complementary in this regard and potentially capable of transforming a broader scope of aryl halides to phenols and derivatives thereof.15 In contrast to the Cu-catalyzed process, the Pd-mediated aryl C–OH bond formation had been challenging due to (1) the slow reductive elimination of the proposed (ligand)Pd(aryl)(hydroxo) intermediate15g and (2) competing arylation of the phenol product to form the corresponding biaryl ether.15g In 2006, we reported the Pd-catalyzed hydroxylation of (hetero)aryl halides using the biarylphosphine ligands tBuXPhos and Me4tBuXPhos (L1 and L2, Figure 2) as the supporting ligands.15a In this example, the use of sterically bulky L1 and L2 was shown to facilitate the selective arylation of hydroxide salts.15a Beller et al. then reported that the use of L3 (Figure 2) promoted the Pd-catalyzed arylation of KOH and CsOH at elevated and room temperatures, respectively.15d,15e Subsequently, Chern et al. demonstrated a microwave-assisted hydroxylation of (hetero)aryl halides using a Pd catalyst based on the Herrmann–Beller palladacycle and L1.15f More recently, Stradiotto et al. reported the room-temperature Pd-catalyzed arylation of CsOH using the BippyPhos ligand L4 (Figure 2).15g
Figure 2.

Bi(hetero)arylphosphine ligands employed for Pd-catalyzed hydroxylation of (hetero)aryl halides.
We recently reported the Pd-catalyzed cross-coupling of methanol with (hetero)aryl halides in 1,4-dioxane to afford a wide range of methyl (hetero)aryl ethers by employing a bulky tBuBrettPhos ligand (L5)16 and its corresponding aminobiphenyl palladacycle precatalyst 1(17) (Figure 2).18 We reasoned that the same catalyst would also promote the cross-coupling of hydroxide salts with (hetero)aryl halides. In addition, the use of L5 as a sole ligand would be operationally more simple and practical for hydroxylation, compared with the previous use of the dual biarylphosphine ligands L1 and L2.15a Herein, we report a modified Pd-catalyzed hydroxylation method of (hetero)aryl halides, utilizing a catalyst system composed of precatalyst 1 and L5. This alternative reaction protocol features the use of a lower Pd-catalyst loading (2 mol %) and the compatibility of a wider scope of heteroaryl halides as coupling partners, compared with our previously reported protocol using catalyst systems based on Pd2dba3 (up to 4 mol % Pd) and both L1 and L2.15a
We began our studies by examining the reaction of 4-chloroanisole with KOH (3 equiv) in 1,4-dioxane at 80 °C, using a Pd catalyst based on 1 (2 mol %) and L5 (2 mol %) (Table 1). In the presence of 20 equiv of water, the reaction occurred smoothly to afford 4-methoxyphenol (2) in 85% yield (Table 1, entry 1). The yield of 2 was decreased when the amount of added water was increased (Table 1, entries 2 and 3). Moreover, varying the stoichiometry of KOH also led to lower yields of 2, as exemplified in entries 4 and 5 (Table 1). Finally, no reaction occurred at 50 °C (Table 1, entry 6).
Table 1. Optimization of Pd-Catalyzed Arylation of KOHa.
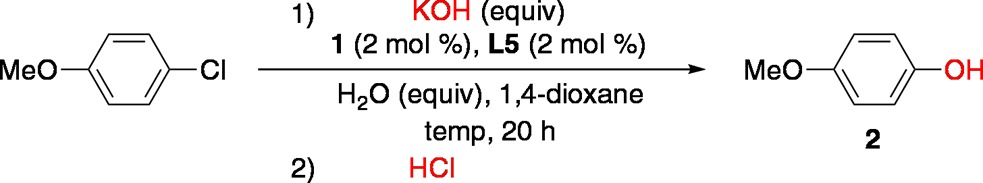
| entry | temp (°C) | KOH (equiv) | H2O (equiv) | conv. (%)b | yield of 2 (%)b |
|---|---|---|---|---|---|
| 1 | 80 | 3 | 20 | 100 | 85 |
| 2 | 80 | 3 | 40 | 100 | 76 |
| 3 | 80 | 3 | 111c | 99 | 76 |
| 4 | 80 | 4 | 20 | 100 | 79 |
| 5 | 80 | 2 | 20 | 100 | 61 |
| 6 | 50 | 3 | 20 | 2 | 0 |
Reaction conditions: 4-chloroanisole (0.25 mmol), KOH (2–4 equiv), H2O (20–111 equiv), 1 (2 mol %), L5 (2 mol %), 1,4-dioxane (0.5 mL), 50–80 °C, 20 h; trace amounts of anisole and biaryl ether were detected by GCMS.
Determined by GC.
Volume ratio of 1,4-dioxane to water = 1:1.
Next, we studied the substrate scope of Pd-catalyzed coupling of KOH with (hetero)aryl halides at 80 °C (Scheme 1).19 Both functionalized and sterically hindered aryl halides could be coupled with KOH to afford the phenols in high to excellent yields (3a–3e). Sterically hindered bromonaphthalenes were also suitable coupling partners (3f, 3g), and a variety of heteroaryl halides could be converted to the corresponding hydroxylated heteroarenes, including quinoxalines (3h), indoles (3i), benzothiophenes (3j), benzofurans (3k), benzo-2,1,3-thiadiazoles (3l), and dibenzothiophenes (3m). Further, the five-membered heterocyclic portion of 3-chloroindazole (3n) was cleanly hydroxylated to provide 3-hydroxyindazole.
Scheme 1. Scope of Pd-Catalyzed Arylation of KOH.
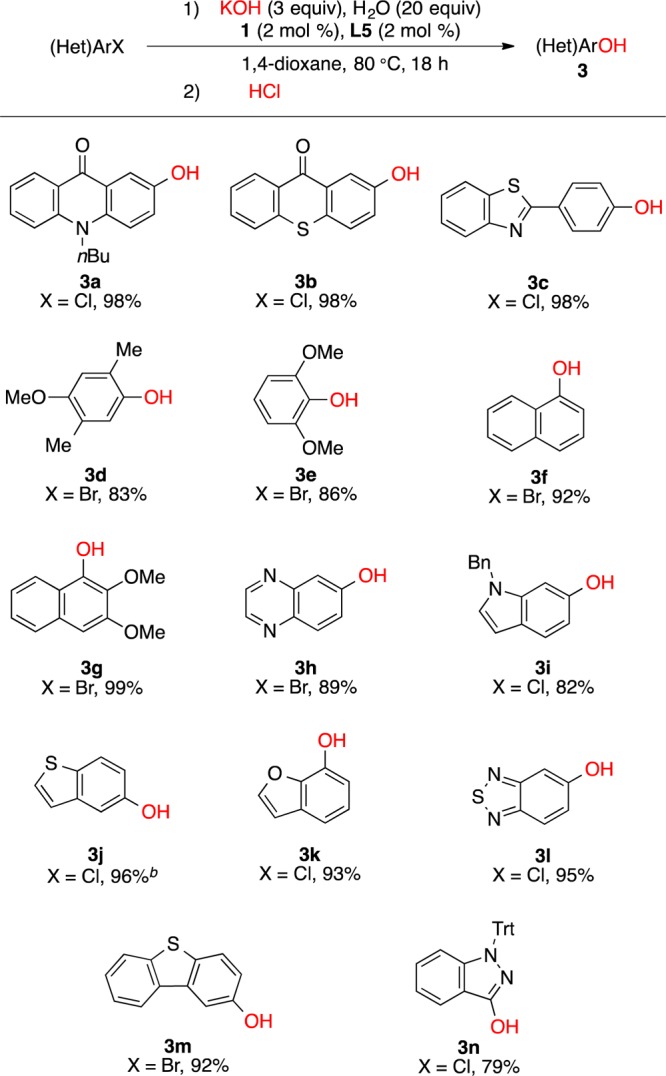
Reaction conditions: (Het)ArX (1 mmol), KOH (3 mmol), H2O (20 mmol), 1 (2 mol %), L5 (2 mol %), 1,4-dioxane (2 mL), 80 °C, 18 h.
100 °C.
We also examined conditions to effect the Pd-catalyzed hydroxylation of aryl halides at ambient conditions. Initial studies focused on the cross-coupling of the test substrate 1-bromo-2,4-dimethylbenzene with CsOH (3 equiv) at room temperature (Table 2), utilizing precatalyst 1 (2 mol %) and ligand L5 (2 mol %). Under these conditions, we found that the addition of 10 equiv of water led to complete conversion of the substrate to provide 2,4-dimethylphenol (4) in the highest yield (Table 2, entry 2). Of note, the use of THF instead of 1,4-dioxane as solvent did not further improve the yield of 4 even though extra water (10 equiv) was added (Table 2, entries 5 and 6). As depicted in Scheme 2, the coupling of both electron-rich and electron-deficient aryl bromides as well as sterically hindered aryl bromides with CsOH could be achieved at ambient temperature.20 A base-sensitive carbonyl and nitrile group were also well-tolerated under the reaction conditions (5b, 5f, Scheme 2).
Table 2. Optimization of Pd-Catalyzed Arylation of CsOH at Room Temperaturea.
| entry | H2O (equiv) | solvent | conv. (%)b | yield of 4 (%)b |
|---|---|---|---|---|
| 1 | 1c | 1,4-dioxane | 18 | 8 |
| 2 | 10 | 1,4-dioxane | 100 | 96 |
| 3 | 20 | 1,4-dioxane | 93 | 89 |
| 4 | 30 | 1,4-dioxane | 57 | 53 |
| 5 | 1c | THF | 28 | 20 |
| 6 | 10 | THF | 60 | 51 |
Reaction conditions: 1-bromo-2,4-dimethylbenzene (0.25 mmol), CsOH (0.75 mmol), H2O (1–30 equiv), 1 (2 mol %), L5 (2 mol %), solvent (0.5 mL), rt, 20 h; trace amounts of anisole and biaryl ether were detected by GCMS.
Determined by GC.
The water was from CsOH·H2O; no extra water was added.
Scheme 2. Scope of Pd-Catalyzed Arylation of CsOH at Room Temperature.
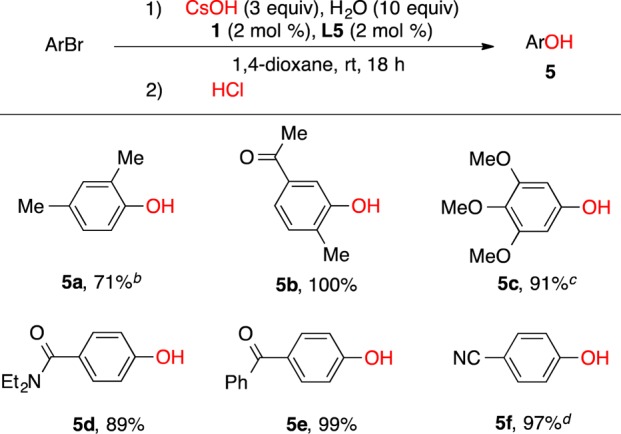
Reaction conditions: ArBr (1 mmol), CsOH (3 mmol), H2O (10 mmol), 1 (2 mol %), L5 (2 mol %), 1,4-dioxane (2 mL), rt, 18 h.
Volatile compound; the GC yield was 96% with 0.25 mmol ArBr.
1 (3 mol %) and L5 (3 mol %).
ArBr (2 mmol), CsOH (6 mmol), H2O (20 mmol), 1 (4 mol %), L5 (4 mol %), and 1,4-dioxane (4 mL).
Experimental Section
General Information
Nuclear magnetic resonance spectra were recorded on a 400 MHz NMR instrument at rt. All 1H NMR spectra were measured in parts per million (ppm) relative to the signals for residual DMSO in DMSO-d6 (2.50 ppm).21 Data for 1H NMR were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qu = quintet, sex = sextet, m = multiplet, ovrlp = overlap, br = broad), coupling constants, and integration. All 13C NMR spectra were reported in ppm relative to DMSO-d6 (39.52 ppm).21 All commercial materials were used as received unless otherwise noted. tBuBrettPhos (L5),16 Pd precatalyst 1,17 and 3-chloro-1-trityl-indazole (S1)18 were prepared according to the literature procedures.
Preparation of Standard Cesium Hydroxide Solution:
(i) Mole Ratio of CsOH to H2O = 3:10
In a nitrogen-filled glovebox, an oven-dried 25 mL round-bottom flask (A) capped with a rubber septum was charged with CsOH·H2O (16.7 g, 99.5 mmol). The flask was then evacuated and backfilled with argon (this process was repeated a total of three times), and degassed deionized water (4.17 mL, 232 mmol) was then added into the flask. The resulting reaction mixture was agitated until all CsOH·H2O dissolved to form a homogeneous solution. The bulk solution was kept under an argon atmosphere. The mole ratio of CsOH and H2O in the standard CsOH solution is 99.5 mmol: [232 mmol (from water added) + 99.5 mmol (from CsOH·H2O)] = 99.5 mmol: 331.5 mmol = 3:10 (or 1:3.33). Thus, the mole ratio of CsOH to water in the standard CsOH solution is always kept at 3:10 for simultaneous transfers of both CsOH and water in a desired mole ratio.
(ii) Mole Ratio of CsOH to H2O = 3:20
Following the procedure of (i), the standard CsOH solution (mole ratio of CsOH:H2O = 3:20) was prepared using CsOH·H2O (520 mg, 3.1 mmol) and deionized water (0.32 mL, 17.8 mmol).
(iii) Mole Ratio of CsOH to H2O = 3:30
Following the procedure of (i), the standard CsOH solution (mole ratio of CsOH:H2O = 3:30) was prepared using CsOH·H2O (520 mg, 3.1 mmol) and deionized water (0.50 mL, 27.8 mmol).
Optimization of Pd-Catalyzed Arylation of KOH (Table 1)
An oven-dried 10 mL resealable screw-cap test tube (A) equipped with a Teflon-coated magnetic stir bar was charged with L5 (2.4 mg, 0.005 mmol, 2 mol %) and KOH (2–4 equiv, 28.1–56.1 mg). Tube A was evacuated and backfilled with argon (this process was repeated a total of three times), and 4-chloroanisole (36 mg, 31 μL, 0.25 mmol, 1 equiv) and degassed deionized water (20–111 equiv, 90 μL to 0.50 mL) were then added into tube A via syringe. A second oven-dried 10 mL resealable screw-cap test tube (B) equipped with a Teflon-coated magnetic stir bar was charged with 1 (4.3 mg, 0.005 mmol, 2 mol %). Tube B was then evacuated and backfilled with argon (this process was repeated a total of three times), and 1,4-dioxane (0.50 mL) was added into tube B via syringe. The reaction mixture in tube B was stirred at rt for ∼1 min to form a homogeneous solution. The precatalyst solution from tube B was transferred into tube A via syringe. The resulting reaction mixture in tube A was stirred at 50 or 80 °C in an oil bath for 20 h. After cooling to rt, the crude product was diluted with EtOAc (2 mL), 1,4-dimethoxybenzene (34.6 mg, 0.25 mmol, 1.0 equiv) was added, and the mixture was then acidified with aqueous HCl solution (1 M, 2 mL). The resulting reaction mixture in the capped test tube was agitated until all of the solid was dissolved into the reaction mixture. The reaction mixture was then neutralized with saturated NaHCO3 solution (2 mL). A small fraction of the upper organic layer was filtered through a plug of silica gel and then subjected to GC analysis to determine the reaction conversion and the GC yields of product and 4-methoxyphenol, using 1,4-dimethoxybenzene as an internal standard.
Optimization of Pd-Catalyzed Arylation of CsOH (Table 2)
An oven-dried 10 mL resealable screw-cap test tube (A) equipped with a Teflon-coated magnetic stir bar was charged with L5 (2.4 mg, 0.005 mmol, 2 mol %) [except for Table 2, entries 1 and 5, in which the tube was then transferred into the N2-filled glovebox, and CsOH·H2O (126 mg, 0.75 mmol, 3 equiv) was added]. Tube A was evacuated and backfilled with argon (this process was repeated a total of three times), and 1-bromo-2,4-dimethylbenzene (46.3 mg, 34 μL, 0.25 mmol) and standard aqueous CsOH solution prepared in the preceding procedures were then added into tube A via syringe (except for Table 2, entries 1 and 5) [For Table 2, entries 2 and 6: CsOH:H2O = 3 equiv:10 equiv; CsOH (112.4 mg, 0.75 mmol, 3 equiv) dissolved in deionized water (45 mg, 2.5 mmol, 10 equiv). For Table 2, entry 3: CsOH:H2O = 3 equiv:20 equiv; CsOH (112.4 mg, 0.75 mmol, 3 equiv) dissolved in deionized water (90 mg, 5.0 mmol, 20 equiv). For Table 2, entry 4: CsOH:H2O = 3 equiv:30 equiv; CsOH (112.4 mg, 0.75 mmol, 3 equiv) dissolved in deionized water (135 mg, 7.5 mmol, 30 equiv)]. A second oven-dried 10 mL resealable screw-cap test tube (B) equipped with a Teflon-coated magnetic stir bar was charged with 1 (4.3 mg, 0.005 mmol, 2 mol %). Tube B was then evacuated and backfilled with argon (this process was repeated a total of three times), and the solvent (1,4-dioxane or THF, 0.50 mL) was added into tube B via syringe. The reaction mixture in tube B was stirred at rt for ∼1 min to form a homogeneous solution. The precatalyst solution from tube B was transferred into tube A via syringe. The resulting reaction mixture in tube A was stirred at rt for 20 h. The crude product was diluted with EtOAc (2 mL), 1,3,5-trimethoxybenzene (42.1 mg, 0.25 mmol, 1.0 equiv) was added, and the mixture was then acidified with aqueous HCl solution (1 M, 2 mL). The resulting reaction mixture in the capped test tube was agitated until all of the solid was dissolved. The reaction mixture was then neutralized with saturated NaHCO3 solution (2 mL). A small fraction of the upper organic layer was filtered through a plug of silica gel and then subjected to GC analysis to determine the reaction conversion and the GC yields of product and 2,4-dimethylphenol, using 1,3,5-trimethoxybenzene as an internal standard.
Substrate Scope for the Pd-Catalyzed Hydroxylation of (Hetero)aryl Halides
General Procedure A
An oven-dried 20 mL resealable screw-cap test tube (A) equipped with a Teflon-coated magnetic stir bar was charged with L5 (9.7 mg, 0.020 mmol, 2 mol %), KOH (168.3 mg, 3.0 mmol, 3 equiv), and (hetero)aryl halide (if solid) (1.0 mmol, 1 equiv). Tube A was evacuated and backfilled with argon (this process was repeated a total of three times), and degassed deionized water (0.36 mL, 20.0 mmol, 20 equiv) and (hetero)aryl halide (if liquid) (1.0 mmol, 1 equiv) were then added into tube A via syringe. A second oven-dried 10 mL resealable screw-cap test tube (B) equipped with a Teflon-coated magnetic stir bar was charged with 1 (17.1 mg, 0.020 mmol, 2 mol %). Tube B was then evacuated and backfilled with argon (this process was repeated a total of three times), and 1,4-dioxane (2.0 mL) was added into tube B via syringe. The reaction mixture in tube B was stirred at rt for ∼1 min to form a homogeneous solution. The precatalyst solution from tube B was transferred into tube A via syringe. The resulting reaction mixture in tube A was stirred at 80 °C in an oil bath for 18 h. After cooling to rt, the crude product was diluted with EtOAc (5 mL) and then acidified with aqueous HCl solution (1 M, 5 mL). The resulting reaction mixture in the capped test tube was agitated until all of the solid was dissolved. The reaction mixture was transferred into a separatory funnel and then neutralized with saturated NaHCO3 solution (5 mL). The organic fraction was isolated, and the aqueous fraction was rinsed with EtOAc (2 × 10 mL). The combined organic fractions were concentrated in vacuo. The crude product residue was purified by flash chromatography using a solvent mixture (EtOAc/hexanes/methanol) as an eluent to afford the isolated product. The reported yields are of the isolated product and averages of two runs.
General Procedure B
An oven-dried 20 mL resealable screw-cap test tube (A) equipped with a Teflon-coated magnetic stir bar was charged with L5 (9.7 mg, 0.020 mmol, 2 mol %) and (hetero)aryl halide (if solid) (1.0 mmol, 1 equiv). Tube A was evacuated and backfilled with argon (this process was repeated a total of three times), and standard aqueous CsOH solution [mole ratio of CsOH to H2O = 3:10; CsOH (450 mg, 3.0 mmol, 3 equiv) dissolved in deionized water (180 mg, 10.0 mmol, 10 equiv) as prepared in the preceding procedures] was then added into tube A via syringe, followed by the addition of (hetero)aryl halide (if liquid) (1.0 mmol, 1 equiv). A second oven-dried 10 mL resealable screw-cap test tube (B) equipped with a Teflon-coated magnetic stir bar was charged with 1 (17.1 mg, 0.020 mmol, 2 mol %). Tube B was then evacuated and backfilled with argon (this process was repeated a total of three times), and 1,4-dioxane (2.0 mL) was added into tube B via syringe. The reaction mixture in tube B was stirred at rt for ∼1 min to form a homogeneous solution. The precatalyst solution from tube B was transferred into tube A via syringe. The resulting reaction mixture in tube A was stirred at rt for 18 h. The crude product was diluted with EtOAc (5 mL) and then acidified with aqueous HCl solution (1 M, 5 mL). The resulting reaction mixture in the capped test tube was agitated until all of the solid was dissolved. The reaction mixture was transferred into a separatory funnel and then neutralized with saturated NaHCO3 solution (5 mL). The organic fraction was isolated, and the aqueous fraction was rinsed with EtOAc (2 × 10 mL). The combined organic fractions were concentrated in vacuo. The crude product residue was purified by flash chromatography using a solvent mixture (EtOAc/hexanes) as an eluent to afford the isolated product. The reported yields are of the isolated product and averages of two runs.
10-n-Butyl-2-hydroxyacridin-9(10H)-one (3a)
Following general procedure A, the title compound was prepared using 10-n-butyl-2-chloroacridin-9(10H)-one (286 mg, 1.0 mmol); EtOAc/hexanes (1:3), then EtOAc; 263 mg, 98%; sunset-yellow solid. m.p.: 239–240 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.70 (br s, 1 H), 8.32 (dd, J = 8.6 Hz, J = 1.2 Hz, 1 H), 7.78–7.71 (ovrlp, 3 H), 7.70 (d, J = 4.0 Hz, 1 H), 7.35 (dd, J = 9.2 Hz, J = 2.8 Hz, 1 H), 7.25 (ddd, J = 8.0 Hz, J = 6.0 Hz, J = 1.6 Hz, 1 H), 4.41 (t, J = 7.6 Hz, 2 H), 1.74 (qu, J = 7.6 Hz, 2 H), 1.49 (sex, J = 7.6 Hz, 2 H), 0.96 (t, J = 7.6 Hz, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 175.9, 152.0, 141.0, 135.2, 133.7, 126.7, 124.1, 122.8, 120.6, 120.4, 117.5, 115.5, 109.2, 45.0, 29.1, 19.4, 13.7. Anal. Calcd for C17H17NO2: C, 76.38; H, 6.41. Found: C, 76.09; H, 6.31. IR (neat cm–1) 3278, 1591, 1568, 1499, 1460, 1357, 1270, 1229, 1171, 861, 810, 752, 684, 638.
2-Hydroxy-9H-thioxanthen-9-one (3b)22
Following general procedure A, the title compound was prepared using 2-chloro-9H-thioxanthen-9-one (247 mg, 1.0 mmol); EtOAc/hexanes (1:4), then EtOAc; 224 mg, 98%; sunset-yellow solid. m.p.: 247–248 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.16 (br s, 1 H), 8.45 (dd, J = 8.4 Hz, J = 1.2 Hz, 1 H), 7.86 (d, J = 2.8 Hz, 1 H), 7.79 (dd, J = 8.0 Hz, J = 0.8 Hz, 1 H), 7.73 (ddd, J = 8.4 Hz, J = 6.8 Hz, J = 1.6 Hz, 1 H), 7.67 (d, J = 8.8 Hz, 1 H), 7.54 (ddd, J = 8.4 Hz, J = 7.2 Hz, J = 1.2 Hz, 1 H), 7.27 (dd, J = 8.8 Hz, J = 2.8 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 178.6, 156.5, 137.0, 132.6, 129.6, 129.0, 128.0, 127.7, 126.5, 126.3, 126.1, 122.7, 113.2. Anal. Calcd for C13H8O2S: C, 68.40; H, 3.53. Found: C, 68.14; H, 3.55. IR (neat cm–1) 3303, 1620, 1568, 1484, 1436, 1341, 1216, 1150, 1121, 1075, 814, 736, 633.
4-(Benzothiazol-2-yl)phenol (3c)23
Following general procedure A, the title compound was prepared using 2-(4-chlorophenyl)benzothiazole (246 mg, 1.0 mmol); EtOAc/hexanes (1:6), then (1:4); 223 mg, 98%; pale-yellow solid. m.p.: 227–229 °C (lit: 228–229 °C).231H NMR (400 MHz, DMSO-d6) δ: 10.23 (br s, 1 H), 8.05 (dd, J = 7.6 Hz, 1 H), 7.98 (dd, J = 8.0 Hz, 1 H), 7.93 (d, J = 8.4 Hz, 2 H), 7.49 (td, J = 8.0 Hz, J = 1.2 Hz, 1 H), 7.39 (td, J = 8.0 Hz, J = 1.2 Hz, 1 H), 6.94 (d, J = 8.8 Hz, 2 H). 13C NMR (100 MHz, DMSO-d6) δ: 167.5, 160.6, 153.7, 134.1, 129.1, 126.4, 124.9, 124.1, 122.3, 122.1, 116.1. Anal. Calcd for C13H9NOS: C, 68.70; H, 3.99. Found: C, 68.83 H, 4.13. IR (neat cm–1) 1604, 1480, 1429, 1283, 1224, 1167, 977, 826, 756, 723.
4-Methoxy-2,5-dimethylphenol (3d)24
Following general procedure A, the title compound was prepared using 1-bromo-4-methoxy-2,5-dimethylbenzene (215.1 mg, 1.0 mmol); EtOAc/hexanes (1:6); 127 mg, 83%; white solid. m.p.: 89–90 °C (lit: 86–88 °C).241H NMR (400 MHz, DMSO-d6) δ: 8.58 (br s, 1 H), 6.62 (s, 1 H), 6.55 (s, 1 H), 3.66 (s, 3 H), 2.08 (s, 3 H), 2.03 (s, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 150.0, 148.5, 123.1, 121.1, 117.1, 113.1, 55.6, 16.0, 15.7. HRMS (ESI) Calcd for C9H13O2 [M + H]: 153.0910. Found: 153.0915. IR (neat cm–1) 3238, 1519, 1452, 1412, 1198, 1103, 1018, 864, 834, 671.
2,6-Dimethoxyphenol (3e)8e
Following general procedure A, the title compound was prepared using 2-bromo-1,3-dimethoxybenzene (217 mg, 1.0 mmol); EtOAc/hexanes (1:6), then (1:4); 134 mg, 86%; pale-brown solid. m.p.: 56–57 °C (lit: 54–56 °C).251H NMR (400 MHz, DMSO-d6) δ: 8.25 (br s, 1 H), 6.69 (t, J = 8.4 Hz, 1 H), 6.59 (d, J = 8.4 Hz, 2 H), 3.74 (s, 6 H). 13C NMR (100 MHz, DMSO-d6) δ: 148.2, 135.7, 118.1, 105.7, 55.9. HRMS (ESI) Calcd for C8H11O3 [M + H]: 155.0703. Found: 155.0706. IR (neat cm–1) 3452, 1612, 1508, 1478, 1438, 1360, 1279, 1210, 1097, 1027, 825, 767, 726, 706, 609.
Naphthalen-1-ol (3f)8c
Following general procedure A, the title compound was prepared using 1-bromonaphthalene (207 mg, 1.0 mmol); EtOAc/hexanes (1:6); 133 mg, 92%; pale-brown solid. m.p.: 95 °C (lit: 91–93 °C).8c1H NMR (400 MHz, DMSO-d6) δ: 10.10 (br s, 1 H), 8.14 (dd, J = 7.2 Hz, J = 1.2 Hz, 1 H), 7.80 (dd, J = 7.2 Hz, J = 1.6 Hz, 1 H), 7.48–7.41 (ovrlp, 2 H), 7.34–7.28 (ovrlp, 2 H), 6.88 (dd, J = 7.2 Hz, J = 1.6 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 153.2, 134.4, 127.4, 126.4, 126.1, 124.6, 124.5, 122.0, 118.3, 108.0. HRMS (ESI) Calcd for C10H9O [M + H]: 145.0648. Found: 145.0646. IR (neat cm–1) 3277, 1598, 1457, 1385, 1268, 1148, 1083, 1043, 1015, 876, 860, 789, 764, 710.
2,3-Dimethoxynaphthalen-1-ol (3g)
Following general procedure A, the title compound was prepared using 1-bromo-2,3-dimethoxynaphthalene (267 mg, 1.0 mmol); EtOAc/hexanes (1:6); 202 mg, 99%; yellow oil. 1H NMR (400 MHz, DMSO-d6) δ: 9.53 (br s, 1 H), 8.01 (d, J = 8.4 Hz, 1 H), 7.69 (d, J = 8.0 Hz, 1 H), 7.35 (ddd, J = 8.0 Hz, J = 6.8 Hz, J = 1.2 Hz, 1 H), 7.28 (ddd, J = 8.4 Hz, J = 6.8 Hz, J = 1.2 Hz, 1 H), 6.87 (s, 1 H), 3.89 (s, 3 H), 3.77 (s, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 152.7, 144.6, 133.8, 130.7, 126.3, 125.5, 122.5, 121.6, 121.1, 98.2, 60.5, 55.5. HRMS (ESI) Calcd for C12H13O3 [M + H]: 205.0859. Found: 205.0852. IR (neat cm–1) 3500, 1739, 1635, 1600, 1509, 1476, 1295, 1265, 1231, 1195, 1120, 1095, 1032, 1015, 975, 859, 812, 735, 700.
Quinoxalin-6-ol (3h)15f
Following general procedure A, the title compound was prepared using 6-bromoquinoxaline (209 mg, 1.0 mmol); EtOAc/MeOH (20:1); 130.5 mg, 89%; brown solid. m.p.: 251–253 °C (lit: 252–254 °C).261H NMR (400 MHz, DMSO-d6) δ: 10.53 (br s, 1 H), 8.77 (d, J = 2.0 Hz, 1 H), 8.68 (d, J = 1.6 Hz, 1 H), 7.93 (d, J = 9.2 Hz, 1 H), 7.41 (dd, J = 9.2 Hz, J = 2.8 Hz, 1 H), 7.27 (d, J = 2.8 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 158.8, 145.4, 144.0, 142.2, 137.6, 130.4, 122.9, 109.5. Anal. Calcd for C8H6N2O: C, 65.75; H, 4.14. Found: C, 65.63; H, 4.12. HRMS (ESI) Calcd for C8H7N2O [M + H]: 147.0553. Found: 147.0548. IR (neat cm–1) 1610, 1478, 1409, 1304, 1231, 1197, 1117, 1036, 941, 831, 750.
1-Benzyl-1H-indol-6-ol (3i)
Following general procedure A, the title compound was prepared using 1-benzyl-6-chloro-1H-indole (241 mg, 1.0 mmol); EtOAc/hexanes (1:4); 182.1 mg, 82%; pale-brown solid. 1H NMR (400 MHz, DMSO-d6) δ: 8.99 (br s, 1 H), 7.35 (d, J = 8.4 Hz, 1 H), 7.30 (t, J = 7.2 Hz, 2 H), 7.26 (d, J = 3.2 Hz, 1 H), 7.24 (t, J = 7.2 Hz, 1 H), 7.15 (d, J = Hz, 2 H), 6.73 (d, J = 2.0 Hz, 1 H), 6.60 (dd, J = 8.4 Hz, J = 2.0 Hz, 1 H), 6.36 (dd, J = 3.2 Hz, J = 0.4 Hz, 1 H), 5.29 (s, 2 H). 13C NMR (100 MHz, DMSO-d6) δ: 153.2, 138.4, 136.9, 128.5, 127.3, 127.2, 126.8, 121.6, 120.9, 109.7, 100.8, 95.4, 49.1. m.p.: 103–104 °C. HRMS (ESI) Calcd for C15H12NO [M – H]: 222.0924. Found: 222.0941. IR (neat cm–1) 3308, 1625, 1580, 1507, 1468, 1354, 1321, 1267, 1168, 1074, 943, 827, 799, 745, 705.
Benzothiophen-5-ol (3j)27
Following general procedure A, the title compound was prepared using 5-chlorobenzothiophene (169 mg, 1.0 mmol) at 100 °C; EtOAc/hexanes (1:6); 143.6 mg, 96%; off-white solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.42 (br s, 1 H), 7.74 (d, J = 8.8 Hz, 1 H), 7.65 (d, J = 5.6 Hz, 1 H), 7.27 (dd, J = 5.6 Hz, J = 0.4 Hz, 1 H), 7.21 (d, J = 2.0 Hz, 1 H), 6.88 (ddd, J = 8.4 Hz, J = 2.0 Hz, J = 0.4 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 154.9, 140.9, 129.4, 128.0, 123.5, 123.1, 114.8, 108.2. m.p.: 105–106 °C (lit: 103–105 °C).28 HRMS (ESI) Calcd for C8H6OS [M]: 150.0134. Found: 150.0133. IR (neat cm–1) 3235, 1599, 1565, 1502, 1422, 1231, 1149, 945, 889, 849, 748, 692.
Benzofuran-7-ol (3k)29
Following general procedure A, the title compound was prepared using 7-chlorobenzofuran (153 mg, 1.0 mmol); EtOAc/hexanes (1:4); 125 mg, 93%; pale-yellow oil. 1H NMR (400 MHz, DMSO-d6) δ: 9.96 (br s, 1 H), 7.91 (d, J = 2.0 Hz, 1 H), 7.06 (d, J = 7.6 Hz, 1 H), 7.02 (t, J = 7.6 Hz, 1 H), 6.87 (d, J = 2.0 Hz, 1 H), 6.74 (d, J = 7.6 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 145.4, 143.4, 142.8, 129.1, 123.5, 111.6, 110.3, 107.0. HRMS (ESI) Calcd for C8H7O2 [M + H]: 135.0441. Found: 135.0440. IR (neat cm–1) 3323, 1593, 1443, 1297, 1217, 1122, 841.
Benzo-2,1,3-thiadiazol-5-ol (3l)
Following general procedure A, the title compound was prepared using 5-chlorobenzo-2-1-3-thiadiazole (171 mg, 1.0 mmol); EtOAc/hexanes (1:6); 145 mg, 95%; pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 10.55 (br s, 1 H), 7.90 (d, J = 9.2 Hz, 1 H), 7.33 (dd, J = 9.2 Hz, J = 2.4 Hz, 1 H), 7.15 (dd, J = 2.4 Hz, J = 0.4 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 159.3, 156.0, 150.1, 125.2, 121.5, 100.4. m.p.: 161–162 °C. HRMS (ESI) Calcd for C6H5N2OS [M + H]: 153.0117. Found: 153.0114. IR (neat cm–1) 3133, 1613, 1495, 1403, 1226, 1187, 1127, 874, 833, 814, 753, 664, 626.
Dibenzothiophen-2-ol (3m)30
Following general procedure A, the title compound was prepared using 2-bromodibenzothiophene (263 mg, 1.0 mmol). After work up, the crude product was purified by flash chromatography using EtOAc/hexanes (1:6) as an eluent to afford dibenzothiophen-2-ol (3m) (185 mg, 92%) as a pale-brown solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.68 (br s, 1 H), 8.23–8.18 (m, 1 H), 7.97–7.91 (m, 1 H), 7.78 (d, J = 8.8 Hz, 1 H), 7.68 (d, J = 2.4 Hz, 1 H), 7.49–7.42 (ovrlp, 2 H), 7.03 (dd, J = 8.4 Hz, J = 2.4 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 155.4, 139.7, 136.3, 135.0, 128.5, 126.8, 124.4, 123.6, 123.0, 121.9, 116.7, 107.4. m.p.: 156–157 °C (lit: 158 °C).31 Anal. Calcd for C12H8OS: C, 71.97; H, 4.03. Found: C, 71.73; H, 4.04. IR (neat cm–1) 3385, 1602, 1470, 1427, 1336, 1187, 849, 810, 755, 733.
1-Trityl-1H-indazol-3-ol (3n)
Following general procedure A, the title compound was prepared using 3-chloro-1-trityl-1H-indazole (S1) (395 mg, 1.0 mmol); EtOAc/hexanes (1:6); 297 mg, 0.79 mmol, 79%; pale-brown solid. m.p.: 196–197 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.98 (s, 1 H), 7.58 (d, J = 8.0 Hz, 1 H), 7.36 (d, J = 7.2 Hz, 6 H), 7.28 (t, J = 7.2 Hz, 6 H), 7.22 (t, J = 7.2 Hz, 3 H), 7.00 (t, J = 7.2 Hz, 1 H), 6.93 (t, J = 7.2 Hz, 1 H), 6.40 (d, J = 8.4 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ: 144.9, 133.2, 132.7, 119.2, 117.6, 116.9, 116.3, 110.1, 109.5, 105.8, 103.6, 67.0. HRMS (ESI) Calcd for C26H19N2O [M – H]: 375.1503. Found: 375.1507. IR (neat cm–1) 1671, 1489, 1450, 1217, 1001, 906, 746, 705, 632.
2,4-Dimethylphenol (5a)32
Following general procedure B, the title compound was prepared using 1-bromo-2,4-dimethylbenzene (185 mg, 1.0 mmol); EtOAc/hexanes (1:6); 87 mg, 71%); volatile, pale-brown oil. The 1H NMR yield of the title product was determined to be 96% based on 0.25 mmol ArBr. 1H NMR (400 MHz, DMSO-d6) δ: 8.93 (br s, 1 H), 6.84 (s, 1 H), 6.76 (dd, J = 8.0 Hz, J = 1.6 Hz, 1 H), 6.64 (d, J = 8.0 Hz, 1 H), 2.15 (s, 3 H), 2.07 (s, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 153.0, 131.1, 127.0, 126.8, 123.4, 114.4, 20.1, 15.9. HRMS (ESI) Calcd for C8H10O [M]: 122.0726. Found: 122.0725. IR (neat cm–1) 3377, 2921, 1506, 1325, 1262, 1204, 1117, 929, 808, 766.
3′-Hydroxy-4′-methylacetophenone (5b)33
Following general procedure B, the title compound was prepared using 3′-bromo-4′-methylacetophenone (213 mg, 1.0 mmol). EtOAc/hexanes (1:2); 150 mg, 100%; off-white solid. m.p.: 121–122 °C (lit: 119–120 °C).331H NMR (400 MHz, DMSO-d6) δ: 9.68 (br s, 1 H), 7.34–7.32 (ovrlp, 2 H), 7.18 (d, J = 8.0 Hz, 1 H), 2.48 (s, 3 H), 2.17 (s, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 197.4, 155.5, 135.9, 130.7, 130.3, 119.6, 113.2, 26.5, 16.2. HRMS (ESI) Calcd for C9H11O2 [M + H]: 151.0754. Found: 151.0752. IR (neat cm–1) 3404, 1659, 1580, 1418, 1349, 1285, 1238, 1129, 899, 884, 813, 712, 688, 608.
3,4,5-Trimethoxyphenol (5c)34
Following general procedure B, the title compound was prepared using 5-bromo-1,2,3-trimethoxybenzene (247 mg, 1.0 mmol), 1 (25.7 mg, 0.03 mmol, 3 mol %), and L5 (14.6 mg, 0.03 mmol, 3 mol %); EtOAc/hexanes (1:1); 168 mg, 91%; pale-brown solid. m.p.: 147–148 °C (lit: 146–147 °C).201H NMR (400 MHz, DMSO-d6) δ: 9.19 (br s, 1 H), 6.05 (s, 2 H), 3.69 (s, 6 H), 3.55 (s, 3 H). 13C NMR (100 MHz, DMSO-d6) δ: 153.8, 153.3, 130.3, 92.8, 60.1, 55.5. HRMS (ESI) Calcd for C9H11O4 [M – H]: 183.0663. Found: 183.0671. IR (neat cm–1) 3254, 1599, 1428, 1218, 1177, 1121, 990, 811, 777.
N,N-Diethyl-4-hydroxybenzamide (5d)35
Following general procedure B, the title compound was prepared using 4-bromo-N,N-diethylbenzamide (256 mg, 1.0 mmol); EtOAc/hexanes (3:1); 173 mg, 89%; pale-brown solid. m.p.: 121–123 °C (lit: 120 °C).351H NMR (400 MHz, DMSO-d6) δ: 9.74 (br s, 1 H), 7.18 (d, J = 8.4 Hz, 2 H), 6.78 (d, J = 8.8 Hz, 2 H), 3.30 (br s, 4 H), 1.08 (t, J = 7.2 Hz, 6 H). 13C NMR (100 MHz, DMSO-d6) δ: 170.2, 158.2, 128.1, 127.7, 114.8, 13.5. HRMS (ESI) Calcd for C11H14NO2 [M – H]: 192.1030. Found: 192.1039. IR (neat cm–1) 3076, 1570, 1434, 1361, 1316, 1277, 1244, 1170, 1101, 837, 764, 597.
4-Hydroxybenzophenone (5e)36
Following general procedure B, the title compound was prepared using 4-bromobenzophenone (261 mg, 1.0 mmol); EtOAc/hexanes (1:4); 196 mg, 99%; pale-brown solid. m.p.: 133–134 °C (lit: 133.5–134.5 °C).361H NMR (400 MHz, DMSO-d6) δ: 10.45 (br s, 1 H), 7.68–7.64 (ovrlp, 4 H), 7.62 (t, J = 7.2 Hz, 1 H), 7.52 (t, J = 7.2 Hz, 2 H), 6.90 (d, J = 8.4 Hz, 2 H). 13C NMR (100 MHz, DMSO-d6) δ: 194.3, 162.0, 138.1, 132.5, 131.8, 129.1, 128.4, 127.9, 115.3. HRMS (ESI) Calcd for C13H9O2 [M – H]: 197.0608. Found: 197.0609. IR (neat cm–1) 3137, 1632, 1599, 1557, 1512, 1444, 1312, 1286, 1232, 1172, 1148, 939, 924, 848, 740, 694, 608.
4-Hydroxybenzonitrile (5f)37
Following general procedure B, the title compound was prepared using 4-bromobenzonitrile (364 mg, 2.0 mmol, 2 equiv), 1 (68.4 mg, 0.08 mmol, 4 mol %), L5 (38.8 mg, 0.08 mmol, 4 mol %), aqueous CsOH solution [CsOH (900 mg, 6.0 mmol, 6 equiv) dissolved in deionized water (360 mg, 20.0 mmol, 20 equiv) as prepared in the preceding procedures], and 1,4-dioxane (4.0 mL); EtOAc/hexanes (1:2); 230 mg, 97%; pale-brown solid. m.p.: 109–110 °C (lit.: 111–112 °C).371H NMR (400 MHz, DMSO-d6) δ: 10.61 (br s, 1 H), 7.62 (d, J = 9.2 Hz, 2 H), 6.90 (d, J = 8.8 Hz, 2 H). 13C NMR (100 MHz, DMSO-d6) δ: 161.7, 134.2, 119.6, 116.4, 101.0. HRMS (ESI) Calcd for C7H9N2O [M + NH4]: 137.0709. Found: 137.0709. IR (neat cm–1) 3263, 2230, 1585, 1507, 1438, 1365, 1281, 1219, 1165, 835, 750, 701.
Acknowledgments
We thank the National Institutes of Health (GM58160) for financial support of this project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to Dr. Christine Nguyen (M.I.T.) and Dr. J. Robb DeBergh (M.I.T.) for help with preparation of this manuscript. We acknowledge Dr. Naoyuki Hoshiya (M.I.T.) for the preparation of L5 and Mr. Nick Bruno (M.I.T.) for the preparation of 1. C.W.C. thanks the Croucher Foundation (Hong Kong) for a postdoctoral fellowship. M.I.T. has patents on some of the ligands and precatalysts used in this work from which S.L.B. and former/current co-workers receive royalty payments.
Supporting Information Available
Experimental and spectroscopic data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): M.I.T. has patents on some of the ligands and precatalysts used in this work from which S.L.B. and former/current co-workers receive royalty payments.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For examples, see:; a Miyashita K.; Imanishi T. Chem. Rev. 2005, 105, 4515–4536. [DOI] [PubMed] [Google Scholar]; b Fan H.; Peng J.; Hamann M. T.; Hu J.-F. Chem. Rev. 2008, 108, 264–287. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nandy J. P.; Prakesch M.; Khadem S.; Reddy P. T.; Sharma U.; Arya P. Chem. Rev. 2009, 109, 1999–2060. [DOI] [PubMed] [Google Scholar]; d Walker S. R.; Carter E. J.; Huff B. C.; Morris J. C. Chem. Rev. 2009, 109, 3080–3098. [DOI] [PubMed] [Google Scholar]; e Bräse S.; Encinas A.; Keck J.; Nising C. F. Chem. Rev. 2009, 109, 3903–3990. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a O’Connor R. D.; Poliks B.; Bolton D. H.; Goetz J. M.; Byers J. A.; Wooley K. L.; Schaefer J. Macromolecules 2002, 35, 2608–2617. [Google Scholar]; b Gustin J.-L. Org. Process Res. Dev. 2006, 10, 1263–1274. [Google Scholar]; c Ogoshi T.; Onodera T.; Yamagishi T.; Nakamoto Y. Macromolecules 2008, 41, 8533–8536. [Google Scholar]; d Tang R.-C.; Tang H.; Yang C. Ind. Eng. Chem. Res. 2010, 49, 8894–8901. [Google Scholar]; e Ryu B.-Y.; Emrick T. Macromolecules 2011, 44, 5693–5700. [Google Scholar]; f Katsoulidis A. P.; Kanatzidis M. G. Chem. Mater. 2011, 23, 1818–1824. [Google Scholar]
- a The approved drugs containing phenol motifs could be searched in the DrugBank (http://www.drugbank.ca/) (accessed on Dec 30, 2013).; b Vitaku E.; Ilardi E. A.; Njarđarson J. T.. Top 200 Pharmaceutical Products by US Retail Sales in 2012 and Top 200 Pharmaceutical Products by US Prescription in 2012. (http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster/) (accessed on Dec 30, 2013).
- a http://www.alanwood.net/pesticides/ (accessed on Dec 30, 2013).; b Cantrell C. L.; Dayan F. E.; Duke S. O. J. Nat. Prod. 2012, 75, 1231–1242. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Bunnet J. F.; Zahler R. E. Chem. Rev. 1951, 49, 273–412. [Google Scholar]; b Miller J.Aromatic Nucleophilic Substitution; Elsevier: New York, 1969. [Google Scholar]; c Terrier F.Nucleophilic Aromatic Displacement. The Influence of the Nitro Group; VCH: New York, 1991. [Google Scholar]; d Imoto M.; Matsui Y.; Takeda M.; Tamaki A.; Taniguchi H.; Mizuno K.; Ikeda H. J. Org. Chem. 2011, 76, 6356–6361. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Smith M. B.; March J.. March’s Advanced Organic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, 2007. [Google Scholar]; b Hanson P.; Jones J. R.; Gilbert B. C.; Timms A. W. J. Chem. Soc., Perkin Trans. 2 1991, 1009–1017. [Google Scholar]
- For examples, see:; a Chakraborti A. K.; Sharma L.; Nayak M. K. J. Org. Chem. 2002, 67, 6406–6414. [DOI] [PubMed] [Google Scholar]; b Ramesh C.; Ravindranath N.; Das B. J. Org. Chem. 2003, 68, 7101–7103. [DOI] [PubMed] [Google Scholar]; c Bartoli G.; Bosco M.; Carlone A.; Locatelli M.; Marcantoni E.; Melchiorre P.; Sambria L. Adv. Synth. Catal. 2006, 348, 905–910. [Google Scholar]; d Zuo L.; Yao S.; Wang W.; Duan W. Tetrahedron Lett. 2008, 49, 4054–4056. [Google Scholar]; e Davidson J. P.; Sarma K.; Fishlock D.; Welch M. H.; Sukhtankar S.; Lee G. M.; Martin M.; Cooper G. F. Org. Process Res. Dev. 2010, 14, 477–480. [Google Scholar]; f Procopio A.; Cravotto G.; Oliverio M.; Costanzo P.; Nardi M.; Paonessa R. Green Chem. 2011, 13, 436–443. [Google Scholar]; g Sergeev A. G.; Webb J. D.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 20226–20229. [DOI] [PubMed] [Google Scholar]
- For the recent examples, see:; a Prakash G. K. S.; Chacko S.; Panja C.; Thomas T. E.; Gurung L.; Rasul G.; Mathew T.; Olah G. A. Adv. Synth. Catal. 2009, 351, 1567–1574. [Google Scholar]; b Inamoto K.; Nozawa K.; Yonemoto M.; Kondo Y. Chem. Commun. 2011, 47, 11775–11777. [DOI] [PubMed] [Google Scholar]; c Molander G. A.; Cavalcanti L. N. J. Org. Chem. 2011, 76, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rayment E. J.; Summerhill N.; Anderson E. A. J. Org. Chem. 2012, 77, 7052–7060. [DOI] [PubMed] [Google Scholar]; e Zhu C.; Wang R.; Falck J. R. Org. Lett. 2012, 14, 3494–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Gogoi A.; Bora U. Synlett 2012, 23, 1079–1081. [Google Scholar]; g Zou Y.-Q.; Chen J.-R.; Liu X.-P.; Lu L.-Q.; Davis R. L.; Jørgensen K. A.; Xiao W.-J. Angew. Chem., Int. Ed. 2012, 51, 784–788. [DOI] [PubMed] [Google Scholar]; h Mulakayala N.; Ismail; Kumar K. M.; Rapolu R. K.; Kandagatla B.; Rao P.; Oruganti S.; Pal M. Tetrahedron Lett. 2012, 53, 6004–6007. [Google Scholar]; i Chen D.-S.; Huang J.-M. Synlett 2013, 24, 499–501. [Google Scholar]; j Gogoi A.; Bora U. Tetrahedron Lett. 2013, 54, 1821–1823. [Google Scholar]
- a Xu J.; Wang X.; Shao C.; Su D.; Cheng G.; Hu Y. Org. Lett. 2010, 12, 1964–1967. [DOI] [PubMed] [Google Scholar]; b Yang H.; Li Y.; Jiang M.; Wang J.; Fu H. Chem.—Eur. J. 2011, 17, 5652–5660. [DOI] [PubMed] [Google Scholar]; c Kaboudin B.; Abedi Y.; Yokomatsu T. Eur. J. Org. Chem. 2011, 6656–6662. [Google Scholar]
- For the recent reports of the preparations of (hetero)arylboronic acids and their derivatives, see: (I) From (hetero)aryl halides:; a Park Y. H.; Ahn H. R.; Canturk B.; Jeon S. I.; Lee S.; Kang H.; Molander G. A.; Ham J. Org. Lett. 2008, 10, 1215–1218. [DOI] [PubMed] [Google Scholar]; b Cho Y. A.; Kim D.-S.; Ahn H. R.; Canturk B.; Molander G. A.; Ham J. Org. Lett. 2009, 11, 4330–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Molander G. A.; Trice S. L. J.; Kennedy S. M.; Dreher S. D.; Tudge M. T. J. Am. Chem. Soc. 2012, 134, 11667–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Molander G. A.; Trice S. L. J.; Kennedy S. M. Org. Lett. 2012, 14, 4814–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Molander G. A.; Cavalcanti L. N.; García-García C. J. Org. Chem. 2013, 78, 6427–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (II) From (hetero)arenes:; f Murphy J. M.; Tzschucke C. C.; Hartwig J. F. Org. Lett. 2007, 9, 757–760. [DOI] [PubMed] [Google Scholar]; g Tzschucke C. C.; Murphy J. M.; Hartwig J. F. Org. Lett. 2007, 9, 761–764. [DOI] [PubMed] [Google Scholar]
- Maleczka R. E. Jr.; Shi F.; Holmes D.; Smith M. R. III J. Am. Chem. Soc. 2003, 125, 7792–7793. [DOI] [PubMed] [Google Scholar]
- For the recent examples of the transition-metal-catalyzed aryl C–H hydroxylations to form phenols, see: (I) Via functional group-directed aryl C–H hydroxylation:; a Kim S. H.; Lee H. S.; Kim S. H.; Kim J. N. Tetrahedron Lett. 2008, 49, 5863–5866. [Google Scholar]; b Makhlynets O. V.; Das P.; Taktak S.; Flook M.; Mas-Ballesté R.; Rybak-Akimova E. V.; Que L. Jr. Chem.—Eur. J. 2009, 15, 13171–13180. [DOI] [PubMed] [Google Scholar]; c Zhang Y.-H.; Yu J.-Q. J. Am. Chem. Soc. 2009, 131, 14654–14655. [DOI] [PubMed] [Google Scholar]; d Yang Y.; Lin Y.; Rao Y. Org. Lett. 2012, 14, 2874–2877. [DOI] [PubMed] [Google Scholar]; e Choy P. Y.; Kwong F. Y. Org. Lett. 2013, 15, 270–273. [DOI] [PubMed] [Google Scholar]; f Yan Y.; Feng P.; Zheng Q.-Z.; Liang Y.-F.; Lu J.-F.; Cui Y.; Jiao N. Angew. Chem., Int. Ed. 2013, 52, 5827–5831. [DOI] [PubMed] [Google Scholar]; g Yang X.; Shan G.; Rao Y. Org. Lett. 2013, 15, 2334–2337. [DOI] [PubMed] [Google Scholar]; h Liu W.; Ackermann L. Org. Lett. 2013, 15, 3484–3486. [DOI] [PubMed] [Google Scholar]; i Zhang H.-Y.; Yi H.-M.; Wang G.-W.; Yang B.; Yang S.-D. Org. Lett. 2013, 15, 6186–6189. [DOI] [PubMed] [Google Scholar]; (II) Via aryl C–H hydroxylation without directing groups:; j Yuzawa H.; Aoki M.; Otake K.; Hattori T.; Itoh H.; Yoshida H. J. Phys. Chem. C 2012, 116, 25376–25387. [Google Scholar]; k Kamata K.; Yamaura T.; Mizuno N. Angew. Chem., Int. Ed. 2012, 51, 7275–7278. [DOI] [PubMed] [Google Scholar]
- For the reviews of the Pd- and Cu-catalyzed hydroxylation of (Het)ArX, see:; a Willis M. C. Angew. Chem., Int. Ed. 2007, 46, 3402–3404. [DOI] [PubMed] [Google Scholar]; b Alonso D. A.; Nájera C.; Pastor I. M.; Yus M. Chem.—Eur. J. 2010, 16, 5274–5284. [DOI] [PubMed] [Google Scholar]; c Enthaler S.; Company A. Chem. Soc. Rev. 2011, 40, 4912–4924. [DOI] [PubMed] [Google Scholar]
- For the recent reports of the Cu-catalyzed hydroxylation of ArX, see:; a Tlili A.; Xia N.; Monnier F.; Taillefer M. Angew. Chem., Int. Ed. 2009, 48, 8725–8728. [DOI] [PubMed] [Google Scholar]; b Zhao D.; Wu N.; Zhang S.; Xi P.; Su X.; Lan J.; You J. Angew. Chem., Int. Ed. 2009, 48, 8729–8732. [DOI] [PubMed] [Google Scholar]; c Yang D.; Fu H. Chem.—Eur. J. 2010, 16, 2366–2370. [DOI] [PubMed] [Google Scholar]; d Paul R.; Ali M. A.; Punniyamurthy T. Synthesis 2010, 24, 4268–4272. [Google Scholar]; e Maurer S.; Liu W.; Zhang X.; Jiang Y.; Ma D. Synlett 2010, 976–978. [Google Scholar]; f Chan C.-C.; Chen Y.-W.; Su C.-S.; Lin H.-P.; Lee C.-F. Eur. J. Org. Chem. 2011, 7288–7293. [Google Scholar]; g Thakur K. G.; Sekar G. Chem. Commun. 2011, 47, 6692–6694. [DOI] [PubMed] [Google Scholar]; h Xu H.-J.; Liang Y.-F.; Cai Z.-Y.; Qi H.-X.; Yang C.-Y.; Feng Y. S. J. Org. Chem. 2011, 76, 2296–2300. [DOI] [PubMed] [Google Scholar]; i Yang K.; Li Z.; Wang Z.; Yao Z.; Jiang S. Org. Lett. 2011, 13, 4340–4343. [DOI] [PubMed] [Google Scholar]; j Songis O.; Boulens P.; Benson C. G. M.; Cazin C. S. J. RSC Adv. 2012, 2, 11675–11677. [Google Scholar]; k Xiao Y.; Xu Y.; Cheon H.-S.; Chae J. J. Org. Chem. 2013, 78, 5804–5809. [DOI] [PubMed] [Google Scholar]
- For the Pd-catalyzed hydroxylation of (Het)ArX, see:; a Anderson K. W.; Ikawa T.; Tundel R. E.; Buchwald S. L. J. Am. Chem. Soc. 2006, 128, 10694–10695. [DOI] [PubMed] [Google Scholar]; b Chen G.; Chan A. S. C.; Kwong F. Y. Tetrahedron Lett. 2007, 48, 473–476. [Google Scholar]; c Gallon B. J.; Kojima R. W.; Kaner R. B.; Diaconescu P. L. Angew. Chem., Int. Ed. 2007, 46, 7251–7254. [DOI] [PubMed] [Google Scholar]; d Schulz T.; Torborg C.; Schäffner B.; Huang J.; Zapf A.; Kadyrov R.; Börner A.; Beller M. Angew. Chem., Int. Ed. 2009, 48, 918–921. [DOI] [PubMed] [Google Scholar]; e Sergeev A. G.; Schulz T.; Torborg C.; Spannenberg A.; Neumann H.; Beller M. Angew. Chem., Int. Ed. 2009, 48, 7595–7599. [DOI] [PubMed] [Google Scholar]; f Yu C.-W.; Chen G. S.; Huang C.-W.; Chern J.-W. Org. Lett. 2012, 14, 3688–3691. [DOI] [PubMed] [Google Scholar]; g Lavery C. B.; Rotta-Loria N. L.; McDonald R.; Stradiotto M. Adv. Synth. Catal. 2013, 355, 981–987. [Google Scholar]
- For the synthesis of L5, see:Hoshiya N.; Buchwald S. L. Adv. Synth. Catal. 2012, 354, 2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the synthesis of 1, see:Bruno N. C.; Buchwald S. L. Org. Lett. 2013, 15, 2876–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. W.; Buchwald S. L. Org. Lett. 2013, 15, 3998–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Under the reaction conditions based on 0.25 mmol of ArX, 4-bromo-3-methylanisole, 5-bromo-1,2,3-trimethoxybenzene, and 5-bromo-2-chlorobenzotrifluoride gave the corresponding phenols in 98, 84, and 90% 1H NMR yields, respectively, via C–Br cleavage.
- Under the reaction conditions based on 0.25 mmol of ArX, 2-bromobenzotrifluoride gave 2-(trifluoromethyl)phenol (76% 1H NMR yield). However, an inseparable mixture of the product and unknown species was isolated by column chromatography.
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. Organometallics 2010, 29, 2176–2179. [Google Scholar]
- Schoevaars A. M.; Hulst R.; Feringa B. L. Tetrahedron Lett. 1994, 35, 9745–9748. [Google Scholar]
- Londhe B. S.; Pratap U. R.; Mali J. R.; Mane R. A. Bull. Korean Chem. Soc. 2010, 31, 2329–2332. [Google Scholar]
- Fausto R.; Chen E. H.; Dershowitz S. J. Am. Chem. Soc. 1959, 81, 4338–4342. [Google Scholar]
- Sharma A.; Kumar R.; Sharma N.; Kumar V.; Sinha A. K. Adv. Synth. Catal. 2008, 350, 2910–2920. [Google Scholar]
- Silk J. A. J. Chem. Soc. 1956, 2058–2063. [Google Scholar]
- Sakairi M.; Kogami M.; Torii M.; Kataoka H.; Fujieda H.; Makino M.; Kataoka D.; Okamoto R.; Miyazawa T.; Okabe M.; Inoue M.; Takahashi N.; Harada S.; Watanabe N. Bioorg. Med. Chem. Lett. 2012, 22, 5123–5128. [DOI] [PubMed] [Google Scholar]
- Martin-Smith M.; Gates M. J. Am. Chem. Soc. 1956, 78, 5351–5357. [Google Scholar]
- Musser J. H.; Chakraborty U.; Bailey K.; Sciortino S.; Whyzmuzis C.; Amin D.; Sutherland C. A. J. Med. Chem. 1987, 30, 62–67. [DOI] [PubMed] [Google Scholar]
- Li H.; Batsanov A. S.; Moss K. C.; Vaughan H. L.; Dias F. B.; Kamtekar K. T.; Bryce M. R.; Monkman A. P. Chem. Commun. 2010, 46, 4812–4814. [DOI] [PubMed] [Google Scholar]
- Nasipuri D.; De Dalal I.; Ghosh S. K. Synthesis 1977, 1, 59–61. [Google Scholar]
- Jia J.; Jiang C.; Zhang X.; Jiang Y.; Ma D. Tetrahedron Lett. 2011, 52, 5593–5595. [Google Scholar]
- Morgan G. T.; Pette A. E. J. Chem. Soc. 1934, 418–422. [Google Scholar]
- Pauson P. L.; Smith B. C. J. Org. Chem. 1953, 18, 1403–1405. [Google Scholar]
- McCabe E. T.; Barthel W. F.; Gertler S. I.; Hall S. A. J. Org. Chem. 1954, 19, 493–498. [Google Scholar]
- Newman M. S.; Pinkus A. G. J. Org. Chem. 1954, 19, 985–991. [Google Scholar]
- Chankeshwara S. V.; Chebolu R.; Chakraborti A. K. J. Org. Chem. 2008, 73, 8615–8618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.