

Abstract
There have been numerous significant advances in catalytic olefin metathesis (OM) during the past two decades. Such progress has transformed this important set of reactions to strategically pivotal processes that generate stereochemical identity while delivering molecules that cannot be easily prepared by alternative routes. In this Perspective, an analysis of the origin of the inception of bidentate benzylidene ligands for Ru-based OM catalysts is first presented. This is followed by an overview of the intellectual basis that culminated in the development of Mo-based diolates and stereogenic-at-Ru complexes for enantioselective OM. The principles accrued from the study of the latter Ru carbenes and Mo alkylidenes and utilized in the design of stereogenic-at-Mo, -W, and -Ru species applicable to enantioselective and Z-selective OM are then discussed. The influence of the recently introduced catalytic OM protocols on the design of synthesis routes leading to complex organic molecules is probed. The impact of a better understanding of the mechanistic nuances of OM toward the discovery of stereoselective catalysts is reviewed as well.
Introduction
Olefin metathesis (OM) is a formidable force in chemistry; it has the inimitable ability to shuffle alkenes; it can convert simple C–C double bonds to those that are difficult to access.1 Otherwise innocuous olefins leap into action in the presence of an OM catalyst. Cross-metathesis (CM) delivers sought-after alkenes by fusing two olefins. Ring-closing metathesis (RCM) transforms linear chains to cyclic olefins of many sizes and shapes, from small rings to macrocycles and from cycloalkenes to macrolactams, macrolactones and peptide rings. OM does not require a base nor does it need an acid; it is typically operative at ambient temperature; it is a rearrangement that imparts intriguing possibilities for development of high impact methods in chemical synthesis. The reverse of RCM, ring-opening metathesis (ROM), a subset of transformations that is customarily coupled with a CM (ring-opening/cross-metathesis or ROCM) or another ROM (ring-opening metathesis polymerization or ROMP), represents an additional fertile dimension.
Catalyst capability fuels all initiatives in reaction development; we go as far as our catalysts take us.2 The same applies to OM, the emergence of which commenced with the arrival of structurally well-defined high oxidation-state alkylidenes (mostly Mo-based) by Schrock,3Mo–1 being the most well-known,4 and Ru-based carbenes exemplified by Ru–1a(5) (Scheme 1) introduced by Grubbs.6 Subsequent progress has brought elevated reactivity; Ru–1c is a widely used complex7 in which a phosphine ligand of Ru–1b(8) is replaced by an N-heterocyclic carbene (NHC).
Scheme 1. Early Complexes Developed for Catalyzing Olefin Metathesis Reactions.

OM can generate olefins while imparting stereochemical identity; in regard to formation of E or Z olefins, this is in contrast to catalytic cross-coupling, where substrates must already possess stereochemical identity. Efficient chiral catalysts for enantioselective OM (EOM) were first reported in 1998, but it was not until 2009 that the discovery of promoters that address the important problem of Z selectivity was disclosed. For years, matters of stereoselectivity were left to the hazards of thermodynamic preferences. The hope that a selective trasnformation might be at hand was limited to when an E-alkene was targeted and then only if a sufficient energy difference separated the two possible isomers and the catalyst was appropriately long lived to bring the equilibration across the finish line.
Here, we dissect the intellectual basis that led to the development of the first instances of enantioselective and Z-selective OM catalysts in our laboratories. A distinctive feature of our program is that it encompasses Ru-based carbenes as well as Mo and W alkylidenes (complexes for OM developed in our laboratories are illustrated in blue; other types of catalysts introduced by us are presented in red). Many advances have been made because of our involvement with several catalyst systems: The lessons learned from efforts in the development of stereoselective Ru-based carbenes guided us in devising selective high oxidation-state alkylidenes. In turn, a deeper comprehension of the mechanistic principles relating to Mo and W complexes enables us to introduce more efficient and selective Ru-based OM catalysts. Such cycles of mechanistic realization/design centered on the two most effective classes of OM catalysts (Figure 1) have been a significant source of cerebral nourishment. The advantages of an inclusive approach to research bring an intimate appreciation of the symbiotic relationship between different types of catalyst systems.
Figure 1.

Continuous cycles of catalyst development.
The Early Phase: OM as an Ancillary Method
OM has the uncanny ability of elevating the utility of a protocol with which it is associated. One of our early encounters with this remarkable set of transformations was in the context of our investigations regarding zirconocene-catalyzed enantioselective allylic alkylations of allylic ethers with alkyl Grignard reagents.9 The advent of well-defined OM catalysts in the early to mid-1990s1,3,6 allowed us to have ready access to a variety of starting materials for the enantioselective C–C bond-forming reactions; preparation of such entities, although feasible by alternative approaches, was otherwise inefficient. An example, reported in 1994,10 is provided in Scheme 2. We showed that an enantiomerically pure 2-substituted dihydropyran [i.e., >99:1 enantiomeric ratio (er)] may be obtained by treatment of a simple racemic allylic ether with 1.0 mol % of Ru–1a for 5.0 h (22 °C), followed by the addition of EtMgCl and 10 mol % of Zr–1a and heating of the mixture at 70 °C for 2.0 h. What was in hand was a one-vessel procedure for preparation of the desired starting material through the use of a Ru-based catalyst that did not hinder the second stage of the process; the need for purification of the cyclic alkene before the alkylation was obviated.11
Scheme 2. Sequential Catalytic Olefin Metathesis/Enantioselective Allylic Alkylation.

OM-Based Rearrangement Processes
OM may be employed to modify the products generated by another enantioselective protocol. The 1998 synthesis of antihypertensive agent nebivolol offers a case in point (Scheme 3).12 Cycloheptadiene oxide was cleaved by an appropriate phenoxide, and the resulting alcohol was masked to afford the syn or anti isomer in the racemic form. Each diastereomer was then kinetically resolved through zirconocene-catalyzed allylic alkylation, providing access to enantiomerically pure materials.13 Subsequent treatment with 4.0 mol % of Mo–1 under an atmosphere of ethylene led to efficient rearrangement of the cycloheptenyl ethers, delivering 2-substituted chromenes,14 which were converted to the target molecule. The latter catalytic isomerization, achieved by a catalytic ring-opening/ring-closing metathesis (RORCM), underscores the ability of catalytic OM to elevate the value of an accompanying method.
Scheme 3. Combination of Catalytic Ring-Opening/Ring-Closing Metathesis and Enantioselective Allylic Alkylation: Total Synthesis of Nebivolol.

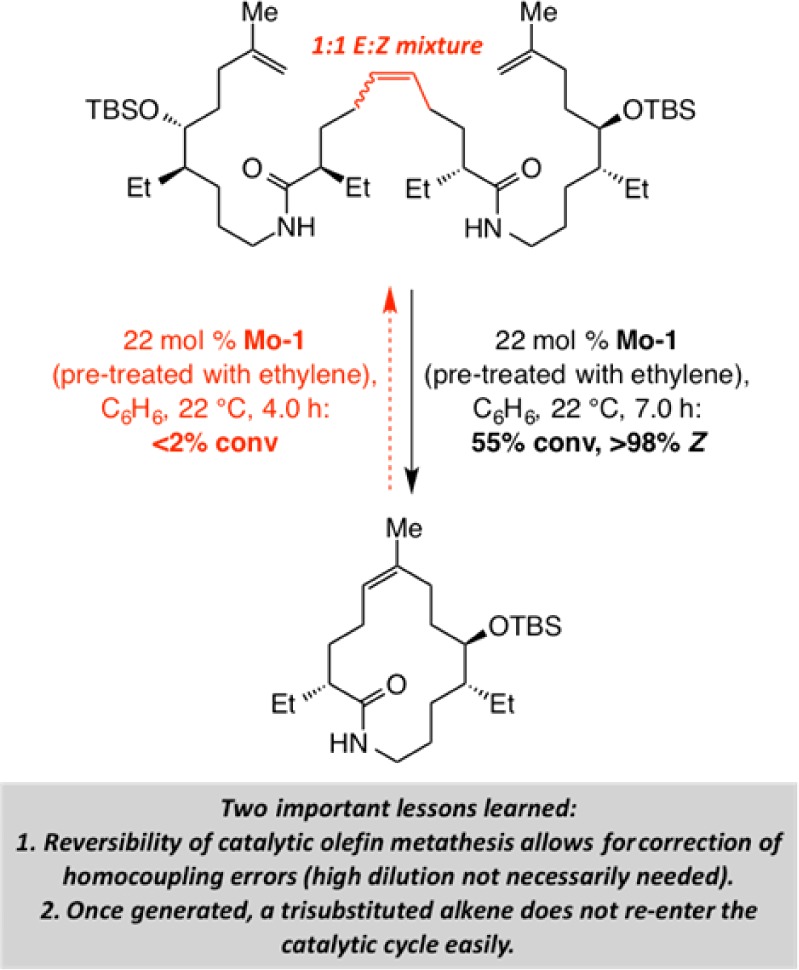
One message gleaned from the sequence shown in Scheme 3 is that we must be mindful of thermodynamic differences between the two sides of a reaction arrow when contemplating the design of a catalytic OM reaction. The high efficiency of the RORCM processes in Scheme 3 is because a cyclohexenyl unit is energetically more favored compared to an unsaturated seven-membered ring; in this way, despite the fact that the product can undergo RCM to regenerate the cycloheptene, the transformation proceeds completely in favor of the smaller ring. Another principle revealed by the nebivolol synthesis relates to the significance of controlling ethylene concentration for achieving the optimal results in a catalytic OM reaction. Initial studies had indicated that without ethylene substantial amounts of the homocoupling product are formed (by reaction of the terminal alkene side chains). With ethylene present, when the product-derived Ru carbene is generated, it reacts rapidly with the more abundant additive to regenerate the terminal olefin. The latter complication, once again, is rooted in the reversible nature of OM; it teaches us that not only the substrate but also the product molecules might contain alkenes that are capable of reacting to regenerate the starting material or undesired side products.
The versatility of catalytic RORCM has been illustrated in natural product synthesis on a number of occasions;15 three examples are provided in Scheme 4. In a 2002 report, Blechert illustrated that a RORCM performed with 5.0 mol % of Ru–1b may be coupled with RCM for assembling the core structure of dihydrocusohygrine.16 The RORCM disclosed four years later by Phillips was capped by a CM to unveil a major portion of cylindramide A.17 The total synthesis of tetracyclic clavilactone A, outlined in 2013 by Takao, entails an initial RORCM promoted by the less active Ru–1b (presumably to minimize cyclobutene-induced oligomerization);18 follow-up treatment with 5.0 mol % of Ru–1c under an atmosphere of ethylene converted the homocoupled product to the desired monomeric entity. The strained cyclodecenyl moiety was later introduced by macrocyclic RCM; fortunately, the desired Z-alkene was obtained stereoselectively almost certainly due to favorable thermodynamic preferences.
Scheme 4. Representative Applications of Catalytic RORCM to Natural Product Synthesis.

An Early Example of Macrocyclic RCM
In 1994, as part of efforts to devise a concise enantioselective total synthesis of antifungal agent fluvirucin B1 (also known as Sch-38516), we had occasion to explore the applicability of several strategic concepts (Scheme 5).19 We showed that the fragments needed for assembling the diene precursor for macrocyclic RCM could be synthesized efficiently by a pathway that relies extensively on catalytic transformations; among them were zirconocene-catalyzed diastereo- and enantioselective carbomagnesation of acyclic and cyclic alkenes, developed previously in our laboratories (Scheme 5). In addition to the better known Cu-catalyzed alkylation of the aziridine and the more widely used catalytic enantioselective directed epoxidation (the primary amine segment) and dihydroxylation reactions (carbohydrate segment), we introduced an alcohol-to-carboxylic acid process as well as a two-stage one-pot catalytic hydrovinyl addition protocol. We demonstrated that by inclusion of sufficient amounts of water in oxidation of an alcohol by (n-Bu)4NRuO4 (tpap) conversion to the carboxylic acid could be achieved; this strategy has subsequently been utilized in other applications and extensively examined.20 We illustrated that the combination of titanocene-catalyzed hydromagnesation of a terminal alkene, performed with an n-alkylmagnesium halide, and a Ni-phosphine-catalyzed cross-coupling with vinyl bromide, constitute a single-vessel catalytic process for net alkene hydrovinyl addition. We used the term “cascade catalysis” to describe the approach, a term that has since become more popular in describing related catalytic approaches.21
Scheme 5. Cascade Catalysis and Macrocyclic RCM in the Mid-1990s: An Early Example of Catalyst-Based Enantioselective Total Synthesis of Fluvirucin B1.

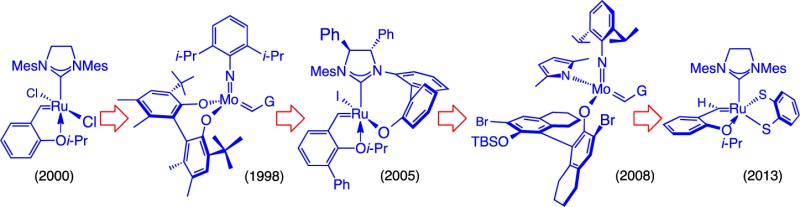
The use of RCM to secure the unsaturated macrocyclic ring of fluvirucin B1 efficiently and stereoselectively is likely the most influential outcome of the two decade-old study.22 The pivotal macrocyclization (Scheme 5) was one of the earliest indications that large ring structures can be accessed efficiently and reliably through OM; contrary to perceptions that remarkably persist today, it demonstrated that Mo alkylidenes remain active in the presence of many Lewis basic functional groups (e.g., a secondary amide or carbamate). Another noteworthy aspect of the RCM in Scheme 5 is the complete control of stereoselectivity (>98% Z), which at the time, we attributed to the strong preference of the unsaturated 14-membered macrolactam ring to exist in one stereoisomeric form (thermodynamic control). In a later discussion, we will examine whether at least some degree of catalyst control might have been in effect (cf. Scheme 33 and related discussion). Regardless of its origin, the exceptional Z selectivity allowed for a simple solution to an otherwise difficult problem in remote stereochemical control: it set the stage for diastereoselective hydrogenation of the trisubstituted alkene that resulted in efficient control of the distal methyl-substituted stereogenic center.
Scheme 33. Z-Selective Ring-Closing Metathesis and Applications to Synthesis of Biologically Active Molecules.

The catalytic macrocyclization performed en route to fluvirucin B1 highlighted several mechanistic principles. One corresponds to the reversibility of catalytic OM. We demonstrated that subjection of the homocoupled product to the RCM conditions leads to efficient formation of the desired 14-membered macrolactam (Scheme 6). The lower reactivity of the cyclic trisubstituted olefin allows the E- or Z-1,2-disubstituted olefin of the homocoupled product to react with the Mo alkylidene to regenerate the precursor to the large ring.23 These observations implied that high dilution may either be superfluous in certain cases or it could be detrimental to efficiency of a macrocyclic RCM that affords a trisubstituted olefin (by lowering RCM rate).
Scheme 6. Significance of the Reversibility of an OM Reaction.
The reversible nature of catalytic OM continues to play a role in catalyst and reaction development. One telling case is that of Smith regarding a total synthesis of the naturally occurring cytotoxic cyclindrocyclophane F (Scheme 7a).24 After a CM and a subsequent macrocyclic RCM, all E-selective processes, the target molecule was obtained through alkene hydrogenation and removal of the methyl ether groups. Mo or Ru complexes shown in Scheme 7a proved to be effective, although it was the former alkylidene (Mo–1) that emerged as the more attractive choice. A notable aspect of the Smith study is the site selectivity with which the tandem CM/RCM takes place: it is exclusively the “head-to-tail” CM product that undergoes macrocyclization. Through a series of experiments (Scheme 7b), it was illustrated that the final high selectivity favoring the “head-to-tail” isomer is probably the result of facile reversibility of the CM reaction and the higher energy of the alternative isomer.
Scheme 7. Reversibility in Catalytic OM: A Notable Example.

A Chelating Ligand for Ru-Based OM Catalysts
The most impactful consequence of our investigations on catalytic RORCM (cf. Scheme 3) was the discovery of Ru complexes with a bidentate benzylidene ligand. As part of our investigations to elucidate the sequence of events leading to the observed products (i.e., ROM followed by RCM or vice versa), we had occasion to treat a mixture of a cyclooctenol ether and 2-ethoxystyrene with 5.0 mol % of Ru–1a (Scheme 8).25 We observed ca. 5% disappearance of the arylalkene and formation of a similar amount of Ru–2a, which, surprisingly, could be isolated and purified by routine silica gel chromatography (unlike the bis-phosphine species). We then prepared the more robust isopropoxy chelated Ru–2b(26) and investigated its use in OM, demonstrating that, unlike the related phosphine systems (i.e., Ru–1a,b), the complex may be recovered and reused. Before long, we reported the synthesis, characterization, and catalytic activity of phosphine-free complex Ru–3a.27 In 2001, we showed that the elements of Ru–3a may be attached to sol–gel glass,28 resulting in a supported system (Ru–3b, Scheme 8) that delivers OM products with minimal Ru contamination. In one case, the bound catalyst was recycled 20 times. We later examined the nuances of the supported system in more detail, establishing that, at least to some extent, the proposed release/return pathway is operative;29 this is a mechanistic angle that remains the subject of some debate.30
Scheme 8. Discovery of Ru-Based Olefin Metathesis Catalysts: Activation by Cross-Metathesis (No Phosphine Release).

Complex Ru–3a is one of the most widely used in OM; phosphine-free Ru carbenes that carry the type of bidentate ligand in Ru–2a,b and Ru–3a have come to dominate the OM scene (see below for examples). Several derivatives have been prepared and examined by others (cf. Scheme 8). The study of 3c–g underlines an attractive property of the bidentate Ru carbenes: through steric (e.g., Ru–3c(31)) or electronic (Ru–3d,e32) attributes of the initiating species, the rate of an OM process can be altered.33 A weaker O→Ru association, caused by a sterically demanding substituent (Ru–3c) or an electron-deficient group (Ru–3d,e), favors formation of the catalytically active, coordinatively unsaturated species that likely undergoes reaction. Similarly, with the less sterically demanding NHC groups (Ru–3f,g34), transformations with bulkier alkenes might be facilitated35 but at the cost of catalyst longevity. Such traits are desirable in situations where the OM process is relatively facile and catalyst durability is less critical; in instances where extended catalyst lifetime is desirable, the parent complex Ru–3a is often the better choice (cf. Scheme 20). Carbene Ru–3h(36) was designed for performing OM in aqueous media.
Scheme 20. A Challenging Metathesis Reaction.

A Versatile Ru-Based Complex
Because of its beneficial attributes, Ru–3a has been used extensively for synthesis of complex organic molecules; three examples are presented in Scheme 9. The RCM reported by Tsantrizos and co-workers at Boehringer-Ingelheim, en route to an antihepatitis C agent BILN 2061 ZW, afforded the desired macrocyclic product 83% yield when 3.0 mol % of Ru–2b was used (0.012 M solution; cf. Scheme 9a);37 this application is one of the earliest examples of its type that was performed at significant scale. The high Z selectivity is likely the result of substrate control.38 Use of phosphine-releasing Ru–1b led to less efficient reactions and epimerization at the neighboring allylic position (cf. highlighted sites); with Ru–1c moderate efficiency was observed. Subsequent studies illustrated that still more efficient macrocyclic RCM could be achieved with Ru–3a or its related derivatives.39 A more recent example is the macrocyclic RCM/hydrogenation sequence reported by scientists at Merck en route to hepatitis C virus inhibitor vaniprevir (also known as MK-7009; Scheme 9b).40 The 20-membered ring was generated in 91% yield with 0.2 mol % Ru–3a on a ∼17 g scale process at a relatively high concentration (0.13 M; simultaneous slow addition). Another case, this time to prepare a cathepsin K inhibitor (SB-462795) and involving the formation of a highly functionalized seven-membered ring, relates to the RCM performed by scientists at GlaxoSmithKline on 80 kg scale, affording nearly 72 kg of the desired product in 96% yield (Scheme 9c); initial studies indicated that other complexes, including phosphine-containing Ru–1c, are much less efficient.41 Use of a tetrahydroxyphosphonium salt allowed removal of the residual transition metal to a substantial degree. The efficient catalytic CM reported by Crimmins in 2006 (Scheme 9d) in the context of an enantioselective total synthesis of cytotoxic natural product mucocin illustrates that Ru–3a can be used to effect the union of two complex fragments;42 with Ru–1c as the catalyst precursor, the CM product was obtained in 53% yield (vs 68% with Ru–3a). The OM reactions in Scheme 9 require elevated temperatures, which might be why the more thermally robust complexes Ru–2b and Ru–3a deliver better results than the more fragile analogues that are activated by the loss of a phosphine ligand.
Scheme 9. Examples of Olefin Metathesis Reactions Performed in the Presence of Bidentate Carbene Ru Complexes 2b and 3a.

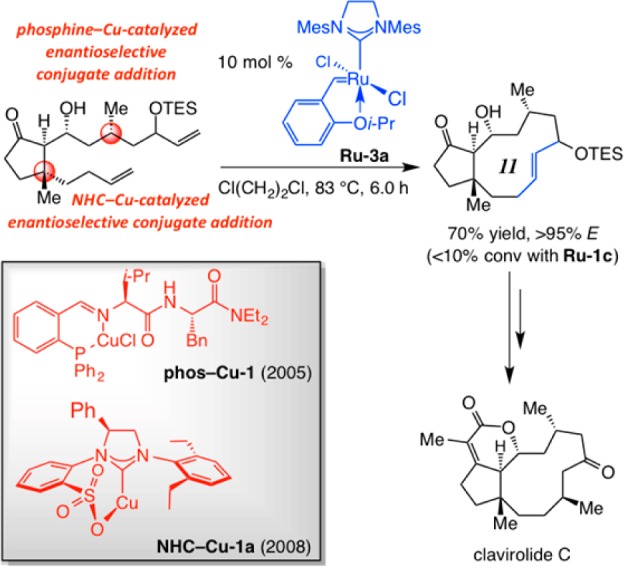
In 2008, we reported the first enantioselective total synthesis of clavirolide C,43 a tricyclic natural product with an 11-membered ring carbocycle (Scheme 10). As was the case with fluvirucin B1 (cf. Scheme 5), other transformations promoted by chiral complexes developed in our laboratories were combined with catalytic OM. There were two enantioselective conjugate additions, one involving an amino acid-based phosphine44 and another a bidentate NHC–Cu complex.45 The medium ring alkene was obtained in 70% yield with 10 mol % of Ru–3a (83 °C, 6.0 h). Reaction in the presence of phosphine-containing Ru–1c, under otherwise identical conditions, led to >90% diene recovery; again, the resulting active carbenes probably do not survive the elevated temperatures needed for the ring formation.
Scheme 10. Applications of Ru-Catalyzed Macrocyclic RCM and Cu-Catalyzed Enantioselective Conjugate Additions to Synthesis of Clavirolide C.
Efficient Catalysts for Enantioselective OM (EOM)
The strategies described thus far involve the use of catalytic OM in conjunction with other enantioselective transformations; a catalytic ROCM or CM or RCM serves to either access the substrates required for a subsequent enantioselective process (see Scheme 3) or, more commonly, utilized to modify an intermediate that has been synthesized in the enantiomerically enriched form (see Schemes 4, 5, 7, 9 and 10). In the mid-1990s, we began to explore the possibility of turning OM into a purveyor of stereochemistry; we set out to develop chiral Mo- as well as Ru-based catalysts that would promote OM transformations enantioselectively.46 One impetus for such initiatives was the realization that many enantiomerically enriched molecules readily accessible by a catalytic enantioselective OM (EOM) would otherwise require a less efficient sequence to prepare.47
Catalytic Enantioselective RCM (ERCM)
In August of 1997, R. R. Schrock and I forged a collaborative initiative with the aim of developing efficient Mo catalysts for EOM.48 The only reported cases of EOM at the time consisted of a limited set of enantioselective RCM (ERCM) processes that involved the use of a chiral Schrock-type Mo alkylidene bearing a nine-membered bidentate hexafluoro-bisalkoxide ligand. The observed selectivities were low in the ERCM-based kinetic resolutions (krel <3),49 and the only reported application to enantioselective desymmetrization proceeded with negligible selectivity [57.5:42.5 enantiomeric ratio (er)].50
In 1998, together with Schrock et al., we introduced chiral Mo diolate Mo–2a as the first efficient complex for kinetic resolution of dienes through ERCM (krel up to >50).51 Soon after, we reported that the corresponding enantioselective triene desymmetrizations furnish cyclopentenyl products in up to >99:1 er (Scheme 11).52 We then established that Mo–3a is especially effective for transformations that generate cyclohexenyl rings.53 Reactions are exceptionally efficient and enantioselective, do not require use of solvent, and can be performed on gram scale. The general stereochemical model in Scheme 11a (cf. I)53b was developed; the more Lewis acidic anti alkylidene isomer1a (alkene substituent oriented away from the imido ligand) reacts in a way that allows the alkene to coordinate with the Mo center while avoiding interaction with the protruding tert-butyl substituent of the bidentate ligand.
Scheme 11. Chiral Mo-Based Diolates for Enantioselective ROM and Comparison with Reactions of Leading Ru-Based OM Catalysts.

The specificity of Mo alkylidenes in promoting different types of ERCM (cf. Scheme 11a) underscore an important principle in catalyst development: an enduringly effective and broadly applicable method in stereoselective transformation frequently entails having the ability to access easily a range of catalysts. Transformations of different substrates, even when distinguished by seemingly subtle differences, might require a complex with a modified structure to proceed with maximum efficiency and selectivity. We would be reminded of this precept later in our investigations (see below).
In 2001, Grubbs and co-workers disclosed the discovery of the first chiral Ru catalysts for EOM; two examples of this beautifully conceived class of carbene complexes, where the stereochemical identity of the NHC moiety controls the orientation of the adjacent NAr groups, are presented in Scheme 11b (Ru–4a,b).54 Comparison of the ERCM reactions promoted by high oxidation-state Mo alkylidenes and Ru carbenes indicates that the former are generally more efficient and provide broader substrate scope: complexes such as Ru–4a,b are not as effective when less substituted alkenes are involved.
Other notable examples of ERCM, catalyzed by Mo diolate alkylidenes, are shown in Scheme 12. In the first instance, Mo–2b, which contains a 2,6-dimethylphenylimido ligand (vs Mo–2a), was used to effect the formation of a 2-alkenyl-substituted piperidine in 83% yield and 93.5:6.5 er; subsequent hydrogenation afforded coniine (Scheme 12a).55 The case in Scheme 12b indicates that the Mo-based chiral catalysts can be employed to generate enantiomerically enriched compounds with a P-substituted stereogenic center.56 An efficient desymmetrization in the presence of Mo–4a, which contains a partially hydrogenated binaphthol ligand, was designed by Ogasawara and Takahashi to form an enantiomerically enriched ferrocene through control of planar stereogenicity (Scheme 12c).57 Finally, the sequence of catalytic kinetic resolutions utilized to access an enantiomerically pure chromium arene based chiral phosphine is noteworthy (Scheme 12c);58 the latter complex promotes Rh-catalyzed enantioselective reaction of arylboronic acids with unsaturated ketones (conjugate additions) and tosylimines. Mo alkylidenes that carry a tetrahydrobinapthol ligand exhibit distinct reactivity compared to those that bear a fully unsaturated binaphthol; the change in the dihedral angle of the bidentate ligand likely causes alterations in the catalyst structure manifested by variations in reactivity and selectivity.59 Such architectural distinctions would play a significant role in the design of stereogenic-at-Mo and W complexes a few years later (see below).
Scheme 12. Representative Applications of Enantioselective Synthesis through RCM with Mo-Based Diolates.

The First Cases of Enantioselective ROCM (EROCM)
The transformations in Scheme 13 offer telling examples of how catalytic EOM can deliver products that are otherwise more difficult to prepare. The tertiary homoallylic silyl ether obtained by enantioselective RORCM (ERORCM) of a cyclopentenol cannot be easily obtained by an alternative approach (Scheme 13a);60 the same applies to the bicyclic tertiary ether that serves as an intermediate in a synthesis of africanol (Scheme 13b),61 or the 2-substituted dihydropyran segment of tipranavir (Scheme 13c).62 As mentioned previously, due to the reversible nature of OM, it is the lower energy of the products (formation of six- vs five-membered rings or release of ring strain in a [2.2.1] bicyclic alkene) that serves as the driving force for the enantioselective rearrangements shown in Scheme 13. Catalytic ERORCM is an efficient protocol for converting relatively simple achiral starting materials to valuable products of high enantiomeric purity.63
Scheme 13. Enantioselective Synthesis through RORCM with Mo-Based Diolates.

Design and Synthesis of Stereogenic-at-Ru Catalysts
In light of strategic advantages of EOM, we initiated a parallel program with the aim of introducing chiral Ru carbenes. The possibility of designing a chiral bidentate NHC ligand appealed to us; this was for several reasons. First, we surmised that such complexes would contain stereogenic Ru centers, an attribute with significant mechanistic implications, as will be described below. We were indeed aware that metal center stereogenicity could lead to difficulties in complex synthesis and generation of diastereomeric mixtures. The possible pitfalls failed to deter us; we convinced ourselves that the stability of phosphine-free complexes to silica gel chromatography might allow us to purify and examine each carbene diastereoisomer. We would soon learn that the initially perceived complications mentioned above are not only inconsequential, the presence of diastereomeric carbenes in a catalytic cycle translates to mechanistically revealing and at times beneficial reactivity/selectivity profiles. Our decision to prepare and investigate the chemistry of stereogenic-at-metal complexes would prove to be propitious in the long run, as the selection of this particular line of catalyst design would exert a critical impact that would lead to the advent of several selective OM catalysts. Furthermore, we would later show that bidentate NHC ligands originally designed to promote EOM, would play a significant role in facilitating the progress of other C–C, C–B as well as C–Si bond forming reactions.
The first stage of our studies led us to develop a completely diastereoselective method for preparation of binaphthyl bridging carbene complex Ru–5 in 2002 (Scheme 14).64 The high stereoselectivity (>98% dr) is mechanistically noteworthy (see below) and was an auspicious finding from the preparative viewpoint. In a limited number of cases, we demonstrated that Ru–5 can promote EROCM selectively.
Scheme 14. Stereogenic-at-Ru Complexes for Enantioselective Olefin Metathesis. Diastereoselective Synthesis, Structure, and Reactivity Profiles.

To improve catalytic activity, we synthesized and investigated a number of related systems where the aryl group of the bidentate isopropxy carbene was modified. Whereas electronic modification gave rise to moderate increase in efficiency (cf. Ru–3d, Scheme 8), incorporation of a phenyl unit ortho to the chelating ether oxygen (Ru–6a, Scheme 14), based on a previous report by Blechert31 (cf. Ru–3c, Scheme 8), enhanced EROCM rate by more than 2 orders of magnitude.65 The latest generation of bidentate chiral Ru carbenes led us to Ru–7a, where, as with the complexes reported by Grubbs in 200154 (cf. Ru–4a,b, Scheme 11), the stereogenic centers of the NHC moiety ensures the formation of a single isomer upon ligand complexation with the Ru center.66 Grubbs and co-workers had shown that the corresponding Ru–iodide, formed in situ by treatment of the chloride with NaI, while less active, gives rise to more enantioselective reactions.54 Based on the aforementioned finding, we prepared, characterized, and investigated complexes Ru–6b and Ru–7b (Scheme 14).
The route in Scheme 14, leading to the formation of Ru–7a, is representative of the procedure used to prepare this class of Ru-based carbenes. Subjection of the appropriate imidazolinium salt to Ag2O afforded head-to-tail dimeric complex Ag–1 in quantitative yield with complete control of diastereoselectivity. Treatment of the air stable silver complex with phosphine complex Ru–2c led to clean ligand exchange and generation of the stereogenic-at-Ru carbene (Ru–7a), which was isolated as a single stereoisomer (>98% exo; see below for a discussion of the significance of this diastereoselectivity).67
Stereogenic-at-Ru Complexes as Chiral OM Catalysts
The findings summarized in Scheme 14 illustrate the utility of the chiral Ru complexes, and underscore the reactivity/selectivity differences between binaphthyl- and biphenyl-bridged carbenes Ru–6 and Ru–7 and their chloro- vs iodo-Ru derivatives. In the first instance, the 4-hydoxypyran is produced with similar efficiency in all cases and with slightly higher enantioselectivity when the earlier generation complexes (Ru–6a,b) are used. The ability of a Ru iodide to enhance enantioselectivity is more evident when the transformations with Ru–7a and Ru–7b are compared (85.5:14.5 vs 92:8 er). The distinctions between different chiral carbenes become more pronounced with the more challenging EROCM processes that yield a ketopyran (Scheme 14). Here, the binaphthyl containing Ru–6a performed less effectively than chloride Ru–7a or iodide Ru–7b (50% conv in 48 h vs >98% conv in 1.5 h or less); use of iodo-carbene Ru–6b led to <2% conversion.
The route leading to enantiomerically pure baconipyrone C, reported by us in 2007,68 constitutes the first example of a total synthesis involving a Ru-catalyzed EOM (Scheme 15). A key step is the EROCM with styrene, carried out in the presence of 2.0 mol % Ru–7b at −15 °C without solvent; desymmetrization of the oxabicycle furnished the fully functionalized pyran in 62% yield and 94:6 er.69 Rupture of the C–O bond took place with complete chemoselectivity at the site proximal to the C2 β-styrene substituent (vs C6 vinyl); this was followed by site-selective protonation exclusively at the benzylic position. The latter transformation provided the desired acyclic diene with the internal alkene at the preferred site such that oxidative cleavage generated the carboxylic ester at the appropriate position. Another highlight is the use of chiral NHC–Cu complex Cu–1b to effect an efficient enantio- and diastereoselectoive double allylic alkylation70 of a bis-allylic phosphate with inexpensive Me3Al. In this way, the stereogenic centers required for the synthesis of the diketo segment of the natural product were obtained in a single operation. An amusing aside is that the bidentate sulfonate bridged NHC ligand was originally designed for applications in Ru-catalyzed OM. We were unable to prepare the targeted complexes, but the derived Cu-based carbenes have emerged as uniquely efficient catalysts for a range of C–C and C–B bond-forming reactions (for another example, see Scheme 10).71
Scheme 15. First Application of Ru-Catalyzed EOM in Natural Product Total Synthesis.

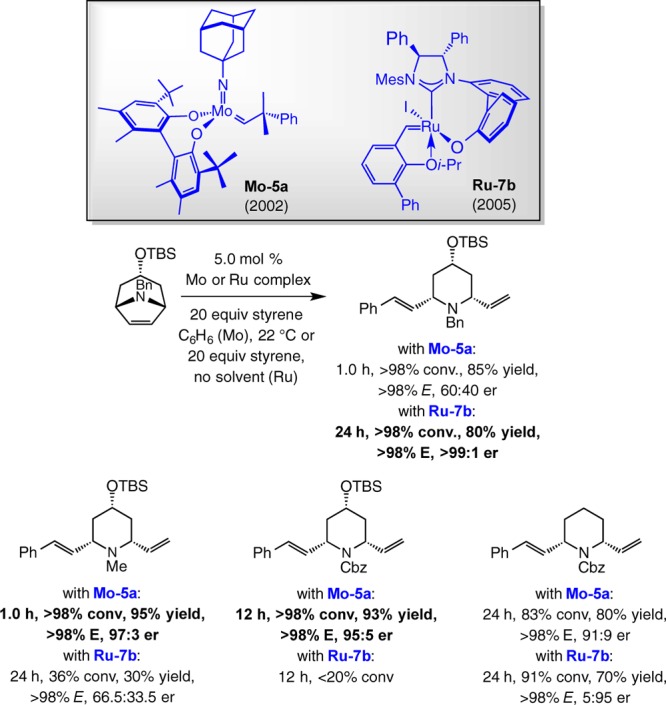
Analysis of the performance of other chiral complexes in promoting the EROCM en route to baconipyrone C is instructive (Scheme 15). Reaction with the adamantylimido Mo-based diolate Mo–5a, which emerged as the most effective alkylidene for the desymmetrization process, was the most efficient (>98% conv in 5.0 h vs 15 h at 22 °C) but took place with diminished enantioselectivity (83.5:16.5 vs 89.5:10.5 er with Ru–7b, respectively).72 The Ru-catalyzed reaction delivered higher selectivity at lower temperature (94:6 er), while there was negligible difference with Mo–5a under the same conditions. As before, binaphthyl-containing carbene Ru–6b, although reasonably enantioselective (90:10 er), occurred in 44% yield after 44 h at 22 °C. None of the Z isomers were detected in any of the transformations (more on this later).
A time-tested credo of catalyst development is that there are no promoter systems that can be considered “universally optimal”—a phrase that must be placed between quotation marks; inviolable reactivity or selectivity “rules” do not exist; the adjectives “general” or “privileged” have at most a tenuous place in a discussion that deals with catalyst performance across a broad range of substrates and/or conditions. An effective solution to a problem in catalysis arrives in the form of a family, or several types, of possible candidates. Earlier we saw an example of catalyst diversity in closely related ERCM reactions in Scheme 11. In Scheme 16, a benzylpiperidene is generated with minimal enantioselectivity (60:40 er) when Mo–5a is present, whereas it is Ru–7b that delivers the same product in >99:1 er and similar efficiency (80% vs 85% yield with Mo–5a).72b When the derived N-methylpiperidene is targeted, the table turns: here, it is Mo–5a that delivers the heterocyclic product in 95% yield (>98% conv) and 97:3 er; use of Ru–7b leads to 36% conv, 30% yield, and nearly racemic product (66.5:33.5 er).72 A Cbz-protected piperidene that contains a C4 silyl ether can be accessed more efficiently and enantioselectively with Mo–5a. If the product is a piperidine that lacks the aforementioned substituent, not only is the EROCM similarly effective when Ru–7b is used, the opposite enantiomer is formed preferentially (Scheme 16).72 Establishing the precise reason for such variations requires comprehensive studies; nonetheless, the data in Scheme 16 do underscore the principle that broad scope solutions in catalysis require that structural diversity within the promoter molecules be readily accessible.
Scheme 16. Complementarity of Stereogenic-at-Ru and Mo Diolate Complexes.
Unique Attributes of Stereogenic-at-Ru Complexes
An intriguing aspect of the catalytic EROCM reactions promoted by chiral bidentate Ru carbenes, such as that utilized for synthesis of baconipyrone (Scheme 15), is that the rate of oxabicyle oligomerization or homocoupling of terminal alkenes is slow even in the absence of solvent. This is in contrast to transformations performed with complexes that contain a monodentate NHC, where side reactions are more prevalent. Cross-metathesis between the two alkene substrates is favored over homometathesis of the starting materials when stereogenic-at-Ru complexes are used. Such chemoselectivity is rooted in the inherent reactivity preferences of diastereomeric carbene complexes, a factor that can made clear through analysis of the catalytic cycle for an EROCM promoted by Ru–7b (Scheme 17). Formation of the coordinatively unsaturated exo carbene II may be followed by reaction with the relatively strained bicyclic alkene, which transpires via olefin complex III followed by metallacyclobutane (mcb) IV(73) to afford alkyl-substituted endo carbene V. Selective reaction of the cyclic alkene with the lower energy exo isomer (II), as suggested by its exclusive formation initially (cf. Scheme 14), is likely driven by strain release. The less sterically accessible but higher energy endo complex (V), on the other hand, reacts more readily with the less substituted alkene; here, the driving force is the release of strain74 of the stereogenic-at-Ru complex through conversion to exo isomer II via olefin complex VI and mcb VII (“side change”).74 Each carbene diastereomer therefore reacts selectively with one type of substrate. These considerations provide a rationale regarding the higher catalytic activity of the biphenyl-bridged complexes derived from Ru–7a,b (vs binapthyl derivatives Ru–6a,b; cf. Scheme 15). The more flexible tether in the former carbene species can better accommodate the strenuous structural demands of a mcb intermediate (cf. IV, Scheme 17).
Scheme 17. Proposed Catalytic Cycle for Sequence-Selective ROCM with Stereogenic-at-Ru Complexes.

DFT calculations confirm the proposition that endo carbene isomers derived from bidentate Ru carbenes are higher in energy (Scheme 18a). We used the reaction of Ru–7b with 3,4-diisopropoxycyclobutene to synthesize, isolate, purify by routine silica gel chromatography endo carbene Ru–9a.75 Our investigations demonstrated that strain release is the driving force for exo-to-endo isomerization (Scheme 18b). We determined that if reaction of the latter higher energy form with an alkene is sufficiently sluggish, non-OM-based polytopal rearrangement76 can occur, leading to conversion of the endo to exo carbenes (cf. Ru–9b and Ru–9c); this disrupts the order of the sequence of interconversions between the two diastereomeric forms (cf. Scheme 17), causing diminution in the yield and enantiomeric purity of the EROCM product.77
Scheme 18. Carbene Diastereomers of Stereogenic-at-Ru Complexes.

Chen and co-workers have exploited similar design principles,74 leading them to introduce a distinguished class of phosphine-containing stereogenic-at-Ru carbenes (e.g., Ru–8a–d, Scheme 19). The resulting complexes have been utilized in preparation of alternating copolymers with impressive selectivity.78
Scheme 19. Stereogenic-at-Ru Complexes Designed by Chen.

The Tale of a Recalcitrant RCM
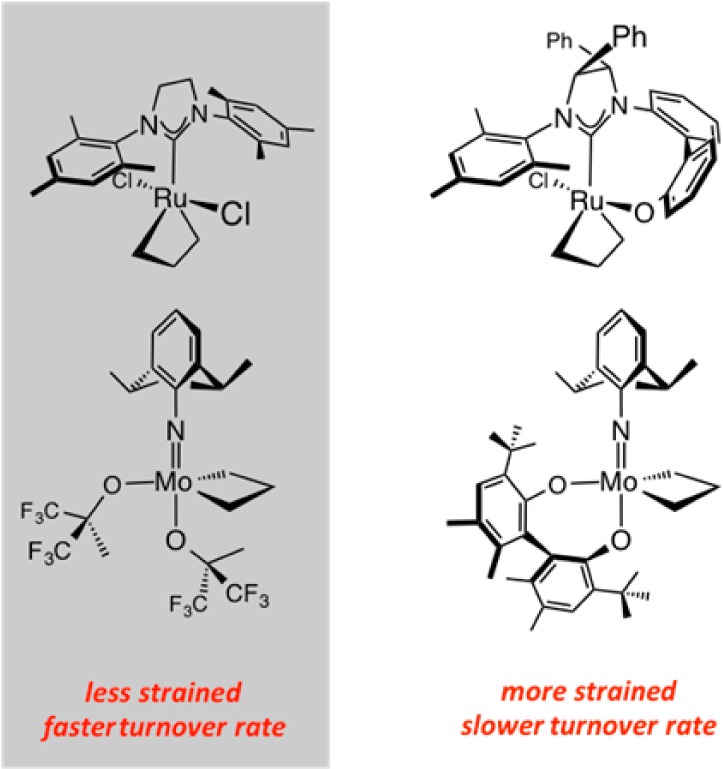
Our studies of OM reactions with stereogenic-at-Ru complexes taught us that the presence of diastereomeric complexes, granted that they do not undergo out-of-sequence interconversion, can lead to group-selective EROCM (cf. Schemes 15–17). These investigations revealed that in-sequence isomerization to the less preferred isomer of a geometrically constrained complex (regardless of whether the Mo or Ru center is stereogenic) with a relatively rigid bidentate ligand can be energetically taxing; in cases where a clear driving force is unavailable (e.g., release of angular strain), the attendant energy barriers might diminish catalytic activity. With the list of inefficient EOM reactions becoming disturbingly long, the need for a general strategy for bypassing the aforementioned energetic hurdles became compelling.
One challenging EOM reaction is one that can lead to the naturally occurring alkaloid quebrachamine (Scheme 20). The proposed ring closure involves enantiotopic alkenes that are adjacent to a quaternary carbon stereogenic center; the transformation would generate a strained tetracyclic structure with a Lewis basic tertiary amine that is proximal to the olefinic site, which is likely where the initial Mo–alkylidene or Ru–carbene would be initially generated. The representative findings in Scheme 20 point out that RCM with achiral carbenes such as Ru–3a, Ru–3c,d and Ru–3f afford the desired product in 36–65% yield.79 Although there was more extensive triene consumption with Ru–3c and Ru–3d (vs the parent Ru–3a), the RCM was not necessarily more efficient: the yield of the isolated product was either lower or nearly the same. Moreover, the less congested Ru–3f proved to be too short-lived to carry the reaction beyond the 50% conversion point, underlining that catalyst longevity and activity are crucial. With 20 mol % Mo–1 there was complete substrate consumption and yet the tetracyclic amine was obtained in 59% yield. The performance of the available chiral catalysts (e.g., Ru–4a, Ru–7a, and Mo–2a) was especially disappointing: even resorting to high catalyst loadings and/or elevated temperatures proved inconsequential.79
The study summarized in Scheme 20 illustrates that the more structurally constrained complexes—those that contain a bidentate ligand—tend to be less effective in promoting OM. We therefore reasoned that the geometrical constraints imposed by the formation of the intermediate mcb complexes might be exacerbated by the added strain (Scheme 21). We pondered whether the solution to conceiving a more effective set of catalysts might lie in entities that are not burdened by a bidentate ligand and are therefore not handicapped by high barriers to “inversion” at the metal center.79
Scheme 21. Bidentate Ligands Can Raise the Activation Barriers to Olefin Metathesis Reactions.
Stereogenic-at-Metal with Only Monodentate Ligands
The above analyses put the spotlight on chiral complexes that contain only monodentate ligands. In contemplating the design of Mo alkylidenes that would fit the bill, we first focused on complexes that carry two identical enantiomerically pure O-based ligands (nonstereogenic-at-metal; cf. Scheme 22a). However, seminal explorations of Eisenstein and Copéret80 led us to adopt a different approach; studies by the talented French team suggested that stereogenic-at-Mo species with a donor and an acceptor ligand (vs two electron acceptor groups as in the abovementioned bisalkoxides) should be considered as legitimate options. The Eisenstein–Copéret paradigm pointed out that the barrier to distortion of a tetrahedral complex (cf. VIII → IX → X, Scheme 22b), an alteration that must occur prior to alkene coordination, would be lower with a donor ligand present. To minimize unfavorable trans influence, the site opposite to the electron-donating unit would thus remain vacant, leading the substrate alkene to approach syn to the acceptor group (cf. IX). Moreover, the proposed ligand arrangement means the intermediacy of mcb XI, where the imido unit resides across from the acceptor group (vs the donor unit being trans to the imido), and trans influence is minimized. Knowledge of the relative arrangement of the acceptor, donor, and imido ligands enabled us to base our catalyst development efforts on solid ground. Replacement of an acceptor ligand by a relatively donating group was attractive, as we intuited that the resulting complexes would probably be longer living (improved turnover number or TON), but at what cost to loss of turnover frequency (TOF)?
Scheme 22. Mechanistic Model Derived from Calculations and Initial Examination of Effectiveness of a MAP Complex.

A corollary to the catalytic cycle in Scheme 22b is that the stereogenic-at-metal complex is converted to its corresponding diastereomer each time a mcb is generated and cleaved in a productive way; such isomerization might be viewed as detrimental to high enantioselectivity (each isomer might favor a different enantiomer). As will be discussed below, the latter complication would prove to be inconsequential for intriguing mechanistic reasons, but we were not yet aware of any nuances of the nascent system. As a result, we convinced ourselves to negotiate the next step with the following logic: Every productive OM reaction entails two mcb complexes with each entailing an inversion at the metal center (Scheme 22c); granted that there are no adventitious out-of-sequence isomerizations, every complete transformation involves a double inversion, resulting in regeneration of the same complex diastereomer.
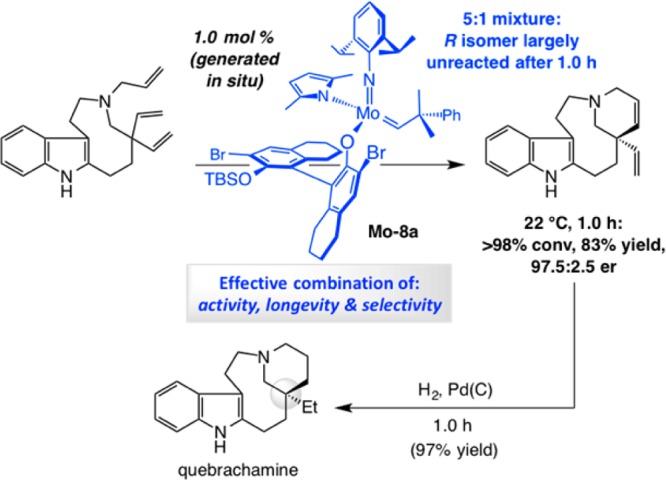
To explore the veracity of the premise that incorporation of a donor ligand might lead to a more effective OM catalyst, we performed the RCM leading to quebrachamine with 1.0 mol % monoaryloxide pyrrolide (MAP) complex Mo–6 (Scheme 22d).81 After 1 h, the tetracyclic amine was isolated in 79% yield (vs 20 mol % bis-alkoxide Mo–1 affording 59% yield after 2 h).79 High efficiency was achieved in spite of the presence of an indole NH and a Lewis basic tertiary amine.
Design and Preparation of an Enantioselective Catalyst
In designing an enantiomerically pure MAP complex, we contemplated a number of possible chiral monodentate alkoxide and aryloxides, entities that have played only a minor role in enantioselective catalysis. We envisioned that an expedient approach to accessing the desired complexes might involve the use of singly protected binaphthol derivatives readily obtained from inexpensive and commercially available binaphthol (either enantiomer can be purchased at low cost). We established that subjection of bis-pyrrolide Mo–7a with an equivalent of a monosilyl protected tetrahydobinaphtol, which contains bromine atoms at its C3 and C3′ sites, leads to efficient formation of a 7:1 mixture of diastereomeric MAP complexes S-Mo–8a and R-Mo–8a (Scheme 23); protonation of the second Mo–N bond is sufficiently slow, presumably due to steric factors, so that only minimal amounts of the bis-aryloxide are generated.79
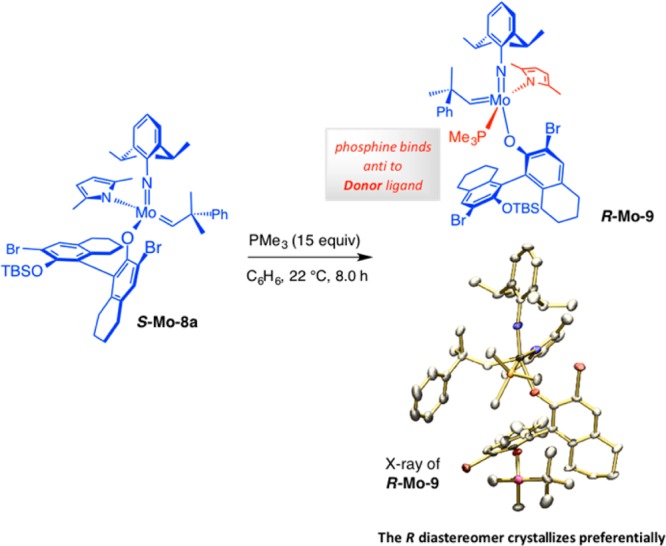
Scheme 23. Diastereoselective Synthesis and Characterization (X-ray) of the First Enantiomerically Pure MAP Complexes.

The X-ray structure of each stereoisomer was then secured (Scheme 23);79a,82 these data showed us that, if alkene association were to occur trans to the pyrrolide ligand (cf. Scheme 22b), then the major diastereomeric form (S-Mo–8a) would probably initiate faster. The aryl oxide group in the alternative (minor) alkylidene complex (R-Mo–8a) is situated in a manner that hinders alkene approach. In the minor isomer, one aryl oxide Br substituent appears to be coordinated to the transition metal, an association that must be disrupted before the Mo–O bond rotates to make available the anti-to-pyrrolide ligation site. Support for the scenario that a donor group binds preferentially trans to the (donor) pyrrolide ligand gained experimental support when a phosphine complex formed through treatment of (S)-Mo–8a with PMe3 yielded to X-ray crystallography; in the crystal structure PMe3 is coordinated trans to the pyrrolide group (cf. R-Mo–9 in Scheme 24).83
Scheme 24. Evidence for Association of Lewis Base Trans to the Pyrrolide.
When a 7:1 mixture of S- and R-Mo–8a, generated and used in situ, was employed in promoting the challenging ERCM en route to quebrachamine, the reaction reached completion in one hour at 22 °C with only 1.0 mol % loading (both diastereomers); the desired product was obtained in 83% yield and 97.5:2.5 er (Scheme 25).79 Other Mo MAP complexes were prepared and examined, including those bearing a dichloro- or diodo-aryloxide ligand or alkylidenes that contain a saturated binol unit as well as other arylimido groups (e.g., 2,6-dimethylphenylimido); we examined derivatives that contain a fully saturated monoprotected binol ligand. None of the alternative species proved superior to Mo–8a. However, we did establish that the protocol involving treatment of an appropriate bis-pyrrolide and an alcohol constitutes an efficient and reliable approach for preparing a wide range of MAP complexes. Such an attribute would prove advantageous later on.
Scheme 25. Application to Enantioselective Synthesis of Quebrachamine.
Mechanistic Nuances of EOM with MAP Complexes
A provocative finding vis-á-vis the above-mentioned ERCM reaction (Scheme 25) was that upon completion of the process (after 1.0 h), spectroscopic analysis indicated that much of the minor diastereomer (R-Mo–8a) remained uninitiated.79a We wondered whether the high er value is because it is solely the S component of the diastereomeric mixture that promotes enantioselective ring closure. To shed light on the differences between the MAP stereoisomers, we performed the experiments shown in Scheme 26.
Scheme 26. Study of Different Diastereomeric MAP Complexes.
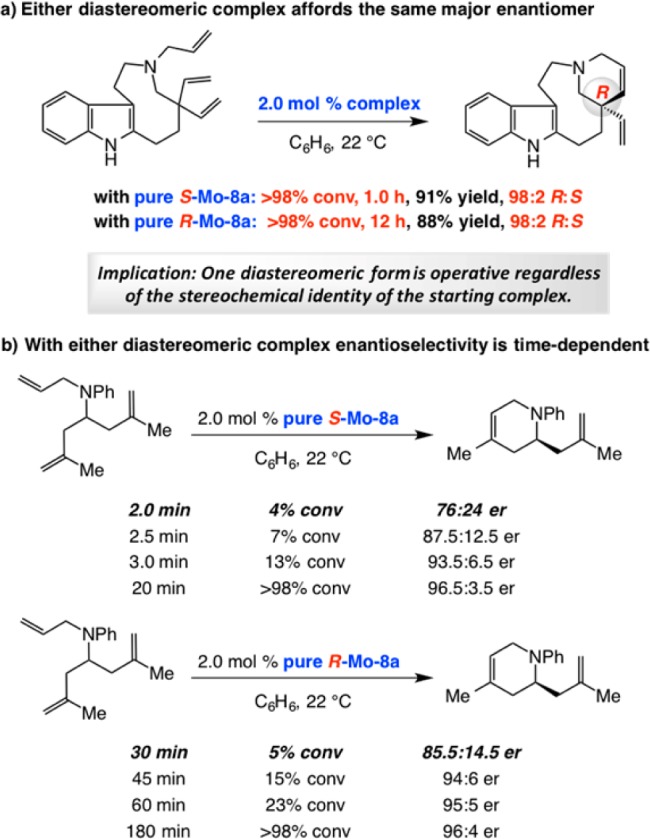
We observed >98% conversion within 1 h for the ERCM leading to quebrachamine when a pure sample of S-Mo–8a was used (>98:2 dr); with R-Mo–8a, under otherwise identical conditions, the transformation required 12 h to proceed to completion (Scheme 26a).82 This indicated that R-Mo–8a initiates significantly slower than S-Mo–8a, as the analysis of the X-ray structures in Scheme 23 and near complete recovery of the minor isomer (cf. Scheme 25) had initially suggested. Furthermore, in either case, it is the same product enantiomer that was generated with identical selectivity (R:S = 98:2). To probe deeper, we performed the time-dependent studies summarized in Scheme 26b with a simpler triene serving as the substrate.82 Reaction with S-Mo–8a was complete in 20 min but the ERCM involving the slower initiating R complex diastereomer needed 3 h. Importantly, er values at the initial stages of the transformation are substantially lower than those for the final product (96.5:3.5 and 96:4 er, respectively): with S-Mo–8a at ∼4% conversion, the dihydropiperidine was generated in 76:24 er and with R-Mo–8a at ∼5% conversion the ERCM product was formed in 85.5:14.5 er.
The data presented above can be explained through the catalytic cycle outlined in Scheme 27.82 The major alkylidene isomer, S-Mo–8a, likely initiates faster than R-Mo–8a. It is, however, the latter slower initiating complex that forms the more swiftly reacting S-Mo–10 that goes on to generate methylidene complex R-Mo–11. On the other hand, although S-Mo–8a enters the catalytic cycle more quickly (vs R-Mo–8a → S-Mo–10), it is the source of the less reactive R-Mo–10, a species that is more hesitant than S-Mo–10 to undergo RCM and is probably precursor to the minor product enantiomer.
Scheme 27. The Crucial Role of Ethylene in Establishing Curtin–Hammett Kinetics.

The finding that at the early stages of reaction with pure S-Mo–8a the product is formed in 76:24 er implies that, due to its slower reaction rate, a significant portion of R-Mo–10 isomerizes to S-Mo–10, leading to the formation of the major dihydropiperidene enantiomer; such isomerization might involve a non-OM-based polytopal rearrangement. It follows that the initial enantioselectivity delivered by the less rapidly initiating R-Mo–8a would be higher (85.5:14.5 er); in this case, the resulting MAP diastereomer promotes ERCM more readily and adventitious isomerization to the higher energy R isomer is less competitive. The formation of the minor enantiomeric product with R-Mo–8 as the starting point might originate from slow polytopal rearrangement of the neophylidene to the faster initiating S-Mo–8.
An implication of the scenario in Scheme 27 is that there would be a major enahancement in enantioselectivity with further transformation; once there is sufficient ethylene, interconversion between methylidene complexes S-Mo–11 and R-Mo–11 becomes faster than the ERCM rate and the process can be funneled through S-Mo–10 (i.e., Curtin–Hammett kinetics predominates).82 The proposed model and the experimental findings further suggest that the rate of reaction of the substrate triene with sterically unhindered methylidene complexes S-Mo–11 and R-Mo–11 is not significantly different. Consistent with the mechanistic model, when ERCM of the model triene is performed under an atmosphere of ethylene (generated in situ by RCM of diallyl ether; eq 1), the er value at the initial stages of the reaction is high (i.e., Curtin–Hammett condition is established right away).82
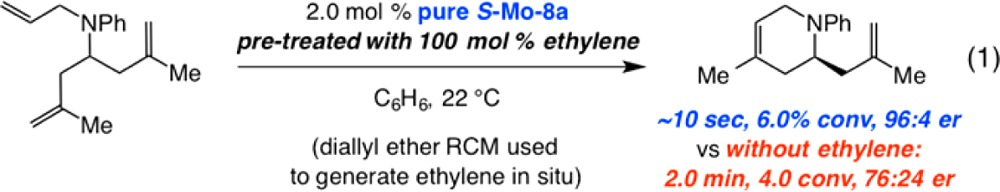 |
1 |
The First Z-Selective OM Catalysts
The MAP complexes described above possess characteristics that render them well suited for addressing a longstanding problem in OM: kinetically controlled generation of the higher energy Z alkenes. The principal attribute is the olefin preferentially binding trans to the pyrrolide ligand, giving rise to mcb intermediates represented by W-1,84 the X-ray structure of which is presented in Scheme 28a; the imido and aryloxide ligands occupy the apical sites of a trigonal bipyramidal complex in such species. Another relevant characteristic is the sizable monodentate aryloxide ligand and its ability to rotate freely, forcing the mcb substituents to orient toward the imido ligand, favoring preferential formation of Z alkenes (Scheme 28b).
Scheme 28. Design of the First Z-Selective Olefin Metathesis Reactions.
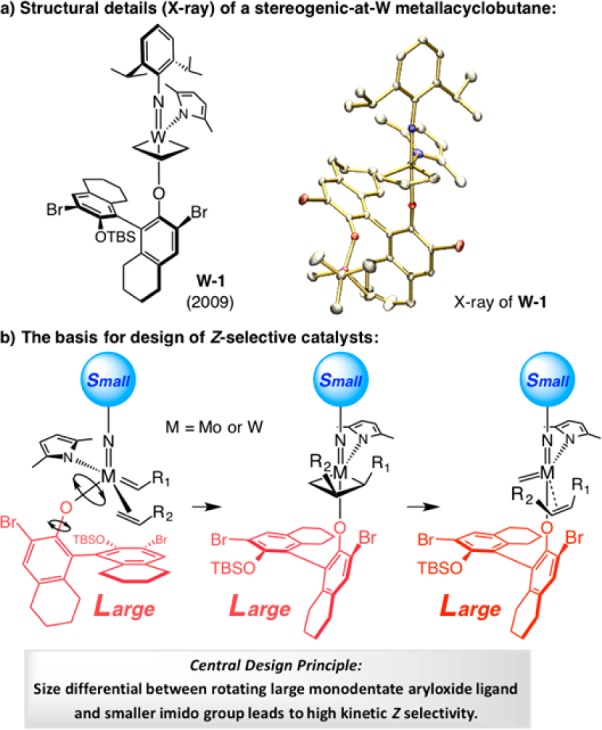
Finding a way to achieve high kinetic Z selectivity in OM is half the battle. The other problem is that a catalyst that prefers to form Z-alkenes also reassociates with the same isomers more readily, increasing the odds of postmetathesis isomerization (Scheme 29). If a trans alkene does form, the chances that it again binds with the Z-selective catalyst are not favorable. Trans 1,2-disubstituted olefins would not coordinate readily with complex that preferentially generates Z-alkenes, which is why E-alkenes are not kinetically favored. The challenge is first to obtain high Z selectivity and then hold on to it.
Scheme 29. Z Alkenes Can Be Formed and Isomerized Faster than E Isomers.
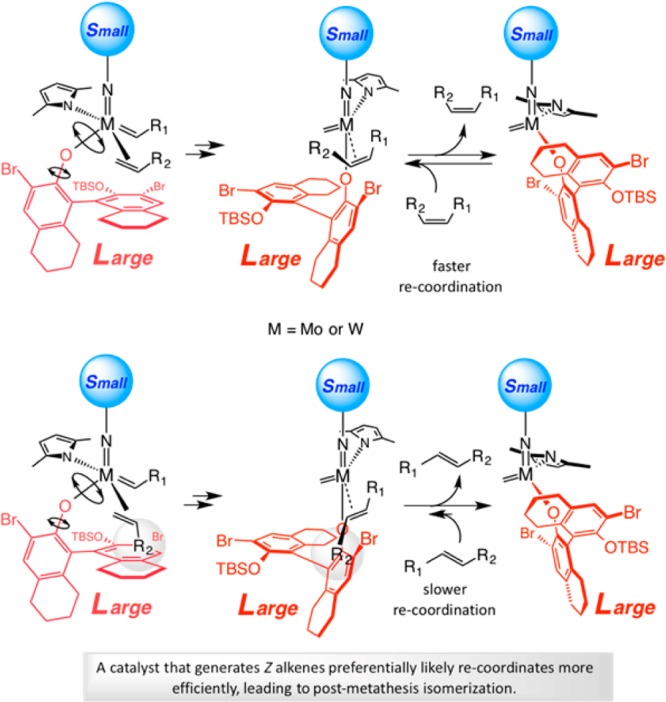
MAP-Catalyzed Z-Selective ROCM
The first successful implementation of the blueprint in Scheme 28b arrived in the form of efficient and Z-selective ROCM carried out with adamantylimido MAP complex Mo–12 (Scheme 30).85 Treatment of oxabicyclic alkenes in the presence of aryl olefins with <1.0 mol % Mo–12 led to complete conversion in 1 h, affording the products in exceptional er and high Z selectivity (Scheme 30a). We later showed that transformations can be carried out with enol ethers with similar efficiency, as well as diastereo- and enantioselectivity.86 Screening studies indicated that complexes bearing the wider spanning 2,6-disubstituted phenylimido ligands do not efficiently catalyze this set of ROCM processes (Scheme 30b); these findings together with the effectiveness of the adamantylimido complex are consistent with the model presented in Scheme 29b. With aliphatic alkenes, susceptible to facile homocoupling, higher yields can be obtained under 7.0 torr of vacuum (Scheme 30b). As will be discussed below, the latter expediency would prove crucial in the development of Z-selective CM.
Scheme 30. First Examples of Catalyst-Controlled Z-Selective Olefin Metathesis.
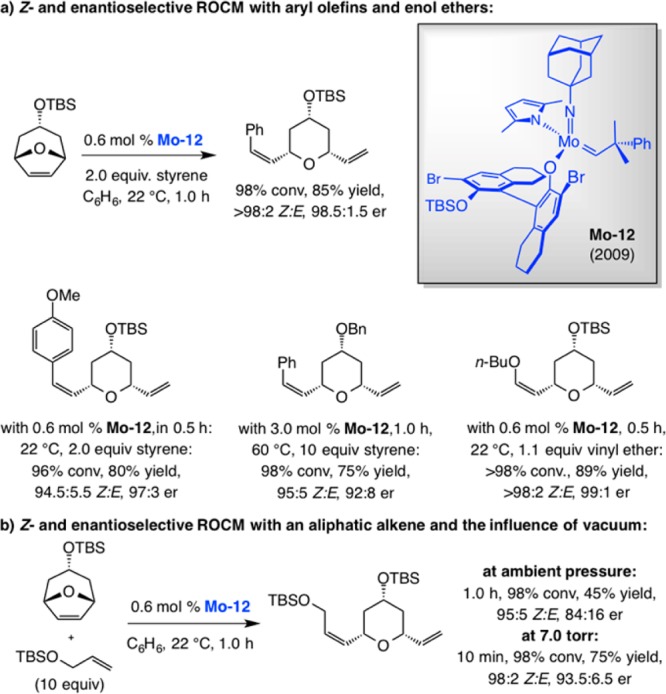
Z-Selective CM Reactions
One of the more striking advances made possible by high-oxidation-state MAP complexes likely relates to their ability to promote efficient Z-selective CM.87 The original examples of this class of reactions are presented in Schemes 31 and 32.88,89 In 2011, we reported that Mo–7b can be used to effect Z-selective CM of enol ethers with terminal alkenes (Scheme 31a); the smaller size of the cross partners renders the 2,6-dimethylimido complex more suitable for this particular application (vs EROCM in Scheme 30). In addition, we showed that allylic amides, including those with sterically hindered protecting units (cf. Scheme 31b), can be used most effectively when the more diminutive adamantylimido complex Mo–12 is utilized (likely because of the larger size of the allylic amides vs enol ethers).88 Not only can MAP-catalyzed CM be performed on gram scale, reaction efficiency improves in such cases (lower catalyst loading). Utility was illustrated by applications to syntheses of biologically active molecules: antioxidant plasmalogen C18 (plasm) 16:0 (PC) in the case of CM with enol ethers and immunostimulant KRN7000 for transformations that afford cis allylic amides. In the first instance, the Z-olefin resides within the target molecule; in the second, it serves as the precursor for a catalytic diastereoselective dihydroxylation.
Scheme 31. Z-Selective Cross-Metathesis Reactions and Applications to Synthesis of Biologically Active Molecules.

Scheme 32. Z-Selective CM with Vinyl–B(pin) and Applications to Synthesis of Biologically Active Molecules.

We later demonstrated that allylic silyl and benzyl ethers can be employed in catalytic Z-selective CM (Scheme 31c).90 The application to falcarindiol, which contains two alkyne units, underlines the utility of the approach for two reasons: First, the CM strategy allows access to molecules that contain a Z olefin and an alkyne group, in which case partial hydrogenation of an acetylene unit cannot be used. Second, we demonstrated that the combination of catalytic CM and catalytic cross-coupling constitutes an attractive combination in chemical synthesis (see below for further discussion).
We have already seen that a strategic angle in catalytic OM relates to controlling the concentration of the ethylene byproduct (cf. Schemes 3 and 27). Unlike ERCM processes, where the presence of ethylene proved to be beneficial, with CM reactions, high concentration of ethylene is often detrimental. When the transformations illustrated in Scheme 31 were performed at ambient pressure, lower efficiency and stereoselectivity was observed. This is likely because, under ambient pressure and elevated ethylene concentration, formation of the corresponding highly reactive and less stable methylidene complex becomes favorable (cf. Mo–11 in Scheme 27). The uncongested methylidene species are comparatively adept at reacting with the Z-disubstituted alkene, facilitating undesired postmetathesis isomerization.
As mentioned earlier, the combination of catalytic stereoselective CM and cross-coupling presents an attractive strategy in organic synthesis; the examples provided in Scheme 32, relating to the preparation and use of Z-alkenyl-(pinacolato)boron [Z-alkenyl–B(pin)] compounds, are illustrative. In the first instance (Scheme 32a), Z-selective CM of commercially available vinyl–B(pin) and an aryl alkene delivered the expected Z-alkenyl–B(pin) in 73% yield and 96:4 Z:E ratio; subsequent coupling with an arylbromide catalyzed by a phosphine–Pd complex [Pd(PPh3)4)] afforded combretastain A-4, an anticancer agent that is 10 000 times more active than its E isomer.91 In another example (Scheme 32b), an aryl-substituted Z-alkenyl–B(pin), obtained in 73% yield and 93:7 Z:E selectivity, was used as the reagent in a site and enantioselective allylic substitution promoted by a sulfonate-based bidentate NHC–Cu complex; the product was converted to natural product nyasol.92
Z-Selective Macrocyclic RCM
Another major consequence of the availability of MAP complexes is the feasibility of carrying out Z-selective macrocyclic RCM.93 The importance of this set of transformations is reflected in the fact that, despite the absence of catalysts that can ensure high stereoselectivity, it has nonetheless been a commonly used transformation for preparation of large rings. On countless occasions, macrocyclic RCM is relied upon to effect a late-stage transformation in multistep sequence of reactions (cf. Schemes 9a,b). Such widespread preference is partly because RCM is a generally reliable process that demands easily accessible and robust alkenes as starting materials. In contrast, and as an example, preparation of a macrolactone through formation of the ester bond would necessitate a hydroxyl unit and a carboxylic acid group that must be masked and differentiated from other related functionalities; this often requires oxidation state adjustments as well as protection/deprotection schemes that increase step counts.
Three cases of catalyst-controlled Z-selective macrocyclic RCM are shown in Scheme 32. With 5.0 mol % mcb W-1, which can be weighed in air (Mo complexes must be handled under inert atmosphere), the macrocyclic RCM generated epilachnene in 82% yield and 91:9 Z:E selectivity (Scheme 33a).94 The ease of handling of unsubstituted mcb W-1 suggests that high ethylene concentration can result in the formation of relatively stable unsubstituted mcb complexes, providing another reason for why reactions should be performed at reduced pressure.
The RCM reactions in Scheme 33b,c, carried out en route to the naturally occurring anticancer agents epothilone C and A and anticancer and antimicrobial natural product nakadomarin A (Scheme 33c) are noteworthy.95 These processes illustrate that MAP-catalyzed macrocyclic RCM can be performed reliably at a late stage in a complicated synthesis scheme (after 16 steps for epothilone C and 12 steps for nakadomarin A). The Z-selective macrocyclization, which is in stark contrast to former attempts where nearly equal mixture96 of nearly impossible-to-separate olefin stereoisomers96g,96i were generated, effectively doubles the overall yield of the total synthesis. In the presence of complex W-2 formation of cyclooctene moiety of nakadomarin A can be performed with minimal isomerization at the sensitive macrocyclic Z-alkene site. An alternative approach involving a Ru carbene had to be performed with slow addition of 40–100 mol % of the less reactive first-generation Ru–1b (involving the corresponding diamide as the substrate).97
Several additional points regarding the transformations in Scheme 33 deserve mention. First, complex W-2, which can also be weighed and used in air, was employed to effect the epothilone RCM on gram scale (83% yield, 95:5 Z:E; Scheme 33b). Second, the efficient cyclization affording the strained Z-alkene within the 15-membered ring in nakadomarin A, which occurs in the presence of two basic tertiary amine units, underscores the tolerance of MAP alkylidenes toward N-containing functional groups. Third, as with Z-selective CM (cf. Schemes 31-32), reactions usually proceed with maximal efficiency and stereoselectivity when run at reduced pressure. Lastly, use of the more active Mo alkylidenes gives rise to lower Z selectivity due to postmetathesis isomerization (cf. Scheme 29), as substantiated by control experiments.94
A Missed Early Opportunity?
If the disparity between the size of the imido and aryloxide ligands in MAP complexes is the main reason for kinetic Z selectivity, should not at least some stereochemical control be observed with bis(hexafluoro-tert-butoxide) Mo–1? Although not as large as a tetrahydrobinaphthol ligand, should not the alkoxide units impose an appreciable degree of stereodifferentiation? The experiments shown in Scheme 34 clarify the above questions: Whereas the RCM en route to epothilone C is probably kinetically E-selective when Ru–3a is used, with Mo–1, the macrocyclization proceeds in ∼70% Z selectivity. Astonishingly, however, postmetathesis isomerization is complete within 20 additional minutes, and the E:Z ratio plunges from 72:28 to 33:67.94 The value reported in 1997 for this macrocyclic RCM, carried out with 20 mol % Mo–1 and after one hour of reaction time, is 33:67 Z:E. Undoubtedly, the equilibrium had been reached and the initial Z selectivity erased.96b The above findings underscore the importance of reversible nature and time dependency of catalytic OM. We can only wonder how the field of stereoselective OM would have progressed if it were noted in the mid to late nineties that Mo–1 is indeed capable of delivering a measurable degree of kinetic Z selectivity.
Scheme 34. Z-Selective Macrocyclic RCM with Mo–1. Selectivity as a Function of Time.

The findings presented in Scheme 34 bear the intriguing implication that the Z selectivity in the macrocyclic RCM en route to fluvirucin B1 may not have been entirely the result of substrate control, as was originally perceived (cf. Schemes 5 and 6). This assertion is supported by the more recent investigations of Urpí and Vilarrasa who discovered that RCM of other structurally similar (e.g., Et-substituted trisubstituted olefin) members of the fluvirucin family performed in the presence of Ru–3a leads to minimal stereoselectivity.98 It is feasible that the high preference for the Z trisubstituted olefin first reported in 1995 may have stemmed from the Mo–1-derived intermediate alkylidene. Together, the above observations emphasize that the efficiency of W-based MAP complexes is not only because of their ability to provide high kinetic Z selectivity, it originates equally from the remarkable degree of chemoselectivity exhibited by the catalysts. While efficient RCM is possible with substrate terminal alkenes, reaction with the product disubstituted olefin, even at high conversion, remains less facile.
Alkene versus Alkyne RCM
A brief discussion is in order regarding the alternative two-step approach to Z-selective macrocyclic RCM, entailing cyclizations of diyne substrates promoted by high oxidation-state alkylidyne complexes followed by Pd-catalyzed partial hydrogenation.99 Primarily due to investigations by Fürstner, the latter strategy has provided a reliable and stereoselective entry to an assortment of complex molecules. Still, the most direct approach to preparation of an alkene is an RCM involving olefins, obviating the need for synthesis of alkynyl substrates and subsequent oxidation state adjustments. Access to diynes is often more cumbersome, likely because of a combination of the higher sensitivity of an alkyne to side reactions and the fact that far fewer alkynes are commercially available. As an example, whereas epilachnene’s diene precursor can be prepared through five transformations, synthesis of the corresponding diyne entails 11 steps.100 Examination of the complexity of synthesis of the requisite diyne for macrocyclic closure that affords the epothilone C intermediate would be similarly revealing.101 High oxidation-state alkylidynes are sensitive to Lewis basic groups: attempts to effect macrocyclic alkyne metathesis en route to nakadomarin A have been unsuccessful102 except with the diamide derivatives,103 partly as the result of the presence of basic amines. Alkyne-containing rings are generally more strained than their alkene counterparts, disfavoring applications to smaller ring structures. Nevertheless, the above analysis should not detract from the strategic value of catalytic alkyne RCM. Macrocyclic alkynes lend themselves to unique functionalization procedures; they allow for synthesis of useful products that are not easily available through reactions of alkenes.104
Synthesis of a Macrocyclic Trisubstituted Z-Alkene
Mother Nature had another surprise in store for us when we set out to identify a catalyst for Z-selective macrocyclic RCM reactions that afford the more congested trisubstituted alkenes. We selected the precursor to epothilones B and D (after alkene epoxidation) as our model systems (Scheme 35), so that direct comparison with our studies vis-á-vis the less substituted congener would be feasible (cf. Scheme 33). When ring closure was performed in the presence of Mo–1 (20 mol %, 22 °C, 24 h, 81% conv) or Ru–3a (20 mol %, 50 °C, 48 h, 59% conv) nearly equal mixtures of isomers were formed.105 In contrast to transformations that afford disubstituted alkenes, those that deliver trisubstituted olefins are unlikely to be subject to postmetathesis isomerization; the latter selectivity levels are probably due to a complete lack of kinetic control. It is surprising then that Mo–1, which is only moderately Z-selective in forming a macrocyclic disubstituted olefin (cf. Scheme 34), fails to promote stereoselective formation of the trisubstituted alkene, where one substituent (likely the Me group here) must be oriented toward the sizable alkoxide group.
Scheme 35. Z-Selective Formation of a Macrocyclic Trisubstituted Alkene by RCM.
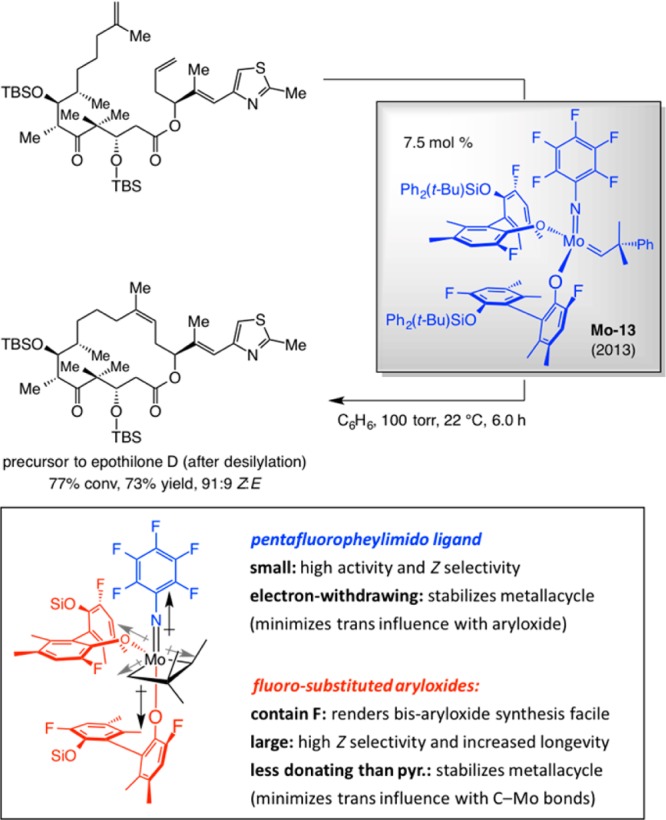
Through screening studies we established that a nonstereogenic-at-metal bis-aryloxide Mo-based alkylidene, with a pentafluorophenylimido ligand, delivers the highest reactivity and selectivity levels (Scheme 35). The trisubstituted macrocyclic alkene was obtained in 73% yield and 91:9 Z:E ratio with 7.5 mol % Mo–13 at 22 °C in only 6 h.106 Use of MAP complexes led to <20% conversion even at 20 mol % loading. Identification of Mo–13 was the result of a serendipitous discovery courtesy of the tendency of the more acidic and less bulky difluoro-substituted monoprotected tetrahydrobinaphthol to undergo a second Mo–pyrrole bond protonation rapidly (vs the dibromo derivatives). The latter discovery was followed was followed by optimization of the imido unit, leading us to determine that the pentafluoroiomido ligand represents the best choice.
We were hardly surprised that the relatively small perfluoro ligand delivered high Z selectivity (cf. stereochemical model in Scheme 29). It was the high reactivity of the sizable bis-aryloxide species that took us unawares. DFT calculations on model systems revealed that mcb cycloreversion is likely the turnover-limiting step of the catalytic cycle and that the lower the energy of the metallacycle, the more accessible the transition structure for conversion to the product. The results of these investigations intimated that two aryloxides and a perfluoroarylimido ligand are collectively responsible for stabilization of the mcb and the less energetically demanding cycloreversion. The positive impact of a second aryloxide and the perfluorophenyl ligands appear to be rooted in minimization of two destabilizing electronic trans influences (cf. Scheme 36). One interaction involves the apical arylimido and aryloxide ligands and the diminution of their electron-donor ability as a result of their electron-withdrawing F atoms; the other corresponds to the electron releasing Mo–C bonds being situated opposite to the more electron-deficient F-substituted aryloxide group (vs a pyrrolide). The DFT studies underline the principle that, as alluded to earlier (cf. Scheme 22 and related discussion), the superior performance of MAP complexes does not originate from a particularly high degree of reactivity per se; rather, it is the relative stability of the monopyrrolide systems combined with sufficient activity that renders the stereogenic-at-Mo and -W systems effective.107
Scheme 36. Z- and Enantioselective ROCM with a Ru-Based Complex.
Z- and Enantioselective Ru-Catalyzed OM
The successful implementation of the design plan outlined in Scheme 28 for achieving high Z selectivity with MAP complexes inspired us to explore the possibility of introducing a stereoselective set of Ru-based catalysts. Considering their complementary reactivity and functional group compatibility profiles compared to Mo or W alkylidenes, efficient and Z-selective OM processes promoted by Ru carbenes are of considerable significance.
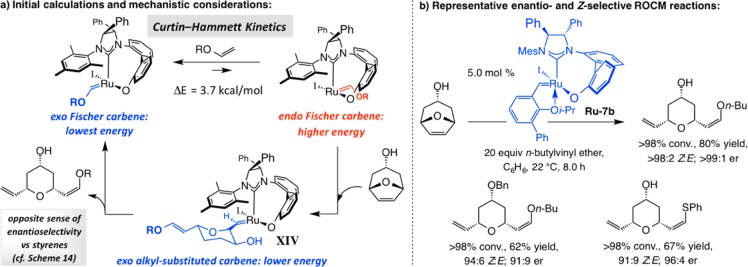
Our first step toward developing Ru-catalyzed Z-selective OM led to the development of the first examples of EROCM with enol ethers (for the MAP-catalyzed version, see Scheme 30). In contrast to Mo alkylidenes (cf. Schemes 30-31), heteroatom-substituted alkenes are seldom ideal substrates for Ru-catalyzed OM. DFT calculations indicated that, in spite of the possibility of strain release, the exo carbene diastereomer is not sufficiently reactive to promote ROM (Scheme 36a): not only would such a “side change” generate a higher energy endo isomer (ΔE = 3.7 kcal/mol), unlike when aryl or alkyl olefins are involved (cf. Schemes 14–17), it would convert a lower energy Fischer-type heteroatom-substituted metal–carbene to a less favorable alkyl-substituted variant. These investigations suggested that exo-to-endo isomerization between the heteroatom-substituted carbenes might be faster than ROCM and that Curtin–Hammett kinetics could be operative: the higher energy endo carbene, if available in sufficient quantities, might promote OM.108 We reasoned that, although reaction via endo heteroatom-substituted carbene would convert a Fischer-type complex to a less stable alkyl-substituted Ru=C bond, the exo-to-endo conversion and the concomitant stain release could provide sufficient impetus for driving the OM process to its conclusion (Scheme 36a). The latter supposition proved to be well founded (Scheme 36b): EROCM products formed efficiently and with the opposite sense of stereochemistry (vs styrenes or alkyl olefins), implicating the involvement of an endo (vs an exo) carbene in the stereochemistry-determining ROCM step (cf. XIV in Scheme 36a).
An unexpected and provocative outcome of these studies was that, contrary to the related transformations with aryl- and alkyl-substituted olefins (cf. Schemes 14–17), the products were uniformly generated with high Z selectivity. Ru-catalyzed EROCM with enol ethers is less facile than when reactions are carried out with Mo-based MAP catalysts (cf. Scheme 30); however, protection of the hydroxyl group is not necessary with Ru carbenes. The results of our mechanistic studies to elucidate the origin of the observed Z selectivity are in progress.
Dithiolate Ru Complexes for Z-Selective OM
The most recent advance emerging from our laboratories relates to Ru-based Z-selective catalysts, the design of which was inspired by our work in connection with MAP alkylidenes (cf. XV vs XVI, Scheme 37a). The principal reasoning that a sufficient size gap between the apical ligands of a trigonal bipyramidal mcb could lead to high stereoselectivity; based on steric factors, intermediacy of complexes such as XVI with an alkene substrate associating syn to the large ligand (L) would deliver Z-alkenes. We were thus led to ponder the ability of bidentate complexes XVII as potential catalyst precursors; in such a system the two anionic ligands (G) would adopt a syn relationship, as opposed to the anti disposition preferred with monodentate variants (minimization of dipole, steric and electron–electron repulsion). Accordingly, we synthesized dithiolate complexes Ru–10a,b by treatment of the commercially available Ru–3a with the disodium salt of dithiols (Scheme 37b).109 The resulting complexes can be easily purified by simple filtration, a trivial task, courtesy of the significant polarity difference between the S-containing carbenes and any unreacted (and E-selective) Ru–3a (cf. μ values in Scheme 38b). The cyanide groups in Ru–10b displace the isopropxy chelate, giving rise to a cyclic hexameric structure, the X-ray structure of which is shown in Scheme 37.
Scheme 37. Ru–Dithiolate Complexes for Z-Selective Olefin Metathesis Reactions.

Scheme 38. Z-Selective Olefin Metathesis with Ru–Dithiolate Complexes.

The Ru-based disulfides promote ring-opening/polymerization reactions with exceptional efficiency and Z selectivity (Scheme 38a). The significance of the 5400 turnovers obtained in 48 h and >98% Z selectivity with cyclooctadiene becomes clearer when compared with the 38 cycles observed after 72 h of reaction time with an alternative, recently discovered, Ru catalyst (further discussed below).110
The dithiolate Ru complexes catalyze efficient and highly Z-selective ROCM as well (Scheme 38b). Sterically demanding cross partners, represented by p-methoxystyrene, can be used, an attribute that is yet to be applicable to other types of Z-selective Ru complexes (see below). ROCM reactions are effective with 1,3-dienes, O- or S-substituted alkenes and with different types of strained disubstituted alkenes (Scheme 38b).111 Of particular note are transformations that involve allylic alcohols (Scheme 38c), including those that furnish sterically hindered alkenes. In the latter instances, we exploit the possibility of internal H-bonding between the hydroxyl unit and the electron-rich (trans to NHC) apical sulfide ligand (see XVIII), a mechanistic aspect that we investigated a few years ago,112 to faciliate OM reactions due to the factors indicated in Scheme 39c. Ru–dithiolates can be manipulated in air. The corresponding Z-selective CM and macrocyclic RCM processes are the focus of ongoing investigations.
Scheme 39. Z-Selective OM Promoted by Stereogenic-at-Ru Complexes Bearing a Bidentate NHC–Alkyl Ligand Developed by Grubbs.

An unanticipated outcome of this chapter of our research efforts is that the corresponding catecholate complexes, although similarly active, promote OM reactions with substantially lower stereoselectivity; the basis for this intriguing disparity in selectivity profiles is being examined in detail. Finally, it merits mention that although carbene complexes Ru–10a,b contain a bidentate dithiolate ligand, we did not expect that the attendant rigidity of the ligand would diminish the rate of inversion at the metal center, hampering the reaction rates (cf. Schemes 21 and 22 and related discussion). The position of the bis-heteroatomic ligand is such that it serves as a pivot around which side changes occur, resulting in little or no impact on the energy of different carbenes.
Other Z-Selective Ru Catalysts for OM
The inaugural set of Z-selective Ru catalysts for OM was introduced by Grubbs in 2011; two of the more recent additions (Ru–11a,b) are illustrated in Scheme 39. Z-Selective homocoupling of unhindered monosubstituted alkenes were the subject of the initial disclosure (Scheme 39a).113 Alcohol-containing substrates can be used but typically if the hyrdoxyl unit is distal to the olefin. A limited number of examples involving a cyclobutene and allylalcohol was reported more recently; however, the scope is presently limited to cyclobutenes.114 In 2013, monothiolate Ru–12 was shown by Jensen to effect Z-selective homocoupling of sterically unhindered terminal alkenes (Scheme 39a; i.e., no allylic or homoallylic substituent).115 As the examples provided indicate, high conversions translate to diminished selectivities as a result of postmetathesis isomerization.
Appreciable reactivities and high Z selectivities have been observed in CM reactions performed with Ru–11a,b; heating and somewhat dilute conditions are typically required (e.g., 0.5 M).116 Various cross partners can be used, including vinyl–B(pin) and allylic acetals (Scheme 39b).117 The corresponding ROCM processes have been reported as well, including those with an enantiomerically pure sample of Ru–11a, which has been obtained in three steps and after a chromatographic separation in ∼30% overall yield.118
The utility of Ru carbenes represented by Ru–11a,b in Z-selective macrocyclic RCM has been explored (Scheme 39d).119 For instance, with 7.5 mol % of Ru–11a at 60 °C, yuzu lactone, a somewhat strained 13-membered ring natural product, was obtained in 40% yield and 86:14 Z:E. In comparison, through the use of 5.0 mol % W–3 in 5.0 mM solution (vs 3.0 mM with Ru–11a), the large ring olefin was isolated in 40% yield and 73% Z selectivity after one hour at ambient temperature.94 Stereoselective synthesis of the 16-membered ring macrolactone and macrolactam offers additional possibilities for probing the effectiveness of Ru carbenes vs high-oxidation-state MAP alkylidenes. The case of ambrettolide is another example where Ru–11a as well as W-3 furnish access the desired macrocycle. In contrast to Z-selective MAP alkylidenes, the utility of Ru catalysts remains to be tested in the context of a late-stage reaction of a multistep sequence (cf. Scheme 33b,c).
On the basis of extensive DFT calculations, Grubbs and Houk have put forward a mechanistic scheme as well as a plausible stereochemical model for Z-selective OM reactions promoted by the aforementioned set of Ru complexes (Scheme 39e).120 A blend of steric and electronic factors77 have been proposed to favor mcb formation syn to the NHC, leading to a preference for Z alkenes due to steric factors originally suggested for high oxidation-state MAP complexes (cf. Scheme 29) and subsequently applied to Ru–dithiolate carbenes (cf. Scheme 37).
Conclusion
Advances in stereoselective OM during the past few years have substantially enhanced the utility of this already powerful class of transformations. Progress has been due to a more extensive appreciation of mechanistic principles and structural factors that result in an efficient catalyst. With a metal center serving as the stereochemical marker, energetic nuances that would otherwise remain undetectable and hence unappreciated can now be examined and understood in detail. One consequential precept pertains to the realization that, unlike other processes, the metal center within an OM catalyst is subject to structural and stereochemical perturbations that have significant energetic implications and can influence the rate and stereoselectivity of an OM reaction. The advent of stereogenic-at-metal OM catalysts has garnered an era of unprecedented efficiency and unique selectivity profiles. The studies discussed here underscore the concept that there is more to chiral catalysts than furnishing enantiomerically enriched products.47 Every reported complex developed thus far for Z-selective (not only enantioselective) OM, Mo-, W-, or Ru-based, has been a stereogenic-at-metal complex.
The recent evolution in catalytic OM should not lead us to presume that every milestone discovery has been made,121 as many types of Z-selective OM reactions of great utility remain undeveloped. Catalytic systems that directly afford α,β-unsaturated ketones, acids, esters, amides, and related derivatives remain to be introduced. Catalyst-controlled stereoselective OM reactions that generate trisubstituted alkenes are scarce. Development of E-selective transformations that will further free us from the grips of thermodynamic control, a force that often leaves us with unattractive and difficult-to-separate isomeric mixture, are badly needed.
Considering the stimulating developments of the last two decades, it is likely that we will witness other exciting advances in the coming years. Additional breakthroughs will benefit from the synergistic relationship between the different classes of complexes that have been, and continue to be, instrumental in our ability to solve additional high-impact problems. It is hoped that this account provides sufficient evidence that, in the case of OM as well as the entire enterprise of catalyst and reaction development, strides fueled by curiosity-driven research remain indispensible.
Acknowledgments
I owe a debt of gratitude to my co-workers whose names appear in the reference section for their steadfast dedication and countless intellectual and experimental contributions to the studies described above. I am indebted to Professor Richard Schrock, my collaborator since August 1997, with whom my co-workers and I have had a wonderful time making discoveries and achieving advances in the area of Mo- and W-based complexes for stereoselective olefin metathesis. The NSF (CHE-1111074) and the NIH (GM-59426 and GM-47480) provided generous financial support for our programs; the Vanderslice Endowment Fund and the LaMattina Graduate Fellowship Fund at Boston College offered additional support. I am grateful to Professor Ilan Marek (Technion, Israel) for helpful discussions regarding this manuscript.
Biography

Amir H. Hoveyda received his B. A. degree at Columbia University in 1981 and his doctoral degree at Yale University in 1986 with Stuart L. Schreiber. He subsequently joined David Evans at Harvard as an NIH and then an American Cancer Society postdoctoral fellow. In 1990, Hoveyda became a member of the faculty at Boston College, where he has been the Patricia and Joseph T. Vanderslice Millennium Professor of Chemistry since 1998. His research interests include the discovery of new catalysts for development of efficient, sustainable, and selective reactions, study of reaction mechanisms, stereoselective catalysis, and synthesis of complex natural products.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- For representative recent reviews on catalytic olefin metathesis (OM), see:; a Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed 2003, 42, 4592–4633. [DOI] [PubMed] [Google Scholar]; b Handbook of Metathesis; Grubbs R. H., Ed.; Wiley–VCH, Weinheim, Germany, 2003. [Google Scholar]; c Diver S. T.; Giessert A. J. Chem. Rev. 2004, 104, 1317–1382. [DOI] [PubMed] [Google Scholar]; d Nicolaou K. C.; Bulger P. G.; Sarlah D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]; e Vougioukalakis G. C.; Grubbs R. H. Chem. Rev. 2010, 110, 1746–1787. [DOI] [PubMed] [Google Scholar]; f Lozano-Vila A. M.; Monsaert S.; Bajek A.; Verpoort F. Chem. Rev. 2010, 110, 4865–4909. [DOI] [PubMed] [Google Scholar]; g Metathesis in Natural Product Synthesis; Cossy J., Arsenyadis S., Meyer C., Eds.; Wiley–VCH: Weinheim, 2010. [Google Scholar]
- Hoveyda A. H.; Zhugralin A. R. Nature 2007, 450, 243–251. [DOI] [PubMed] [Google Scholar]
- Schrock R. R. Angew. Chem., Int. Ed. 2006, 45, 3748–3759. [DOI] [PubMed] [Google Scholar]
- a Murdzek J. S.; Shrock R. R. Organometallics 1987, 6, 1373–1374. [Google Scholar]; b Schrock R. R.; Murdzek J. S.; Bazan G. C.; Robbins J.; DiMare M.; O’Regan M. J. Am. Chem. Soc. 1990, 112, 3875–3886. [Google Scholar]
- Nguyen S. T.; Johnson L. K.; Grubbs R. H.; Ziller J. W. J. Am. Chem. Soc. 1992, 114, 3974–3975. [Google Scholar]
- Grubbs R. H. Angew. Chem., Int. Ed. 2006, 45, 3760–3765. [DOI] [PubMed] [Google Scholar]
- a Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Org. Lett. 1999, 1, 953–956. [DOI] [PubMed] [Google Scholar]; For a closely related complex, see:; b Huang J.; Stevens E. D.; Nolan S. P.; Petersen J. L. J. Am. Chem. Soc. 1999, 121, 2674–2678. [Google Scholar]
- Schwab P.; Grubbs R. H.; Ziller J. W. J. Am. Chem. Soc. 1996, 118, 100–110. [Google Scholar]
- a Morken J. P.; Didiuk M. T.; Hoveyda A. H. J. Am. Chem. Soc. 1993, 115, 6997–6998. [Google Scholar]; For an overview of related zirconocene-catalyzed enantioselective processes, see:; b Hoveyda A. H.; Morken J. P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1262–1284. [Google Scholar]
- Morken J. P.; Didiuk M. T.; Visser M. S.; Hoveyda A. H. J. Am. Chem. Soc. 1994, 116, 3123–3124. [Google Scholar]
- For other illustrative examples involving the combination of catalytic OM and zirconocene-catalyzed enantioselective alkylation, see:Visser M. S.; Heron N. M.; Didiuk M. T.; Sagal J. F.; Hoveyda A. H. J. Am. Chem. Soc. 1996, 118, 4291–4298. [Google Scholar]
- Johannes C. W.; Visser M. S.; Weatherhead G. S.; Hoveyda A. H. J. Am. Chem. Soc. 1998, 120, 8340–8347. [Google Scholar]
- Visser M. S.; Harrity J. P. A.; Hoveyda A. H. J. Am. Chem. Soc. 1996, 118, 3779–3780. [Google Scholar]
- a Harrity J. P. A.; Visser M. S.; Gleason J. D.; Hoveyda A. H. J. Am. Chem. Soc. 1997, 119, 1488–1489. [Google Scholar]; b Harrity J. P. A.; La D. S.; Cefalo D. R.; Visser M. S.; Hoveyda A. H. J. Am. Chem. Soc. 1998, 120, 2343–2351. [Google Scholar]
- For representative examples, see:; a Stragies R.; Blechert S. J. Am. Chem. Soc. 2000, 122, 9584–9591. [Google Scholar]; b Nicolaou K. C.; Vega J. A.; Vassilikogiannakis G. Angew. Chem., Int. Ed. 2001, 40, 4441–4445. [DOI] [PubMed] [Google Scholar]; c Henderson J.; Phillips A. J. Angew. Chem., Int. Ed. 2008, 47, 8499–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jackson K. L.; Henderson J. A.; Motoyoshi H.; Phillips A. J. Angew. Chem., Int. Ed. 2009, 48, 2346–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapper C.; Blechert S. J. Org. Chem. 2002, 67, 6456–6460. [DOI] [PubMed] [Google Scholar]
- Hart A. C.; Phillips A. J. J. Am. Chem. Soc. 2006, 128, 1094–1095. [DOI] [PubMed] [Google Scholar]
- Takao K.-i.; Nanamiya R.; Fukushima Y.; Namba A.; Yoshida K.; Tadano K.-i. Org. Lett. 2013, 15, 5582–5585. [DOI] [PubMed] [Google Scholar]
- Houri A. F.; Xu Z.; Cogan D. A.; Hoveyda A. H. J. Am. Chem. Soc. 1995, 117, 2943–2944. [Google Scholar]
- a Hu T.; Panek J. S. J. Org. Chem. 1999, 64, 3000–3001. [DOI] [PubMed] [Google Scholar]; b Schmidt A.-K. C.; Stark C. B. W. Org. Lett. 2011, 13, 4164–4167. [DOI] [PubMed] [Google Scholar]
- We originally used “cascade catalysis” to define a synthesis sequence where a series of catalytic processes are used, including some that are performed in the same vessel (e.g., the net hydrovinyl addition in Scheme 5). Subsequently, the term has been used to describe the same or similar applications of catalytic reactions. For representative cases, see:; a Huang Y.; Walji A. M.; Larsen C. H.; MacMillan D. W. C. J. Am. Chem. Soc. 2005, 127, 15051–15053. [DOI] [PubMed] [Google Scholar]; b Yang T.; Ferrali A.; Campbell L.; Dixon D. J. Chem. Commun. 2008, 2923–2925. [DOI] [PubMed] [Google Scholar]; c Zhang X.; Zhang S.; Wang W. Angew. Chem., Int. Ed. 2010, 49, 1481–1484. [DOI] [PubMed] [Google Scholar]; d Ozboya K. E.; Rovis T. Chem. Sci. 2011, 2, 1835–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Huff C. A.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18122–18125. [DOI] [PubMed] [Google Scholar]; f Candish L.; Lupton D. W. Chem. Sci. 2012, 3, 380–383. [Google Scholar]; g Li L.; Herzon S. B. Nature Chem. 2014, 6, 22–27. [DOI] [PubMed] [Google Scholar]; For a recent review, see:; h Volla C. M. R.; Atodiresei I.; Rueping M. Chem. Rev. 2013, 113, 2390–2431. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Johannes C. W.; Salman S. S.; Hoveyda A. H. J. Am. Chem. Soc. 1996, 118, 10926–10927. [Google Scholar]
- Xu Z.; Johannes C. W.; Houri A. F.; La D. S.; Cogan D. A.; Hofilena G. E.; Hoveyda A. H. J. Am. Chem. Soc. 1997, 119, 10302–10316. [Google Scholar]
- Smith A. B.; Adams C. M.; Kozmin S. A.; Paone D. V. J. Am. Chem. Soc. 2001, 123, 5925–5937. [DOI] [PubMed] [Google Scholar]
- For additional details regarding the discovery of this class of Ru-based OM catalysts, see:Hoveyda A. H.; Gillingham D. G.; Van Veldhuizen J. J.; Kataoka O.; Garber S. B.; Kingsbury J. S.; Harrity J. P. A. Org. Biomol. Chem. 2004, 2, 8–23. [DOI] [PubMed] [Google Scholar]
- Kingsbury J. S.; Harrity J. P. A.; Bonitatatebus P. J.; Hoveyda A. H. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar]
- Garber S. B.; Kingsbury J. S.; Gray B. L.; Hoveyda A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar]
- a Kingsbury J. S.; Garber S. B.; Giftos J. M.; Gray B. L.; Okamoto M. M.; Farrer R. A.; Fourkas J. T.; Hoveyda A. H. Angew. Chem., Int. Ed. 2001, 40, 4251–4256. [DOI] [PubMed] [Google Scholar]; For a review on supported OM catalysts, see:; b Kingsbury J. S.; Hoveyda A. H. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser M. R., Ed.; Wiley-VCH: Weinheim, 2003, pp 467–502. For notable recent advances, see: [Google Scholar]; c Allen D. P.; Van Wingerden M. W.; Grubbs R. H. Org. Lett. 2009, 11, 1261–1264. [DOI] [PubMed] [Google Scholar]; d Mayer C.; Gillingham D. G.; Ward T. R.; Hilvert D. Chem. Commun. 2011, 47, 12068–12070. [DOI] [PubMed] [Google Scholar]; e Showerski K.; Wierzbicka C.; Szczepaniak G.; Gulajski L.; Bieniek M.; Grela K. Green Chem. 2012, 4, 3264–3268. [Google Scholar]
- Kingsbury J. S.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 4510–4517. [DOI] [PubMed] [Google Scholar]
- Vorfalt T.; Wannowius K. J.; Thiel V.; Plenio H. Chem. Eur. J. 2010, 16, 12312–12315and references cited therein. [DOI] [PubMed] [Google Scholar]
- Wakamatsu H.; Blechert S. Angew. Chem., Int. Ed. 2002, 41, 2403–2405. [DOI] [PubMed] [Google Scholar]
- a Grela K.; Harutyunyan S.; Michrowska A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. [DOI] [PubMed] [Google Scholar]; b Zhang Z.-Y. J. US Patent UP2007/0043180 A1.
- For a discussion on the suitability of different Ru complexes to various OM reaction types, see:Bieniek M.; Michrowska A.; Usanov D. L.; Grela K. Chem. Eur. J. 2008, 14, 806–818. [DOI] [PubMed] [Google Scholar]
- a Complex Ru–3f:Stewart I. C.; Ung T.; Pletnev A. A.; Berlin J. M.; Grubbs R. H.; Schrodi Y. Org. Lett. 2007, 9, 1589–1592. [DOI] [PubMed] [Google Scholar]; b Complex Ru–3g:Berlin J. M.; Campbell K.; Ritter T.; Funk T. W.; Chlenov A.; Grubbs R. H. Org. Lett. 2007, 9, 1339–1342. [DOI] [PubMed] [Google Scholar]
- For an example, see:Enquist J. A.; Stoltz B. M. Nature 2008, 453, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. H.; Grubbs R. H. J. Am. Chem. Soc. 2006, 128, 3508–3509. [DOI] [PubMed] [Google Scholar]
- Nicola T.; Brenner M.; Donsbach K.; Kreye P. Org. Proc. Res. Dev. 2005, 9, 513–515. [Google Scholar]
- Poirer M.; Aubry N.; Boucher C.; Ferland J.-F.; LaPlante S.; Tsantrizos Y. S. J. Org. Chem. 2005, 70, 10765–10773. [DOI] [PubMed] [Google Scholar]
- Yee N. K.; Farina V.; Houpis I. N.; Haddad N.; Frutos R. P.; Gallou F.; Wang X.; Wei X.; Simpson R. D.; Feng X.; Fuchs V.; Xu Y.; Tan J.; Zhang L.; Xu J.; Smith-Keenan L. L.; Vitous J.; Ridges M. D.; Spinelli E. M.; Johnson M. J. Org. Chem. 2006, 71, 7133–7245. [DOI] [PubMed] [Google Scholar]
- Kong J.; Chen C.; Balsells-Padros J.; Cao Y.; Dunn R. F.; Dolman S. J.; Janey J.; Li H.; Zacuto M. J. J. Org. Chem. 2012, 77, 3820–3828. [DOI] [PubMed] [Google Scholar]
- Wang H.; Matsuhashi H.; Doan B. D.; Goodman S. N.; Ouyang X.; Clark W. M. Tetrahedron 2009, 65, 6291–6303. [Google Scholar]
- Crimmins M. T.; Zhang Y.; Diaz F. A. Org. Lett. 2006, 8, 2369–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. K.; Hoveyda A. H. J. Am. Chem. Soc. 2008, 130, 12904–12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. K.; Degrado S. J.; Hoveyda A. H. Angew. Chem., Int. Ed. 2005, 44, 5306–5310. [DOI] [PubMed] [Google Scholar]
- May T. L.; Brown M. K.; Hoveyda A. H. Angew. Chem., Int. Ed. 2008, 47, 7358–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review on catalytic EOM, see:Hoveyda A. H.; Malcolmson S. J.; Meek S. J.; Zhugralin A. R. In Catalytic Asymmetric Synthesis; Ojima I., Ed.; Wiley-VCH: Weinheim, 2010; pp 739–770. [Google Scholar]
- For a discussion of advantages offered by catalytic EOM, see:Hoveyda A. H.; Malcolmson S. J.; Meek S. J.; Zhugralin A. R. Angew. Chem., Int. Ed. 2010, 49, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an early overview of enantioselective OM reactions, see:Hoveyda A. H.; Schrock R. R. Chem. Eur. J. 2001, 7, 945–950. [DOI] [PubMed] [Google Scholar]
- Fujimura O.; Grubbs R. H. J. Am. Chem. Soc. 1996, 118, 2499–2500. [Google Scholar]
- Fujimura O.; Grubbs R. H. J. Org. Chem. 1998, 63, 824–832. [DOI] [PubMed] [Google Scholar]
- Alexander J. B.; La D. S.; Cefalo D. R.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1998, 120, 4041–4042. [Google Scholar]
- La D. S.; Alexander J. B.; Cefalo D. R.; Graf D. D.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1998, 120, 9720–9721. [Google Scholar]
- a Zhu S. S.; Cefalo D. R.; La D. S.; Jamieson J. Y.; Davis W. M.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1999, 121, 8251–8259. [Google Scholar]; b Weatherhead G. S.; Houser J. H.; Ford J. G.; Jamieson J. H.; Schrock R. R.; Hoveyda A. H. Terahedron Lett. 2000, 41, 9553–9559. [Google Scholar]
- a Seiders T. J.; Ward D. W.; Grubbs R. H. Org. Lett. 2001, 3, 3225–3228. [DOI] [PubMed] [Google Scholar]; b Funk T. W.; Berlin J. M.; Grubbs R. H. J. Am. Chem. Soc. 2006, 128, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattely E. S.; Cortez G. A.; Moebius D. C.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 8526–8533. [DOI] [PubMed] [Google Scholar]
- Harvey J. S.; Malcolmson S. J.; Dunne K. S.; Meek S. J.; Thompson A. L.; Schrock R. R.; Hoveyda A. H.; Gouverneur V. Angew. Chem., Int. Ed. 2009, 48, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ogasawara M.; Watanabe S.; Fan L.; Nakajima K.; Takahashi T. Organometallics 2006, 25, 5201–5203. [Google Scholar]; b Ogasawara M.; Watanabe S.; Nakajima K.; Takahashi T. J. Am. Chem. Soc. 2010, 132, 2136–2137. [DOI] [PubMed] [Google Scholar]
- Ogasawara M.; Wu W-Y.; Arae S.; Watanabe S.; Morita T.; Takahashi T.; Kamikawa K. Angew. Chem., Int. Ed. 2012, 51, 2951–2955. [DOI] [PubMed] [Google Scholar]
- a Aeilts S. L.; Cefalo D. R.; Bonitatebus P. J.; Houser J. H.; Hoveyda A. H.; Schrock R. R. Angew. Chem., Int. Ed. 2001, 40, 1452–1456. [PubMed] [Google Scholar]; b Schrock R. R.; Jamieson J. Y.; Dolman S. J.; Miller S. A.; Davis W. M.; Hoveyda A. H.; Schrock R. R. Organometallics 2002, 21, 409–417. [Google Scholar]
- Teng X.; Cefalo D. R.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2002, 124, 10779–10784. [DOI] [PubMed] [Google Scholar]
- Weatherhead G. S.; Cortez A. G.; Schrock R. R.; Hoveyda A. H. Proc. Nat. Acad. Sci. U.S.A. 2004, 101, 5805–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cefalo D. R.; Kiely A. F.; Wuchrer M.; Jamieson J. Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2001, 123, 3139–3140. [Google Scholar]
- For the first reported example of catalytic ERORCM, see:Weatherhead G. S.; Ford J. G.; Alexanian E. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2000, 122, 1828–1829. [Google Scholar]
- Van Veldhuizen J. J.; Garber S. B.; Kingsbury J. S.; Hoveyda A. H. J. Am. Chem. Soc. 2002, 124, 4954–4955. [DOI] [PubMed] [Google Scholar]
- Van Veldhuizen J. J.; Gillingham D. G.; Garber S. B.; Kataoka O.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 12502–12508. [DOI] [PubMed] [Google Scholar]
- Van Veldhuizen J. J.; Campbell J. E.; Giudici R. E.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 6877–6882. [DOI] [PubMed] [Google Scholar]
- The term exo indicates that the C=Ru is oriented below the mesityl substituent of NHC moiety, whereas the term endo is indicative of a Ru=C that is situated within the cavity generated by the biaryl bridge.
- Gillingham D. G.; Hoveyda A. H. Angew. Chem., Int. Ed. 2007, 46, 3860–3864. [DOI] [PubMed] [Google Scholar]
- For the original report on catalytic EROCM of oxabicyclic alkenes with terminal olefins, see:Gillingham D. G.; Kataoka O.; Garber S. B.; Hoveyda A. H. J. Am. Chem. Soc. 2004, 126, 12288–12290. [DOI] [PubMed] [Google Scholar]
- For representative recent reports regarding the use of sulfonate-bridged bidentate NHC–Cu complexes in catalytic enantioselective allylic substitution reaction, see:; a Lee Y.; Akiyama K.; Gillingham D. G.; Brown M. K.; Hoveyda A. H. J. Am. Chem. Soc. 2008, 130, 446–447. [DOI] [PubMed] [Google Scholar]; b Akiyama A.; Gao F.; Hoveyda A. H. Angew. Chem., Int. Ed. 2010, 49, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gao F.; McGrath K. P.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]; d May T. L.; Dabrowski J. A.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Dabrowski J. A.; Gao F.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 4778–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Jung B.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 1490–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Dabrowski J. A.; Haeffner F.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 52, 7694–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]; h McGrath K. P.; Hoveyda A. H. Angew. Chem., Int. Ed. 2014, 53, 1910–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For applications of sulfonate-bridged NHC–Cu complexes to enantioselective C–B bond-forming reactions, see:; a Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3160–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guzman-Martinez A.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10634–10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cortez G. A.; Baxter C. A.; Schrock R. R.; Hoveyda A. H. Org. Lett. 2007, 9, 2871–2874. [DOI] [PubMed] [Google Scholar]; b Cortez G. A.; Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed. 2007, 46, 4534–4538. [DOI] [PubMed] [Google Scholar]
- For investigations in connection with spectroscopic characterization of ruthenacyclobutane intermediates in OM reactions, see:; a Romero P. E.; Piers W. E. J. Am. Chem. Soc. 2005, 127, 5032–5033. [DOI] [PubMed] [Google Scholar]; b Wenzel A. G.; Grubbs R. H. J. Am. Chem. Soc. 2006, 128, 16048–16049. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Romero P. E.; Piers W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. [DOI] [PubMed] [Google Scholar]; d van der Eide E. F.; Romero P. E.; Piers W. E. J. Am. Chem. Soc. 2008, 130, 4485–4491. [DOI] [PubMed] [Google Scholar]; e van der Eide E. F.; Piers W. E. Nature Chem. 2010, 2, 571–576. [DOI] [PubMed] [Google Scholar]; f Wenzel A. G.; Blake G.; VanderVelde D. G.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 6429–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The principle of “side change” of a carbene ligand in a Ru-based complex, which becomes equal to inversion at the metal center in stereogenic-at-metal complexes, was first proposed by Chen and co-workers in the context of kinetic isotope effect studies. See:; a Adlhart C.; Hinderling C.; Baumann H.; Chen P. J. Am. Chem. Soc. 2000, 122, 8204–8214. [Google Scholar]; b Adlhart C.; Chen P. Angew. Chem., Int. Ed. 2002, 41, 4484–4487. [DOI] [PubMed] [Google Scholar]
- Khan R. K. M.; Zhugralin A. R.; Torker S.; O’Brien R. V.; Lombardi P. J.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 12438–12441. [DOI] [PubMed] [Google Scholar]
- For early work on the mechanism of polytopal rearrangements, see:; a Berry R. S. J. Chem. Phys. 1960, 32, 933–938. [Google Scholar]; b Mutterties E. L. J. Am. Chem. Soc. 1969, 91, 1636–1643. [Google Scholar]; c Gillespie P.; Hoffman P.; Klusacek H.; Marquarding D.; Pfohl S.; Ramirez F.; Tsolis E. A.; Ugi I. Angew. Chem., Int. Ed. Engl. 1971, 10, 687–715. [Google Scholar]; For recent reviews on polytopal rearrangements in pentacoordinated complexes, see:; d Couzijn E. P. A.; Slootweg J. C.; Ehlers A. W.; Lammertsma K. J. Am. Chem. Soc. 2010, 132, 18127–18140. [DOI] [PubMed] [Google Scholar]; e Moberg C. Angew. Chem., Int. Ed. 2011, 50, 10290–10292. [DOI] [PubMed] [Google Scholar]
- Torker S.; Khan R. K. M.; Hoveyda A. H. J. Am. Chem. Soc. 2014, 136, 1968–1972. [DOI] [PubMed] [Google Scholar]
- a Bornand M.; Chen P. Angew. Chem., Int. Ed. 2005, 44, 7909–7911. [DOI] [PubMed] [Google Scholar]; b Bornand M.; Torker S.; Chen P. Organometallics 2007, 26, 3585–3596. [Google Scholar]; c Torker S.; Müller A.; Sigrist R.; Chen P. Organometallics 2010, 29, 2735–2751. [Google Scholar]; d Torker S.; Müller A.; Chen P. Angew. Chem., Int. Ed. 2010, 49, 3762–3766. [DOI] [PubMed] [Google Scholar]; e Jovic M.; Torker S.; Chen P. Organometallics 2011, 30, 3971–3980. [Google Scholar]
- a Malcolmson S. J.; Meek S. J.; Sattely E. S.; Schrock R. R.; Hoveyda A. H. Nature 2008, 456, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sattely E. S.; Meek S. J.; Malcolmson S. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Solans-Monfort X.; Clot E.; Copéret C.; Eisenstein O. J. Am. Chem. Soc. 2005, 127, 14015–14025. [DOI] [PubMed] [Google Scholar]; b Poater A.; Solans-Monfort X.; Clot E.; Copéret C.; Eisenstein O. J. Am. Chem. Soc. 2007, 129, 8207–8216. [DOI] [PubMed] [Google Scholar]
- Singh R.; Schrock R. R.; Müller P.; Hoveyda A. H. J. Am. Chem. Soc. 2007, 129, 12654–12655. [DOI] [PubMed] [Google Scholar]
- Meek S. J.; Malcolmson S. J.; Li B.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 16407–16409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinescu S.; Schrock R. R.; Li B.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 58–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang A. J.; Simpson J. H.; Müller P.; Schrock R. R. J. Am. Chem. Soc. 2009, 131, 7770–7780. [DOI] [PubMed] [Google Scholar]
- a Ibrahem I.; Yu M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3844–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a follow-up study regarding Z-selective ring-opening metathesis polymerization (ROMP), see:; b Flook M. M.; Jiang A. J.; Schrock R. R.; Müller P.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 7962–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Ibrahem I.; Hasegawa M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 2788–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an overview of catalytic CM and its utility in natural product synthesis, see:Prunet J.; Grimaud L. In Metathesis in Natural Product Synthesis; Cossy J., Arsenyadis S., Meyer C., Eds.; Wiley-VCH: Weinheim, 2010; pp 287–312. [Google Scholar]
- Meek S. J.; O’Brien R. V.; Llaveria J.; Schrock R. R.; Hoveyda A. H. Nature 2011, 471, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related catalytic Z-selective homocoupling of terminal alkenes, see:; a Jiang A. J.; Zhao Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 16630–16631. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marinescu S. C.; Schrock R. R.; Müller P.; Takase M. K.; Hoveyda A. H. Organometallics 2011, 30, 1780–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Townsend E. M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 11334–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selective MAP-catalyzed ethenolysis reactions, see:; d Marinescu S. C.; Schrock R. R.; Müller P.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 10840–10841. [DOI] [PubMed] [Google Scholar]; e Marinescu S. C.; Levine D. S.; Zhao Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 11512–11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann T. J.; Speed A. W. H.; Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 52, 8395–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter E. T.; O’Brien R. V.; Yu E. C.; Meek S. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2013, 135, 6026–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gao F.; Carr M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2014, 136, 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a related approach involving phosphine–Ir-catalyzed EAS reactions with Z-alkenyl–B(pin) reagent, see:; b Hamilton J. Y.; Sarlah D.; Carreira E. M. J. Am. Chem. Soc. 2013, 135, 994–997. [DOI] [PubMed] [Google Scholar]
- For overviews regarding macrocyclic RCM reactions and their utility in natural product synthesis, see:; a Gradillas A.; Perez-Castells J. Angew. Chem., Int. Ed. 2006, 45, 6086–6101. [DOI] [PubMed] [Google Scholar]; b Gradillas A.; Pérez-Castells J. in Metathesis in Natural Product Synthesis; Cossy J., Arsenyadis S., Meyer C., Eds.; Wiley-VCH: Weinheim, 2010; pp 149–182. [Google Scholar]
- Wang C.; Yu M.; Kyle A. F.; Jakubec P.; Dixon D. J.; Schrock R. R.; Hoveyda A. H. Chem.—Eur. J. 2013, 19, 2726–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Wang C.; Kyle A. F.; Jakubec P.; Dixon D. J.; Schrock R. R.; Hoveyda A. H. Nature 2011, 479, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For previous studies regarding epothilones A and C, see:; a Nicolaou K. C.; He Y.; Vourloumis D.; Vallberg H.; Roschangar F.; Sarabia F.; Ninkovic S.; Yang Z.; Trujillo J. I. J. Am. Chem. Soc. 1997, 119, 7960–7973. [Google Scholar]; b Meng D.; Bertinato P.; Balog A.; Su D.-S.; Kamenecka T.; Sorensen E. J.; Danishefsky S. J. J. Am. Chem. Soc. 1997, 119, 10073–10092. [Google Scholar]; c Schinzer D.; Bauer A.; Böhm O. M.; Limberg A.; Cordes M. Chem.—Eur. J. 1999, 5, 2483–2491. [Google Scholar]; d Sinha S. C.; Sun J.; Miller G. P.; Wartmann M.; Lerner R. A. Chem.—Eur. J. 2001, 7, 1691–1702. [DOI] [PubMed] [Google Scholar]; For enantioselective total syntheses of nakadomarin A, see:; e Nagata T.; Nakagawa M.; Nishida A. J. Am. Chem. Soc. 2003, 125, 7484–7485. [DOI] [PubMed] [Google Scholar]; f Ono K.; Nakagawa M.; Nishida A. Angew. Chem., Int. Ed. 2004, 43, 2020–2023. [DOI] [PubMed] [Google Scholar]; g Young I. S.; Kerr M. A. J. Am. Chem. Soc. 2007, 129, 1465–1469. [DOI] [PubMed] [Google Scholar]; h Jakubec P.; Cockfield D. M.; Dixon D. J. J. Am. Chem. Soc. 2009, 131, 16632–16633. [DOI] [PubMed] [Google Scholar]; i Cheng B.; Wu F.; Yang X.; Zhou Y.; Wan X.; Zhai H. Chem.—Eur. J. 2011, 17, 12569–12572. [DOI] [PubMed] [Google Scholar]
- a Kyle A. F.; Jakubec P.; Cockfield D. M.; Cleator E.; Skidmore J.; Dixon D. J. Chem. Commun. 2011, 47, 10037–10039. [DOI] [PubMed] [Google Scholar]; For a total synthesis of nakadomarin A entailing the use of macrocyclic alkyne RCM, see:; b Nilson M. G.; Funk R. L. Org. Lett. 2010, 12, 4912–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llàcer E.; Urpí F.; Vilarrasa J. Org. Lett. 2009, 11, 3198–3201. [DOI] [PubMed] [Google Scholar]
- For reviews on catalytic alkyne metathesis reactions, see:; a Fürstner A.; Davis P. W. Chem. Commun. 2005, 2307–2320. [DOI] [PubMed] [Google Scholar]; b Zhang W.; Moore J. S. Adv. Synth. Catal. 2007, 349, 93–120. [Google Scholar]; c Fürstner A. Angew. Chem., Int. Ed. 2013, 52, 2794–2819. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Guth O.; Rumbo A.; Seidel G. J. Am. Chem. Soc. 1999, 121, 11108–11113. [Google Scholar]
- Fürstner A.; Mathes C.; Lehmann C. W. Chem.—Eur. J. 2001, 7, 5299–5317. [DOI] [PubMed] [Google Scholar]
- a Reference (97a).; b Jakubec P.; Kyle A. F.; Calleja J.; Dixon D. J. Tetrahedron Lett. 2011, 52, 6094–6097. [Google Scholar]
- Nilson M. G.; Funk R. L. Org. Lett. 2010, 12, 4912–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For example, see:; a Fürstner A.; Radkowski K. Chem. Commun. 2002, 2182–2183. [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Castanet A.-S.; Radkowski K.; Lehmann C. W. J. Org. Chem. 2003, 68, 1521–1528. [DOI] [PubMed] [Google Scholar]; c Fürstner A.; Lorionov O.; Flügge S. Angew. Chem., Int. Ed. 2007, 46, 5545–5548. [DOI] [PubMed] [Google Scholar]; d Fürstner A.; Flügge S.; Lorionov O.; Takahashi Y.; Kubota T.; Kobayashi J. Chem.—Eur. J. 2009, 15, 4011–4029. [DOI] [PubMed] [Google Scholar]; e Micoine K.; Fürstner A. J. Am. Chem. Soc. 2010, 132, 14064–14066. [DOI] [PubMed] [Google Scholar]
- For previous studies regarding macrocyclic RCM en route to epothilones B and D, see:; a Reference (96b).; b Reference (96d).
- Wang C.; Haeffner F.; Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 52, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solans-Monfort X.; Copéret C.; Eisenstein O. J. Am. Chem. Soc. 2010, 132, 7750–7757. [DOI] [PubMed] [Google Scholar]
- Khan R. K. M.; O’Brien R. V.; Torker S.; Li B.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 12774–12779. [DOI] [PubMed] [Google Scholar]
- Khan R. K. M.; Torker S.; Hoveyda A. H. J. Am. Chem. Soc. 2013, 135, 10258–10261. [DOI] [PubMed] [Google Scholar]
- Keitz B. K.; Fedorov A.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 2040–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh M. J.; Khan R. K. M.; Torker S.; Hoveyda A. H. Angew. Chem., Int. Ed. 2014, 53, 1968–1972. [DOI] [PubMed] [Google Scholar]
- Hoveyda A. H.; Lombardi P. J.; O’Brien R. V.; Zhugralin A. R. J. Am. Chem. Soc. 2009, 131, 8378–8379. [DOI] [PubMed] [Google Scholar]
- a Endo K.; Grubbs R. H. J. A. Chem. Soc. 2011, 133, 8525–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Keitz B. K.; Endo K.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 133, 9686–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Keitz B. K.; Endo K.; Patel P. R.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rosebrugh L. E.; Herbert M. B.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung J.; Grubbs R. H. Angew. Chem., Int. Ed. 2014, 53, 3885–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhipinti G.; Hansen F. R.; Törnroos K. W.; Jensen V. R. J. Am. Chem. Soc. 2013, 135, 3331–3334. [DOI] [PubMed] [Google Scholar]
- Herbert M. B.; Marx V. M.; Pederson R. L.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley B. L.; Grubbs R. H. Chem. Sci. 2014, 5, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung J.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10183–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. M.; Herbert M. B.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464–1467. [DOI] [PubMed] [Google Scholar]; b Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464–1467. [DOI] [PubMed] [Google Scholar]; See also:; c Deng Y.; Wang Z.-H.; Wang X. Organometallics 2012, 31, 7222–7234. [Google Scholar]
- For an overview of challenges in catalytic OM reactions, see:; a Reference (2).; b Fürstner A. Science 2013, 341, 1357–1364. [DOI] [PubMed] [Google Scholar]