

Abstract

Fluorescent N-phenyl-4-aminoquinazoline probes targeting the ATP-binding pocket of the ERBB family of receptor tyrosine kinases are reported. Extension of the aromatic quinazoline core with fluorophore “arms” through substitution at the 6- position of the quinazoline core with phenyl, styryl, and phenylbutadienyl moieties was predicted by means of TD-DFT calculations to produce probes with tunable photoexcitation energies and excited states possessing charge-transfer character. Optical spectroscopy identified several synthesized probes that are nonemissive in aqueous solutions and exhibit emission enhancements in solvents of low polarity, suggesting good performance as turn-on fluorophores. Ligand-induced ERBB2 phosphorylation assays demonstrate that despite chemical modification to the quinazoline core these probes still function as ERBB2 inhibitors in MCF7 cells. Two probes were found to exhibit ERBB2-induced fluorescence, demonstrating the utility of these probes as turn-on, fluoroescent kinase inhibitors.
Introduction
Small molecule kinase inhibitors are cornerstones of advanced chemotherapy. In their ideal embodiment, they may selectively target a validated signaling pathway that is uniquely dysregulated in cancer cells; in reality, multiple factors complicate this strategy. First, the structural and functional homology of kinases combined with the abundance of ATP-binding folds throughout the cell make off-target binding events unavoidable.1,2 Second, compensatory or alternate modes of pathway activation can lead to the development of resistance, requiring additional antineoplastic drugs or a different chemotherapy regimen.3,4 Ideally the observation of compensatory shifts in signaling on a systemic and live cell level could facilitate the early development of second-generation drugs with larger barriers or at least anticipated paths to adaptation. In this context, assaying changes in receptor kinase states (i.e., level of expression, oligomerization, localization, conformation, phosphorylation) remains a challenge, particularly in live cells or tissue preparations.
In an effort to develop molecular probes capable of interrogating kinase signaling pathways, we have introduced the concept of “turn-on” fluorescent ligands that target the ATP-binding pocket of the EGFR/ERBB family of receptor tyrosine kinases.5 These molecular probes are built upon the 4-aminoquinazoline scaffold, which is an established EGFR/ERBB pharmacophore;6−10 examples include gefitinib or erlotinib (Figure 1A,C), so-called type I inhibitors that preferentially binding the active kinase conformation, and lapatinib (Figure 1B,D), a type II inhibitor targeting the inactive conformation.10 The preference for active and inactive conformations is addressed through substitution at the 4-amino position (Figure 1E). While the quinazoline core conveys binding to the nucleotide pocket, but with limited specificity for ERBB-type receptors, the 4-amino aryl arm further increases specificity and contributes discrimination between activation states. This selectivity reflects increased access to the hydrophobic pocket adjacent to the nucleotide binding site in the inactive conformation. Together, the quinazoline core and N-aryl arm constitute the pharmacophore. Crystal structures of the kinase domain of EGFR with either erlotinib (1M17)15 or lapatinib (1XKK)9 (Figure 1A,B) show that while the pharmacophore arm is oriented deep in the binding pocket, the 6-position is amenable to chemical modification without perturbing the key conserved contacts of the binding pocket. The structure of lapatinib demonstrates this point as the aromatic core is extended by the addition of a furan ring. Thus, a potential strategy for synthesizing fluorescent kinase inhibitors is to modify the 4-aminoquinazoline core with fluorphore arms (Figure 1E) at the 6-position. In principle, probes targeting specific kinases (i.e., IGF1R vs EGFR) or activation states could be encoded with unique optical outputs by tuning the excitation and emission energies through the fluorophore arm.
Figure 1.
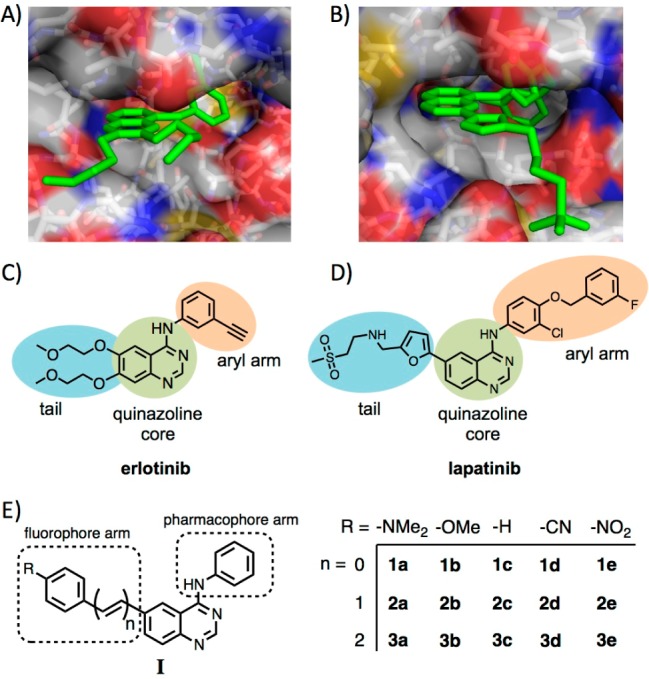
Crystal structures of the EGFR ATP-binding pocket with (A) erlotinib (PDB ID: 1M17)15 and (B) lapatinib (PDB ID: 1XKK)9 reveal the inhibitor binding modes. The arms at the 6-position of the quinazoline core (in blue; C, D) may be replaced by fluorophore arms without disturbing the key binding contacts. (E) General structure and substituent key for the synthesized fluorescent quinazolines.
Herein, we investigate the effect of conjugation length and auxochrome substitution on the optical properties of a family of N-phenyl-4-aminoquinazoline probes. We find that extension of the π framework effectively lowers the excitation and emission energies, yielding fluorescent probes that compare favorably with other fluorescent adenosine analogues.11−14 The introduction of strong electron donors or strong electron-withdrawing groups generates donor–acceptor systems that are highly sensitive to solvent polarity. As a result, several probes exhibit high fluorescence ON/OFF ratios, a feature that is key to their performance as self-reporting fluorescent kinase inhibitors. Despite the modifications to the quinazoline core, we also found that several of these probes inhibit ERBB2 phosphorylation in a live cell setting, demonstrating that binding to the ATP pocket and cell permeability are preserved. Overall, these fluorescent adenosine mimics compare favorably with other nucleobase analogues as they possess tunable optical properties and high fluorescence turn-on ratios and compete effectively with the native substrate to inhibit tyrosine phosphorylation.
Results and Discussion
Design and Synthesis
The optical properties of kinase-binding probes of general structure I can be addressed through the fluorophore arm depicted in Figure 1E. We envisaged tuning two parameters: (1) the electron-donating ability of the substituent on the phenyl ring and (2) the conjugation length between the phenyl ring and the quinazoline core. TD-DFT calculations, vide infra, guided the selection of the electron-donating and electron-withdrawing substituents that span the range from the strongly donating dimethylamino group (e.g., 1a–3a) to strongly electron-withdrawing nitro substitution (1e–3e).
The 15-membered family of quinazolines, 1a–3e (Figure 1), was synthesized from a common intermediate, 6-iodo-N-phenyl-4-quinazolin-amine (4), by Suzuki coupling of the appropriate arylboronic acid (1a–1e, Scheme 1, route A) or by Heck coupling (Scheme1, route B) of the styryl (2a–2e) and phenyl butadiene (3a–3e) arms. Following reaction workup, flash chromatography and crystallization from 2-propanol/ethyl acetate mixtures, the products were isolated in moderate yields as microcrystals that appeared colorless (e.g., 1c and 3c) to bright orange–yellow (e.g., 1a and 3e).
Scheme 1. Synthesis Routes to Fluorescent Quinazolines 1a–3e.

Representative structures 1a and 3d are shown.
Quantum Chemical Calculations
Two photophysical properties, the fluorescence ON/OFF ratio and the optical band gap, of the kinase-binding probes can simultaneously be addressed by varying the donor or acceptor substitution and the conjugation length. Ideally, the probes should be nonemissive or off in solution, but upon binding in the solvent excluding and geometrically confined ATP pocket, emission should be enhanced or turned on. To achieve this emission behavior, a probe should possess an excited state with a high degree of charge-transfer (CT) character and possibly access at twisted intramolecular charge-transfer (TICT) excited state. TD-DFT calculations (6-31G*, CHCl3, SMD solvent model)16 show that both strong electron donors and strong electron acceptors will lead to an S1 state with CT character. The S1 state is accessible via the allowed one-electron transition between the HOMO and LUMO for all 15 members, with energies ranging from 2.5 eV (500 nm) in the case of 3e to 3.6 eV (345 nm) in the case of 1c (Table 1). The addition of each vinylene bridge lowers the S1 transition energy by approximately 0.25 eV. The introduction of a moderately electron-donating or -withdrawing group (i.e., −OMe or −CN) lowers the energy of the S0 → S1 transition energy by 0.1 to 0.2 eV when compared to that of isostructural members of the unsubstituted series (i.e., 1c–3c). The presence of the strong electron-donating dimethylamino group more effectively lowers the HOMO–LUMO band gap by 0.5–0.3 eV when comparing 1a–3a and 1a–3c, whereas the strongly electron-withdrawing nitro group has the greatest effect, with differences of 0.8 to 0.5 eV when comparing 1a–3c and 1a–3e.
Table 1. Calculated (TDDFT/CAM-B3LYP/6-31G*)a S1 Energies and S0 → S1 Oscillator Strength.
| 1a | 1b | 1c | 1d | 1e | 2a | 2b | 2c | 2d | 2e | 3a | 3b | 3c | 3d | 3e | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE S0 → S1(eV) | 3.9 | 4.0 | 4.1 | 4.0 | 3.9 | 3.6 | 3.8 | 3.8 | 3.7 | 3.5 | 3.3 | 3.5 | 3.6 | 3.4 | 3.3 |
| f | 0.7 | 0.7 | 0.7 | 0.8 | 0.8 | 1.7 | 1.5 | 1.4 | 1.6 | 1.5 | 2.3 | 2.1 | 2.0 | 2.2 | 2.0 |
CHCl3, SMD solvent model.16
In addition to manipulating the optical band gap, the presence of an electron-donating or electron-withdrawing substituent on the pendant phenyl arm influences the CT character of the excited state; the polarization and spatial segregation17 of the HOMO and LUMO is directly linked to the identity of the substituent. In the series of dimethylamino-substituted compounds (e.g., 2a; Figure 2), the HOMO is largely localized to the fluorophore arm, whereas the LUMO is polarized toward the quinazoline core. The presence of a strong electron-withdrawing group (i.e., −NO2 or −CN) on the fluorophore arm reverses the frontier molecular orbital distribution. In the case of 2e (Figure 2), the HOMO is polarized toward the quinazoline core, whereas the LUMO is largely localized to the styryl arm; an excited state with high CT character still results from the promotion of an electron from the HOMO to the LUMO. Compounds 2b–2d show a gradual redistribution of the HOMO and LUMO densities between the two extreme cases, 2a and 2e. Similar polarization of the frontier molecular orbitals is also observed in the 1a–1e (Figure S16) and 3a–3e series (Figure S17).
Figure 2.
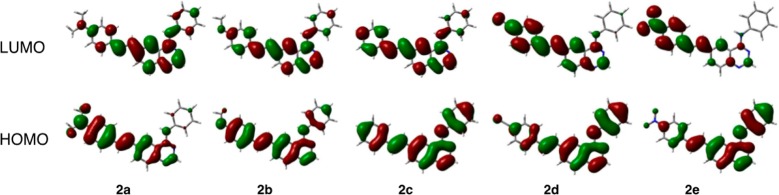
Frontier molecular orbitals of 2a–2e calculated at the CAM-B3LYP/6-31G* level: the polarization of the HOMO and LUMO shifts across the series (compare 2a and 2e); strong CT character is expected for both 2a and 2e, although the localization of charge should be reversed.
Optical Spectroscopy
Probes 1a–3c were designed as turn-on fluorescent ligands and thus were expected to be nonemissive in solvents of high polarity (e.g., water and methanol) and emissive in less polar solvents. This behavior can be qualitatively observed by eye as in chloroform (ET(30) = 39.1 kcal/mol)18 solutions; most of the probes appear bright, with emissions ranging from blue (2b and 3c) to green (1a and 2a) to yellow or orange (3a and 3e), Figure 3, whereas in aqueous solutions, weak or no emission was observed.
Figure 3.

Chloroform solutions of selected probes (5 μM) under UV illumination (354 nm); aqueous solutions showed weak or no emission (vials not shown).
To investigate their optical properties quantitatively, we obtained their UV–vis and fluorescence spectra in chloroform. The absorption maxima (Figure 4 and Table 2) correlate very well with the predicted values both in terms of transition energies as well as oscillator strength. The transition energies progress in a clear trend, as λmax,abs increases with increasing conjugation length. The presence of an auxochrome also serves to modulate the absorption wavelengths, as is evident in the lower transition energies for compounds possessing the dimethylamino (1a–3a) or nitro substituent (1e–3e); these modifications enhance CT character and lead to a longer wavelength absorption band lacking vibronic structure. Some vibronic structure is visible in the absorption spectra of compounds lacking an auxochrome or possessing the methoxy or cyano groups; however, these compounds possess some degree of CT in the excited state, as their emission spectra are largely devoid of vibronic progressions. Despite the presence of CT character, the allowability of the S0 → S1 transition is relatively high for most compounds, as is evident from the good to moderate molar absorptivities. Increasing the conjugation length increases the extinction coefficient in a stepwise fashion: compounds 2a–2e possess molar absorptivities roughly twice the values observed for 1a–1e, whereas compounds 3a–3c have molar absorptivities approximately three times greater than those of 1a–1e. This trend, which reflects the relative magnitudes of transition dipoles, can be directly linked to the spatial overlap of the contributing molecular orbitals, in this case the HOMO and LUMO. In terms of optical compatibility, all of the synthesized probes are compatible with DAPI, Hoechst 33342, or blue fluorescent protein filter sets for epifluorescence microscopy, whereas 2a, 2e, and 3a–3e are optimally matched to the 405 nm diode laser.
Figure 4.
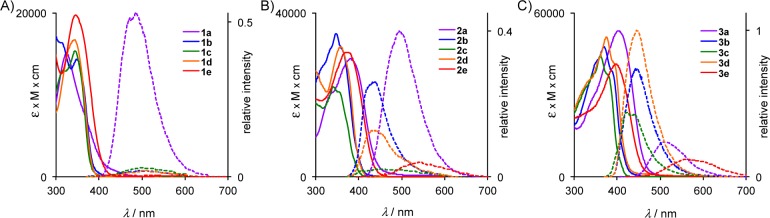
Absorption (solid lines) and emission (dashed lines) spectra of (A) 1a–1e, (B) 2a–2e, and (C) 3a–3e in CHCl3. Emission intensities are given relative to 3d, which has the highest quantum yield (see Table 2).
Table 2. Photophysical Data for 1a–3e.
| compd | λmax,abs (nm) | ε (M–1 cm–1) | λmax,em (nm) | ϕema | Δν (cm–1) | τ1 (ns) | f1 | τ2 (ns) | f2 | χ2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 325 | 15 000 | 485 | 0.37 | 10 200 | 1.22 | 0.03 | 6.54 | 0.97 | 2.46 |
| 1b | 348 | 14 300 | 510 | 0.02 | 9130 | 1.39 | 0.70 | 6.76 | 0.30 | 5.89 |
| 1c | 344 | 15 400 | 502 | 0.03 | 9150 | 0.56 | 0.06 | 1.60 | 0.94 | 2.88 |
| 1d | 343 | 16 700 | 508 | 0.02 | 9470 | 0.17 | 0.18 | 1.29 | 0.82 | 1.47 |
| 1e | 346 | 19 700 | 446 | 0.006 | 6480 | 0.11 | 0.74 | 1.56 | 0.26 | 10.2 |
| 2a | 381 | 28 800 | 495 | 0.28 | 6050 | 0.69 | 0.08 | 2.49 | 0.92 | 3.65 |
| 2b | 347 | 34 900 | 440 | 0.15 | 6090 | 0.89 | 0.57 | 1.50 | 0.43 | 2.17 |
| 2c | 341 | 21 900 | 455 | 0.02 | 7350 | 0.05 | 0.11 | 1.75 | 0.89 | 4.86 |
| 2d | 358 | 31 700 | 436 | 0.09 | 5000 | 0.12 | 0.30 | 1.46 | 0.70 | 0.57 |
| 2e | 372 | 30 400 | 539 | 0.04 | 8330 | 0.38 | 0.83 | 1.60 | 0.17 | 1.77 |
| 3a | 403 | 53 300 | 510 | 0.17 | 5210 | 0.80 | 0.77 | 2.48 | 0.23 | 1.47 |
| 3b | 369 | 47 500 | 443 | 0.42 | 4530 | 0.77 | 0.43 | 1.90 | 0.57 | 1.24 |
| 3c | 361 | 44 300 | 422 | 0.25 | 4000 | 0.54 | 0.32 | 1.78 | 0.68 | 1.33 |
| 3d | 375 | 51 000 | 445 | 0.57 | 4200 | 0.23 | 0.21 | 1.57 | 0.26 | 0.86 |
| 3e | 391 | 41 200 | 561 | 0.11 | 7750 | 0.40 | 0.37 | 1.04 | 0.63 | 1.16 |
±10%.
The emission spectra of 1a–3c in chloroform are shown in Figure 4; most of the probes exhibit good to moderate quantum yields (Table 2). Emission intensities are enhanced in less polar solvents such as toluene and are reduced in more polar solvents such as acetonitrile, supporting the existence of excited states with significant CT character (see Supporting Information, Figure S18). Although many of the probes exhibit emission on the blue end of the visible spectrum, several probes show longer-wavelength emission owing to their longer conjugation length and/or the presence of strong electron-donating or -withdrawing groups. Of the probes exhibiting strong emission, 3c has the bluest emission maximum (42 nm), whereas 3e has the reddest emission maximum (561 nm). Surprisingly, strong CT character does not necessarily equate to poor quantum yields, as seen for probes 1a and 2a. Indeed, 1a is the only member of the 4-phenylquinazoline series (1a–1e) of compounds that exhibits an appreciable degree of fluorescence (ϕem = 0.37). In the 4-styrylquinazoline (2a–2e) series, compounds possessing electron-donating groups (2a and 2b) also exhibit the highest quantum yields. With longer conjugation lengths, quantum yields of fluorescence are markedly higher and the identity of the auxochrome influences ϕem to a lesser degree.
The overall brightness (ε·ϕem) is one important parameter when considering the utility of the probes as optical reporters. An additional parameter, the turn-on ratio, should also be considered as a measure of the probes’ responsiveness to changes in their chemical microenvironment.19 Water (ET(30) = 63.1 kcal/mol, η = 0.89 mPa·s) and octanol (ET(30) = 48.3 kcal/mol, η = 7.24 mPa·s) represent two distinct environments that can be used to assess the physical properties and distribution of organic molecules.18,20 The ratios of emission intensities obtained in octanol and water are shown in Figure 5. Compounds 1a, 2a, and 3e showed the largest on/off ratios with enhancements greater than 50-fold; moderate ratios, between 20 and 40 were observed for 2d, 3a, 3b, and 3d. Moderate enhancements were found for the remaining members of the 2 and 3 series, whereas the remaining 6-arylquinazolines (series 1) showed essentially no emission enhancements.
Figure 5.
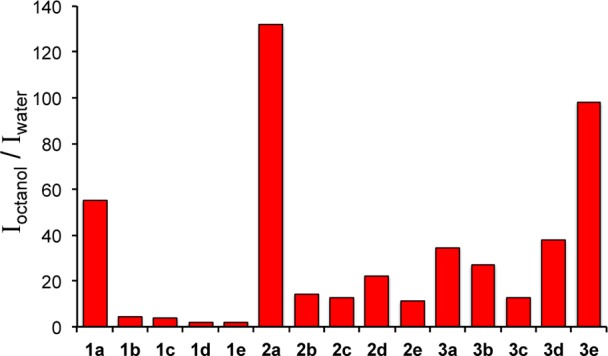
Emission intensities obtained in octanol and water reveal that several probes are highly responsive to changes in their chemical microenvironment and possess high ON/OFF ratios.
Inhibition Studies
The extension of the aromatic system via the 6-position of the quinazoline core was not anticipated to affect the key binding contacts between the probes and the ATP-binding fold of the ERBB family. The crystallographically determined binding modes of gefitinib,8 erlotinib,15 and lapatinib9 show that the 6-position is amenable to substitution and, in some cases, decreases koff of inhibitors.21 To test this assumption, we evaluated selected probes as inhibitors of ERBB2 phosphorylation in MCF7 cells initially using two probe concentrations, 10 μM and 100 nM (Figure 6). MCF7 cells are a well-established model system for the ligand-induced activation of ERBB2–ERBB3 heterocomplexes by ligands of the neuregulin family (NRGβ1 in this case). Compounds 1a, 2a, 2d, 3d, and 3e were selected because they showed the highest turn-on ratios of their respective series. All five probes showed inhibition of NRGβ1-induced phosphorylation of ERBB2 at 10 μM; however, little to no inhibitory action was observed at 100 nM. These results demonstrate two key features of these fluorescent quinazoline probes: First, despite chemical modification, the probes remain membrane permeable and are able to access the intracellular kinase binding domain of ERBB2. Second, the pharmacophore remains an inhibitor of ERBB2 phosphorylation despite the presence of bulky extensions of the aromatic system.
Figure 6.
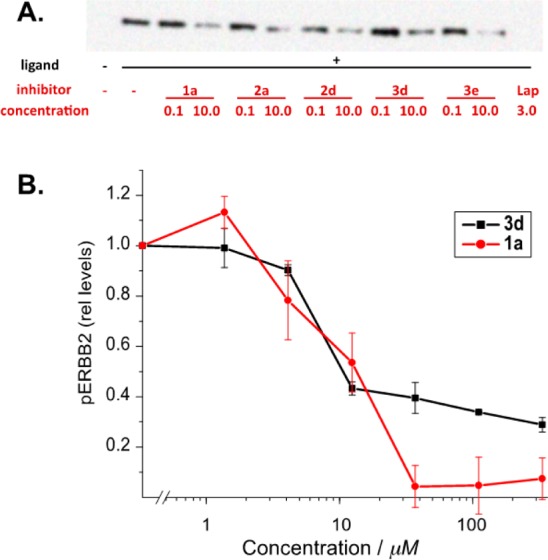
(A) Comparative inhibitory properties of selected probes: the inhibition of NRGβ1-induced ERBB2 tyrosine phosphorylation in MCF7 cells was evaluated at 0.1 and 10 μM concentrations of the indicated compounds. For comparison, lapatinib was used at its clinically used serum concentration of 3 μM. (B) Compounds 3d and 1a show comparable affinity in inhibition studies but a difference in the maximum achievable inhibition.
We next obtained the inhibition curves of 1a, which possesses the smallest modification at the 6-position, and 3d, which possesses one of the longest arms. The Ki for these probes was similar with values of 10 μM for 1a and 9 μM for 3d. A direct comparison with actual binding constants for a type I inhibitor such as gefitinib in a cellular setting are difficult to obtain. Recombinant EGFR kinase domains have yielded an in vitro Kd of approximately 1 nM.22 Equilibrium models in live cells treated with 10 μM Gefitinib have yielded Kd estimates of 2 to 3 nM after competition with cellular ATP was taken into account.23 By this standard, the derivatives used in our study show comparable potency. Although 1a exhibited a typical inhibition curve profile, with complete inhibition of ERBB2 phosphorylation at approximately 20 μM, higher concentrations of 3d do not lead to complete inhibition. While 3d is a larger molecule, hydrophobicity does not appear to play a role, as 1a and 3d have similar octanol/water partition coefficients, with log Poctanol/water = 1.50 and 1.55, respectively. The lack of complete inhibition may reflect alternative binding modes for the inhibitors, as similar partial inhibition resulting from competing cellular ATP has been described for gefitinib when compared to the complete inhibition by the type II inhibitor, lapatinib.23 We are presently evaluating the binding kinetics of these probes and will report their binding modes separately.
Binding-Induced Emission Studies
Emission spectroscopy identified several probes (1a, 2a, and 3e) that exhibit large turn-on ratios, and the phosphorylation inhibition studies demonstrate that the modified N-phenyl quinazolines are capable of accessing the ATP-binding pocket of ERBB2. To determine if turn-on emission is observed upon binding to ERBB2, we obtained the fluorescence spectra of 1a, 2a, and 3e in PBS in the presence and absence of the soluble ERBB2 kinase domain. In PBS alone, the emission of the probes is largely quenched, whereas the addition of the ERBB2 kinase domain produced substantial emission enhancements for both of the dimethylamino-substituted probes. In the case of 1a, the emission enhancement was 4-fold when comparing all emission wavelengths, with a maximum enhancement of 10 at 435 nm (Figure 7). For 2a, the emission was increased by a factor of 8 when comparing all emission wavelengths, with a maximum enhancement of 12 at 560 nm. No emission enhancement was observed for 3e despite the fact that it also exhibits solvent sensitivity and was shown to inhibit ATP binding to ERBB2. The lack of binding-induced emission enhancement may be a result of the longer conjugated arm projecting beyond the binding pocket and being exposed to the polar solvent environment.
Figure 7.
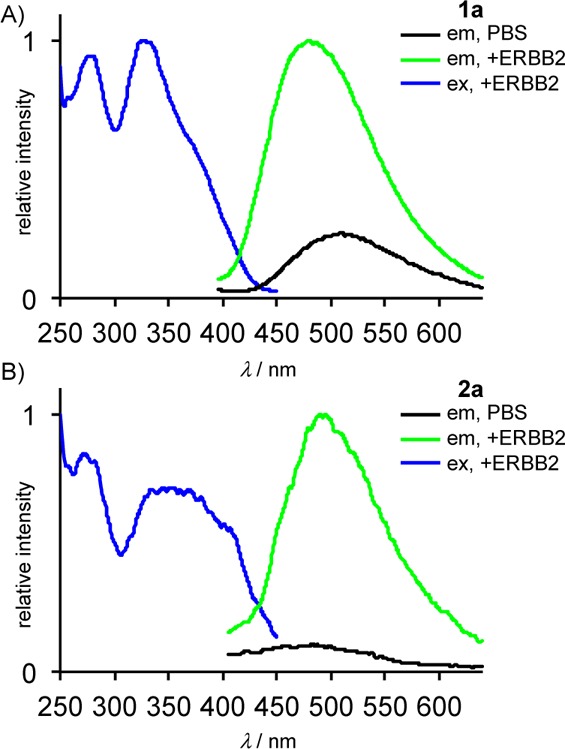
(A) 1a and (B) 2a exhibit turn-on emission upon binding to soluble ERBB2 kinase domain. In PBS alone, emission (in black) is largely quenched, whereas in the presence of ERBB2 kinase, emission (in green) is significantly enhanced; conditions: [1a] = [2a] = 1 μM; [ERBB2] = 100 nM, λex = 370 nm.
Conclusions
We have synthesized a family fluorescent of quinazoline probes targeting the ATP-binding pocket of ERBB2 and evaluated the influence of extended conjugation as well as substituent effects through the 6-position of the quinazoline core. Our results demonstrate that the optical band gap can be manipulated by varying the conjugation length and auxochrome substitution. Depending on the auxochrome identity, the quinazoline core can function either as an electron acceptor or an electron donor to achieve polar CT excited states. The strongest electron-donating and -withdrawing groups (e.g., dimethylamine, cyano, and nitro) yield high on/off ratios, suggesting that they are the best candidates for designing future turn-on probes. Importantly, the presence of a fluorophore arm at the 6-position of the quinazoline does not significantly attenuate the ability of the probes to function as inhibitors of ERBB2. Two probes, 1a and 2a, were successfully demonstrated as turn-on probes that can report binding to the ERBB2 kinase domain in solution. One limitation of the current family of fluorescent inhibitors is their relatively low solubility in aqueous solutions, although this is not unique to their modified structure, as lapatinib shows significant aggregation at physiologically relevant concentrations.24 Our future work will focus on improving the solubility of the probes through chemical modification at either the 7-position of the core (Figure 8) or on the fluorophore arm.
Figure 8.

R1, R2, and R3 are possible sites for chemical modification of 1a and 2a to enhance aqueous solubility.
Experimental Section
General Methods
Reagents and solvents were obtained from commercial suppliers and used without further purification. 1H and 13C NMR spectra were recorded on a 500 MHz spectrometer. Absorbance spectra were obtained using probe concentrations of 10 μM. Fluorescence studies were performed using probe concentrations of 1 μM. For determination of ϕem, solutions were prepared to an optical density of 0.05 or less in order to minimize inner filter effects. Perylene in cyclohexane was used as a reference for quantum yields.25
Computational Methods
Quantum chemical calculations were carried out utilizing the Gaussian ′09 suite of electronic structure modeling software.26 Ground-state geometries of the dyes were optimized by DFT with the B3LYP/6-31G(d) method using Truhlar’s SMD solvation model.16 Vertical transition energies were obtained by TD-DFT calculations with the B3LYP/6-31G(d) with the SMD model. Molecular orbitals were visualized using the GaussView 5 program. The coordinates of optimized geometries are provided in the Supporting Information.
Inhibition of Ligand-Induced Receptor Activation
MCF cells were seeded with equal quantity (200 000/well) in six-well plates. After 48 h, cells were pretreated with small molecule inhibitors of various concentrations for 1 h before induction by neuregulin (NRGβ1, 20 nM, 15 min). Cell lysates were generated immediately by SDS lysis. Equal aliquots were subjected to SDS-PAGE and western blot analysis. ERBB2 phosphorylation was evaluated for Tyr1239 located close to the extreme cytoplasmic C-terminus of the receptor (validated by pan-TyrP detection (4G10)). The signal obtained for pTyr1239 relative to the ERBB2 receptor levels was determined as the relative receptor phosphorylation.
Representative Synthesis via Route A: 6-(4-(Dimethylamino)phenyl)-N-phenylquinazolin-4-amine (1a)
Three hundred forty seven milligrams (1.0 mmol) of 6-iodo-N-phenyl-4-quinazolin-amine (4), 248 mg (1.5 mmol) of boronic acid, 425 mg of CsCO3 (1.3 mmol), and 3 mL of DMF were placed in a 25 mL Schlenk flask with a stirrer bar under argon purge. The reaction mixture was degassed for a further 20 min under a slow stream of argon, at which point 25 mg of a 1:2 mixture of PdCl2(PPh3)2 and PPh3 was added. The reaction was heated at 80 °C for 24 h, cooled, poured into 100 mL of H2O, and extracted with EtOAc (3 × 50 mL). The organic layer was concentrated and purified over silica (100% CH2Cl2 to 100% EtOAc) followed by crystallization from 2-propanol/EtOAc to afford 1a (47 mg, 14%); mp 257–259 °C (dec); IR νmax (cm–1): 2957.1, 1934.3, 1600.9, 1568.8, 1523.8, 1499.1, 1446.5, 1403.8, 1357.4, 1327.4, 1290.7, 1228.1, 810.1, 744.4, 692.5; 1H NMR (500 MHz, DMSO-d6): δ 2.98 (s, 6H), 6.87 (d, 2H, J = 8.9 Hz), 7.15 (t, 1H, J = 7.4 Hz), 7.41 (t, 2H, J = 8.3 Hz), 7.78 (t, 3H 8.4 Hz), 7.86 (d, 2H, J = 8.4 Hz), 8.14 (dd, 1H, J = 8.7, 1.8 Hz), 8.53 (s, 1H), 8.73 (s, 1H), 9.88 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 40.4, 112.9, 116, 118.6, 123, 124.1, 126.8, 128, 128.6, 128.9, 131.4, 138.7, 139.6, 148.7, 150.5,.154.2, 158.1; HR-ESI (Q-TOF) m/z: calcd for C22H21N4+ [M + H]+, 341.1766; found, 341.1770.
Representative Synthesis via Route B: (E)-6-(4-Methoxystyryl)-N-phenylquinazolin-4-amine (2b)
Five hundred milligrams (1.44 mmol) of 6-iodo-N-phenyl-4-quinazolin-amine (4), 289 mg (2.2 mmol) of 4-vinylanisole, 0.38 mL of Et3N (2.87 mmol), and 5 mL of DMF were placed in a 50 mL Schlenk flask with a stirrer bar under argon purge. The reaction mixture was degassed for a further 20 min under a slow stream of argon, at which point 48 mg of Pd(OAc)2 and 56 mg of PPh3 and the subsequent mixture was further degassed and heated at 80–85 °C for 24 h, cooled, poured into 100 mL of H2O, and extracted with EtOAc (3 × 50 mL). The organic layer was concentrated and purified over silica (100% CH2Cl2 to 100% EtOAc) followed by crystallization from 2-propanol to afford 2b (111 mg, 22%); mp: 238–241 °C (dec); IR νmax (cm–1): 3034.4, 1600.9, 1568.2, 1524.6, 1512.3, 1495.0, 1445.8, 1409.0, 1358.4, 1272.8, 1250.3, 1174.0, 837.0, 751.3, 688.5; 1H NMR (500 MHz, DMSO-d6): δ 3.80 (s, 3H), 7.00 (d, 2H, J = 8.7 Hz), 7.15 (t, 1H, J = 7.3 Hz), 7.24 (d, 1H, J = 16.4 Hz), 7.40–7.45 (m, 3H), 7.60 (d, 2H, J = 8.6 Hz), 7.76 (d, 1H, J = 8.6 Hz), 7.86 (d, 2H, J = 7.6 Hz), 8.10 (d, 1H, J = 8.0 Hz), 8.55 (s, 1H), 8.67 (s, 1H), 9.79 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 55.6, 114.8, 115.9, 120.2, 122.8, 124.2, 125.7, 128.3, 128.5, 128.9, 129.9, 130.1, 131.6, 136.0, 139.6, 149.6, 154.5, 158.0, 159.7; HR-ESI (Q-TOF) m/z: calcd for C23H21N3O+ [M + H]+, 354.1606; found,: 354.1621.
6-(4-Methoxyphenyl)-N-phenylquinazolin-4-amine (1b)
Yield: 52 mg, 16%; mp 245–247 °C; IR νmax (cm–1): 2999.8, 2835.4, 1937.5, 1600.6, 1570.2, 1522.6, 1496.5, 1445.4, 1400.9, 1358.3, 1270.5, 1247.2, 828.7, 818.3 753.3; 1H NMR (500 MHz, DMSO-d6): δ 3.83 (s, 3H), 7.11 (d, 1H, J = 8.7 Hz), 7.15 (t, 2H, J = 7.4 Hz), 7.42 (t, 2H, J = 8.1 Hz), 7.81–7.86 (m, 5H), 8.15 (dd, 1H, J = 8.7, 1.5 Hz), 8.57 (s, 1H), 8.79 (s, 1H), 9.93 (s, 1H); 13C NMR (125 MHz, DMSO-d6, TFA) δ 55.7, 114.6, 115.0, 120.8, 122.1, 124.9, 126.8, 128.8, 129.2, 130.7, 134.3, 137.4, 139.7, 140.1, 151.7, 159.8, 160.2; HRMS HR-ESI (Q-TOF) m/z: calcd for C21H18N3O+ [M + H]+, 328.1449; found,: 328.1453.
N,6-Diphenylquinazolin-4-amine (1c)
Yield: 41 mg, 14%; mp 258–262 °C; IR νmax (cm–1): 3067.3, 1939.0, 1600.5 1567.8, 1528.9, 1488.6, 1445.3, 1405.6, 1359.8, 1325.9, 1295.0, 840.7, 748.5, 689.2; 1H NMR (500 MHz, DMSO-d6): δ 7.16 (t, 1H, J = 7.3 Hz), 7.40–7.46 (m, 3H), 7.56 (t, 2H, J = 7.6 Hz), 7.85–7.91 (m, 5H), 8.20 (d, 1H, J = 8.7 Hz), 8.59 (s, 1H), 8.86 (s, 1H), 9.96 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 115.8, 120.9, 123.1, 124.3, 127.6, 128.4, 128.8, 128.9, 129.5, 132.2, 138.4, 139.5, 139.6, 149.5, 155.0, 158.3; HR-ESI (Q-TOF) m/z: calcd for C C20H16N3+ [M + H]+, 298.1344; found, 298.1352.
4-(4-Phenylamino)quinazolin-6-yl)benzonitrile (1d)
Yield: 48 mg, 15%; mp 242–245 °C; IR νmax (cm–1): 3107.2, 2228.4, 1624.1, 1596.7, 1571.4, 1527.8, 1496.9, 1443.8, 1403.4, 1326.6, 1255.8, 1106.2, 829.1, 747.3, 690.4; 1H NMR (500 MHz, DMSO-d6): δ 7.31 (t, 1H, J = 7.4 Hz), 7.57 (t, 2H, J = 8.3 Hz), 7.98 (d, 2H, J = 7.8 Hz), 8.02 (d, 1H, J = 8.7 Hz), 8.19 (d, 2H, J = 8.5 Hz), 8.26 (d, 2H, J = 8.4 Hz), 8.41 (dd, 1H, J = 8.7, 1.9 Hz), 8.75 (s, 1H), 9.09 (s, 1H), 10.15 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 110.8, 115.8, 118.4, 119.2, 121.9, 123.2, 124.5, 128.3, 129.0, 132.0, 133.3, 136.3, 139.3, 144.0, 150.2, 155.5, 158.4; HR-ESI (Q-TOF) m/z: calcd for C21H15N4+ [M + H]+, 323.1296; found, 323.1305.
6-(4-Nitrophenyl)-N-phenylquinazolin-4-amine (1e)
Yield: 55 mg, 16%; mp 262–264 °C; IR νmax (cm–1): 3089.6, 1934.3, 1597.5, 1569.0, 1531.1, 1514.6, 1488.0, 1449.5, 1401.4, 1417.5, 1341.8, 1271.0, 834.7, 752.1, 693.2; 1H NMR (500 MHz, DMSO-d6): δ 7.32 (t, 1H, J = 7.4 Hz), 7.57 (t, 2H, J = 8.1 Hz), 7.98 (d, 2H, J = 7.6 Hz), 8.05 (d, 1H, J = 8.6 Hz), 8.33 (d, 2H, J = 8.8 Hz), 8.44 (dd, 1H, J = 8.7, 1.7 Hz), 8.55 (d, 2H, J = 8.8 Hz), 8.76 (s, 1H), 9.14 (d, 1H, J = 1.3 Hz), 10.20 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 115.8, 122.3, 123.2, 124.5, 128.6, 128.9, 129.1, 132.1, 135.8, 139.3, 145.9, 147.3, 150.3, 155.7, 158.5; HR-ESI (Q-TOF) m/z: calcd for C20H15N4O2+ [M + H]+, 343.1195; found, 343.1194.
(E)-6-(4-Dimethylaminostyryl)-N-phenylquinazolin-4-amine (2a)
Yield: 130 mg, 25%; mp 220 °C (dec); IR νmax (cm–1): 3206.0, 2940.8, 1646.0, 1599.4, 1564.6, 1521.6, 1493.6, 1443.9, 1403.9, 1382.3, 1363.2, 1254.4, 957.2, 752.1, 691.3; 1H NMR (400 MHz, DMSO-d6): δ 2.94 (s, 6H), 6.76 (d, 2H, J = 8.7 Hz), 7.10–7.14 (m, 2H), 7.34–7.42 (m, 3H), 7.48 (d, 2H, J = 8.7 Hz), 7.73 (d, 1H, J = 8.7 Hz), 7.87 (d, 2H, J = 7.8 Hz), 8.07 (d, 1H, J = 8.7 Hz), 8.52 (s, 1H), 8.60 (s, 1H), 9.78 (s, 1H); 13C NMR (125 MHz, DMSO): δ 112.3, 115.5, 119.1, 122.4, 123.8, 124.6, 127.7, 128.0, 128.5, 130.5, 131.1, 136.2, 139.3, 148.7, 150.3, 153.8, 157.6; HR-ESI (Q-TOF) m/z: calcd for C24H23N4+ [M + H]+, 367.1923; found, 367.1929.
(E)-N-Phenyl-6-styrylquinazolin-4-amine (2c)
Yield: 74 mg, 16%; mp 248–252 °C (dec); IR νmax (cm–1): 3048.8, 1949.9, 1622.7, 1600.8, 1570.5, 1523.4, 1494.9, 1446.5, 1409.5, 1385.9, 1360.5, 1332.6, 1254.5, 827.7, 747.8, 687.8; 1H NMR (500 MHz, DMSO-d6): δ 7.16 (t, 1H, J = 7.2 Hz), 7.32 (t, 1H, J = 7.2 Hz), 7.38–7.51 (m, 6H), 7.66 (d, 2H, J = 7.4 Hz), 7.78 (d, 1H, J = 8.6 Hz), 7.87 (d, 2H, J = 7.6 Hz), 8.14 (d, 1H, J = 8.3 Hz), 8.56 (s, 1H), 8.72 (s, 1H), 9.81 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 115.8, 120.8, 122.9, 124.2, 127.0, 128.0, 128.4, 128.6, 128.9, 129.3, 130.3, 131.7, 135.6, 137.2, 139.6, 149.8, 154.8, 158.1; HR-ESI (Q-TOF) m/z: calcd for C22H18N3+ [M + H]+, 324.1500; found, 324.1516.
4-((E-2-(4-(Phenylamino)quinazoline-6-yl)vinyl)benzonitrile (2d)
Yield: 98 mg, 20%; mp 250–254 °C; IR νmax (cm–1): 3071.4, 2221.6, 1599.8, 1571.8, 1498.4, 1441.7, 1403.6, 1362.3, 964.8, 838.4, 752.4, 693.8; 1H NMR (500 MHz, DMSO-d6): δ 7.15 (t, 1H, J = 7.4 Hz), 7.42 (t, 2 H, J = 8.3 Hz), 7.56 (d, 1H, J = 16.4 Hz), 7.59 (d, 1H, J = 16.4 Hz), 7.79–7. 89 (br m, 7H), 8.17 (dd, 1H, J = 8.7, 1.7 Hz), 8.57 (s, 1H), 8.76 (s, 1H), 9.88 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 110.2, 115.8, 119.3, 121.8, 122.9, 124.2, 127.5, 128.5, 129.1, 129.2, 131.9 133.1, 134.7, 139.5, 141.9, 150.2, 155.1, 158.2; HR-ESI (Q-TOF) m/z: calcd for C23H17N4+ [M + H]+, 349.1453; found, 349.1453.
(E)-6-(4-Nitrostyryl)-N-phenylquinazolin-4-amine (2e)
Yield: 128 mg, 24%; mp 240 °C (dec); IR νmax(cm–1): 3085.1, 1932.2, 1602.1, 1591.5, 1571.4, 1527.7, 1501.7, 1328.8, 1404.1, 1443.2, 1365.5, 1255.4, 1105.2, 968.0, 749.5, 684.1, 694.9; 1H NMR (500 MHz, DMSO-d6): δ 7.30 (t, 1H, J = 7.3 Hz), 7.56 (t, 2H, J = 7.8 Hz), 7.72 (d, 1H, J = 16.5 Hz), 7.76 (d, 1H, J = 16.5 Hz), 7.93 (d, 1H, J = 8.6 Hz), 8.01 (t, 4H, J = 7.8 Hz), 8.30, (d, 1H, J = 8.6 Hz), 8.40 (d, 2H, J = 8.4 Hz), 8.71 (s, 1H), 8.91(s, 1H), 9.98 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 115.8, 122.1, 122.9, 124.3, 124.6, 127.8, 128.1, 128.7, 128.9, 131.9, 132.7, 134.7, 139.4, 144.1, 146.8, 150.3, 155.2, 158.2; HR-ESI (Q-TOF) m/z: calcd for C22H17N4O2+ [M + H]+, 369.1351; found, 369.1346.
6-(1E,3E)-(4-(4-Dimethylaminophenyl)buta-1,3-dienyl)-N-phenylquinazolin-4-amine (3a)
Yield: 90 mg, 16%; mp 255–258 °C (dec); IR νmax (cm–1): 2980.6, 1596.3, 1570.0, 1521.7, 1496.4, 1445.6, 1409.3, 1385.4, 1354.0, 1186.4, 987.4, 801.0, 746.0, 689.8; 1H NMR (500 MHz, DMSO-d6): δ 2.94 (s, 6H), 6.71 (d, 2H, J = 8.7 Hz), 6.77 (d, 2H, J = 15.6 Hz), 6.95 (dd, 1H, J = 15.2, 11.7 Hz), 7.15 (t, 1H, J = 7.0 Hz), 7.27 (dd, 1H, J = 15.0, 11.1 Hz), 7.38–7.43 (m, 4H), 7.72 (d, 1H, J = 8.4 Hz), 7.87 (d, 2H, J = 7.3 Hz), 8.00 (d, 1H, J = 8.3 Hz), 8.54 (d, 2H, J = 15.6 Hz), 9.8 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 112.6, 115.9, 119.9, 122.9, 124.2, 125.0, 125.2, 128.1, 128.5, 128.9, 129.2, 131.2, 132.2, 134.7, 136.2, 139.6, 149.5, 150.5, 154.4, 158.0; HR-ESI (Q-TOF) m/z: calcd for C26H25N4+ [M + H]+, 393.2079; found, 393.2088.
6-(1E,3E)-4-(4-(Methoxyphenyl)buta-1,3-dienyl)-N-phenylquinazolin-4-amine (3b)
Yield: 70 mg, 13%; mp 245–248 °C (dec); IR νmax (cm–1): 3029.9, 1939.7, 1599.1, 1568.7 1525.3, 1498.5, 1446.4, 1410.1, 1385.1, 1357.7, 1300.1, 1248.8, 988.5, 806.7, 740.2, 690.7; 1H NMR (500 MHz, DMSO-d6): δ 3.84 (s, 3H), 6.77 (d, 1H, J = 15.5 Hz), 6.85 (d, 1H, J = 15.5 Hz), 6.95 (d, 2H, J = 8.6 Hz), 7.07 (dd, 1H, J = 15.4, 10.7 Hz), 7.15 (t, 1H, J = 7.3 Hz), 7.29 (dd, 1H, J = 15.5, 10.7 Hz), 7.42 (t, 2H, J = 7.9 Hz), 7.51 (d, 2H, J = 8.5 Hz), 7.73 (d, 1H, J = 8.6 Hz), 7.86 (d, 2H, J = 7.7 Hz), 8.04 (d, 1H, J = 8.6 Hz), 8.54 (s, 1H), 8.59 (s, 1H), 9.84 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 55.6, 114.7, 115.9, 120.4, 122.9, 124.2, 127.5, 128.3, 128.6, 128.9, 130.0, 130.8, 131.3, 131.7, 133.7, 135.9, 139.6, 149.6, 154.6, 158.0, 159.6; HR-ESI (Q-TOF) m/z: calcd for C25H22N3O+ [M + H]+, 380.1763; found, 380.1766.
N-Phenyl-6-((1E,3E)-4-phenylbuta-1,3-dienyl)quinazolin-4-amine (3c)
Yield: 70 mg, 14%; mp 258–260 °C (dec); IR νmax (cm–1): 3259.1, 3055.1, 1924.2, 1604.2, 1569.2, 1528.3, 1496.2, 1440.3, 1406.5, 1388.2, 1363.4, 1329.5, 1310.3, 1253.8, 978.0, 901.4, 750.3, 686.5; 1H NMR (500 MHz, DMSO-d6): δ 6.81 (d, 1H, J = 15.2 Hz), 6.91 (d, 1H, J = 15.2 Hz), 7.11 (t, 1H, J = 7.2 Hz), 7.19–7.32 (m, 3H), 7.38 (d, 2H, J = 7.4 Hz), 7.39 (d, 2H, J = 7.4 Hz), 7.56 (d, 2H, J = 7.6 Hz), 7.68 (d, 1H, J = 8.5 Hz), 7.80 (d, 2H, J = 7.6 Hz), 8.01 (d, 1H, J = 8.5 Hz), 8.47 (s, 1H), 8.58 (s, 1H), 9.84 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 116.1, 121.1, 123.0, 123.8, 126.9, 128.2, 128.4, 128.8, 129.2, 129.8, 131.0, 132.3, 133.7, 135.3, 137.4, 140.7, 149.9, 154.9, 158.1; HR-ESI (Q-TOF) m/z: calcd for C24H20N3+ [M + H]+, 350.1657; found, 350.1673.
4-((1E,3E-4-(4-(Phenylamino)quinazolin-6-yl)buta-1,3-dienyl)benzonitrile (3d)
Yield: 134 mg, 25%; mp 242–245 °C (dec); IR νmax (cm–1): 3365.8, 3028.7, 2224.5, 1598.1, 1568.6, 1525.6, 1496.8, 1443.5, 1402.4, 1387.6, 1361.7, 1252.8, 983.0, 746.2, 848.0, 687.4; 1H NMR (500 MHz, DMSO-d6): δ 7.02 (d, 1H, J = 15.2 Hz), 7.16 (d, 1H, J = 15.1 Hz), 7.30 (t, 1H, J = 7.4 Hz), 7.46–7.60 (m, 4H), 7.90 (d, 3H, J = 8.5 Hz), 7.96 (d, 2H, J = 8.35 Hz), 8.00 (d, 2H, J = 7.65 Hz), 8.22 (dd, 1H, J = 8.7, 1.0 Hz), 8.69 (s, 1H), 8.79 (s, 1H), 9.98 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 109.9, 115.8, 119.4, 121.4, 122.9, 124.2, 127.5, 128.7, 128.9, 130.6, 131.3, 131.8, 133.0, 133.5, 134.4, 135.2, 139.5, 142.1, 150.1, 154.9, 158.1; HR-ESI (Q-TOF) m/z: calcd for C25H19N4+ [M + H]+, 375.1609; found, 375.1610.
6-((1E,3E)-4-(4-Nitrophenyl)buta-1,3-dienyl)-N-phenylquinazolin-4-amine (3e)
Yield: 113 mg, 20%; mp 273–275 °C (dec); IR νmax (cm–1): 3421.3, 3020.7, 1927.9, 1603.3, 1585.6, 1571.5, 1557.7, 1535.3, 1494.4, 1444.3, 1331.5, 1253.8, 1109.3, 987.1, 743.6, 804.3; 1H NMR (500 MHz, DMSO-d6): δ 6.92 (d, 1H, J = 15.4 Hz), 7.03 (d, 1H, J = 15.3 Hz), 7.13 (t, 1H, J = 7.2 Hz), 7.32–7.41 (m, 3H), 7.48 (dd, 1H, J = 15.4, 10.7 Hz), 7.73 (d, 1H, J = 8.6 Hz), 7.81 (d, 2H, J = 8.8 Hz), 7.83 (d, 2H, J = 8.8 Hz), 8.08 (d, 1H, J = 8.6 Hz), 8.20 (d, 2H, J = 8.6 Hz), 8.53 (s, 1H), 8.64 (s, 1H), 9.86 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 115.8, 121.5, 123.0, 124.3, 124.4, 127.6, 128.7, 128.9, 130.6, 131.4, 132.0, 134.5, 135.1, 135.2, 139.5, 144.2, 146.6, 150.1, 155.0, 158.1; HR-ESI (Q-TOF) m/z: calcd for C24H19N4O2+ [M + H]+, 395.1508; found, 395.1503.
Acknowledgments
This work was supported by the National Cancer Institute Innovative Molecular Analysis Technologies Program, CA182341-01 (J.N.W. and R.L.). J.N.W. also acknowledges the support of the Bankhead-Coley New Investigator Research Program, 3BN08. R.L. also acknowledges the support of National Cancer Institute, CA98881-05, and the Braman Family Breast Cancer Institute.
Supporting Information Available
1H and 13C NMR spectra, emission spectra in toluene and acetonitrile, frontier molecular orbitals, and Cartesian coordinates of optimized geometries. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Nat. Biotechnol. 2011, 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Nat. Biotechnol. 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Cortot A. B.; Repellin C. E.; Shimamura T.; Capelletti M.; Zejnullahu K.; Ercan D.; Christensen J. G.; Wong K. K.; Gray N. S.; Janne P. A. Cancer Res. 2013, 73, 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware K. E.; Marshall M. E.; Heasley L. R.; Marek L.; Hinz T. K.; Hercule P.; Helfrich B. A.; Doebele R. C.; Heasley L. E. PLoS One 2010, 5, e14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard R.; Dhuguru J.; Liu W.; Patel N.; Landgraf R.; Wilson J. N. Bioorg. Med. Chem. Lett. 2012, 22, 5532–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rewcastle G. W.; Denny W. A.; Bridges A. J.; Zhou H.; Cody D. R.; McMichael A.; Fry D. W. J. Med. Chem. 1995, 38, 3482–3487. [DOI] [PubMed] [Google Scholar]
- Bridges A. J.; Zhou H.; Cody D. R.; Rewcastle G. W.; McMichael A.; Showalter H. D.; Fry D. W.; Kraker A. J.; Denny W. A. J. Med. Chem. 1996, 39, 267–276. [DOI] [PubMed] [Google Scholar]
- Yun C. H.; Boggon T. J.; Li Y.; Woo M. S.; Greulich H.; Meyerson M.; Eck M. J. Cancer Cell 2007, 11, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E. R.; Truesdale A. T.; McDonald O. B.; Yuan D.; Hassell A.; Dickerson S. H.; Ellis B.; Pennisi C.; Horne E.; Lackey K.; Alligood K. J.; Rusnak D. W.; Gilmer T. M.; Shewchuk L. Cancer Res. 2004, 64, 6652–6659. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. Nat. Chem. Biol. 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Krueger A. T.; Lu H.; Lee A. H. F.; Kool E. T. Acc. Chem. Res. 2006, 40, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins M. E.; Pfleiderer W.; Jungmann O.; Balis F. M. Anal. Biochem. 2001, 298, 231–240. [DOI] [PubMed] [Google Scholar]
- Sinkeldam R. W.; Greco N. J.; Tor Y. Chem. Rev. 2010, 110, 2579–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secrist J. A. III; Barrio J. R.; Leonard N. J.; Villar-Palasi C.; Gilman A. G. Science 1972, 177, 279–280. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. J. Biol. Chem. 2002, 277, 46265–46275. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- Zucchero A. J.; McGrier P. L.; Bunz U. H. F. Acc. Chem. Res. 2010, 43, 397–408. [DOI] [PubMed] [Google Scholar]
- Reichardt C. Angew. Chem., Int. Ed. Engl. 1979, 18, 98–110. [Google Scholar]
- Pitter D. R.; Wigenius J.; Brown A. S.; Baker J. D.; Westerlund F.; Wilson J. N. Org. Lett. 2013, 15, 1330–1333. [DOI] [PubMed] [Google Scholar]
- Danielsson L.-G.; Zhang Y.-H. Trends Anal. Chem. 1996, 15, 188–196. [Google Scholar]
- Ban H. S.; Tanaka Y.; Nabeyama W.; Hatori M.; Nakamura H. Bioorg. Med. Chem. 2010, 18, 870–879. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Kleiman L. B.; Maiwald T.; Conzelmann H.; Lauffenburger D. A.; Sorger P. K. Mol. Cell 2011, 43, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen S. C.; Doak A. K.; Wassam P.; Shoichet M. S.; Shoichet B. K. ACS Chem. Biol. 2012, 7, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlman I.Handbook of Fluorescence Spectra of Aromatic Molecules, 2nd ed.; Academic Press: New York, 1971. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian ′09, Revision A.1; Gaussian, Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.