

Abstract
Background
Amyotrophic lateral sclerosis (ALS) is relatively rare, yet the economic and social burden is substantial. Having accurate incidence and prevalence estimates would facilitate efficient allocation of healthcare resources.
Objective
To provide a comprehensive and critical review of the epidemiologic literature on ALS.
Methods
MEDLINE and EMBASE (1995–2011) databases of population-based studies on ALS incidence and prevalence reporting quantitative data were analyzed. Data extracted included study location and time, design and data sources, case ascertainment methods, and incidence and/or prevalence rates. Medians and inter-quartile ranges (IQRs) were calculated, and ALS case estimates derived using 2010 population estimates.
Results
In all, 37 articles met inclusion criteria. In Europe, the median (IQR) incidence rate (/100,000 population) was 2.08 (1.47–2.43), corresponding to an estimated 15,355 (10,852–17,938) cases. Median (IQR) prevalence (/100,000 population) was 5.40 (4.06–7.89), or 39,863 (29,971–58,244) prevalent cases.
Conclusions
Disparity in rates among ALS incidence and prevalence studies may be due to differences in study design or true variations in population demographics, such as age, and geography, including environmental factors and genetic predisposition. Additional large-scale studies that use standardized case ascertainment methods are needed to more accurately assess the true global burden of ALS.
Keywords: Amyotrophic lateral sclerosis, Case ascertainment, Demographics, Diagnostic criteria, Epidemiology, Incidence, Prevalence, Temporal- and age-related trends
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease (MND) characterized by degenerative changes to upper and lower motor neurons [1, 2]. Patients experience signs and symptoms of progressive muscle atrophy and weakness, increased fatigue, and problems with swallowing, which typically lead to respiratory failure and death [1, 2]. Progressive functional deficits lead to an overall loss of independence [3]. Median survival is 2 to 4 years from onset [4, 5]; only 5–10% of patients survive beyond 10 years [4].
Although ALS is relatively rare, the personal, societal, and economic burden is substantial [6, 7]. Therefore, understanding the size and age distribution of the ALS population will assist in evaluating the extent to which new drug therapies and clinical interventions may improve overall quality of life and increase life expectancy among affected patients. Studies of the epidemiology of ALS are numerous, and incidence and prevalence rates vary widely. It is important to understand the strengths and limitations of the literature and by doing so, attempt to account for the geographic and demographic diversity of the global ALS population. However, current global epidemiologic data are not available in the literature. The most recent systematic review, published in 2009, evaluated the incidence and prevalence of ALS in Canada [8]; the most recent worldwide review was published 5 years ago [9]. Seminal papers include those published by Chancellor and Warlow [10] and Román [11]. Our objective is to provide an updated and comprehensive review of the descriptive epidemiology of ALS, with an emphasis on incidence and prevalence of the disease.
Methods
MEDLINE (via PubMed) and EMBASE were searched for English-language articles published between January 1995 and November 2011. Only population-based, observational studies reporting quantitative data on ALS incidence and/or prevalence were selected for further review. Systematic reviews of observational studies describing the epidemiology of ALS were also considered if they were published in 2008 or later. Because the homogeneity of diagnostic criteria is essential for meaningful comparisons among different studies, studies conducted before the publication of the El Escorial criteria (ie, before 1995) were excluded from the search. Information from multiple publications on the same patient population was pooled to avoid double-counting, and the article reporting the most up-to-date information was used as the primary study for data extraction. Search results from EMBASE and MEDLINE were combined, and duplicate articles were removed.
Study-specific information extracted from each article included: study design (prospective or retrospective), site(s) or location(s) and time period(s), catchment area population size, data collection method, and data sources. After a thorough review of the relevant articles, 4 data collection methods were identified: (1) medical records review, (2) ALS registries, (3) survey, and (4) database. These mutually exclusive categories adequately accounted for the different data collection methods represented and left no uncertainty with regard to category assignment. The key distinction between a registry and a database is that the former is designed for a predetermined clinical or research purpose, and information about individual patients is collected in a systematic and comprehensive way [12]. By contrast, health-related databases are not designed for research purposes, although they can be used for research [12]. In this report, nearly every ALS registry used at least 1 health system database (e.g. hospital discharge data [HDD], death certificates) as a source of patient information. Importantly, for each collection method, any number of contributing data sources could have been used (as appropriate) and in this case, data sources were not mutually exclusive for data collection method categories. Examples of contributing data sources included hospital discharge records, death certificates, government records, as well as records from neurology departments and clinics, ALS centers, and rehabilitation facilities.
Patient-related information extracted included sample size, age (mean or median, range) and gender distributions, age of symptom onset and/or ALS diagnosis (mean or median), and mean duration between onset and diagnosis. Details regarding the ALS case ascertainment methods were summarized qualitatively. Important aspects of case ascertainment included diagnostic criteria, age thresholds, other documentation of clinical signs and symptoms, and whether other confirmatory testing and/or examinations were performed.
For each article, the following information for ALS incidence and prevalence was extracted: number of incident and prevalent cases identified, overall crude incidence and prevalence, and the denominator used to calculate these rates. For standardized and/or age- and gender-adjusted rates, the year and population used were recorded. For studies that evaluated incidence and prevalence among different strata of the population or study period, the age groups and time periods were examined and the results for each were also extracted.
Using the reported crude incidence and prevalence, we calculated the median and interquartile range (IQR) for incidence and prevalence in Europe, North America (i.e. United States and Canada), Asia, and the Pacific region. Using these statistics, we estimated the number of ALS cases in these regions by applying their respective population estimates for 2010 obtained online from the Population Division of the Department of Economic and Social Affairs at the United Nations (http://esa.un.org/unpd/wpp/unpp/panel_indicators.htm).
Results
In total, 2,219 articles were identified and 492 duplicate articles were removed, leaving 1,727 publications. After applying a priori inclusion and exclusion criteria, 1,572 additional articles were excluded. The most common reasons for article exclusion included: type of study (eg, animal studies, in vitro studies, clinical trials), type of article (eg, letters, conference proceedings, editorials), publication date before 1995, publication language other than English, and disease state other than ALS. We retrieved 155 articles and, of these, 118 were excluded for not meeting the inclusion/exclusion criteria after in-depth review (fig. 1). Thus, 37 articles were included in the review. The number of studies by region included 25 in Europe (11 in Italy), 5 in North America, 6 in Asia and the Pacific, and 1 in Uruguay, South America. A comprehensive summary of the key characteristics of the included studies by region is provided in supplementary table S1; studies are listed in chronological order according to the index year (i.e. first year data were collected).
Fig. 1.

Flow diagram. I/P = incident/prevalence.
There were comparable numbers of prospective (n = 17) [13–29] and retrospective (n = 20) studies [30–49]; study design appeared to be independent of time of study. Study durations ranged from 1 year [15] to 74 years [46]. There was a 34-year study [38] and 4 studies of 20 to 22 years [22, 26, 33, 41]. Most studies, however, were conducted using 5-year [14, 29, 30, 43, 47, 48] or 10-year intervals [16, 19, 20, 32, 39, 42, 44]. The most frequently used data collection method was medical records review (n = 16) [13, 19, 22, 31–34, 37–40, 42, 44–46, 48], followed by ALS registries (n = 11) [14, 16–18, 20, 21, 23, 27, 30, 35, 49], surveys (n = 5) [15, 17, 18, 21, 25, 27–30, 43, 45, 47], and databases (n = 5) [24, 26, 36, 41, 47]. Most of the studies that used the medical records review approach were retrospective (81%); by contrast, most of the studies using ALS registries were prospective (70%).
For each mode of data collection, and with few exceptions, multiple contributing data sources were used to identify ALS cases. Five studies used only 1 data source [13, 24, 34, 36, 46]; 4 studies used 8 or more sources [28, 44, 48, 49]. Most studies (68%) used between 2 and 6 different contributing data sources, and most of these were retrospective in design. The study by Logroscino et al. [49] is not included in many of the results presented below because the study design was sufficiently different from the others in that data were collected prospectively from 6 ALS registries in 4 European countries (i.e. England, Ireland, Italy, and Scotland).
Overall, the most commonly used data sources were HDD, MND associations, and neurology departments, each accounting for 12% of the total. Information collected from hospital and/or Emergency Department (ED) records, death certificates, and other clinics each accounted for 11% of the sources. The type and number of data sources differed depending on the data collection method. For example, for medical records reviews, information from hospital and ED records was the most commonly used source; for ALS registries, information from MND associations was most often used. In Logroscino et al. [49], each ALS registry used multiple sources of information to ensure complete case ascertainment; however, details regarding the nature of these sources were not provided.
The majority of included studies reported the mean or median age of disease onset, mean or median age at ALS diagnosis, and mean time interval between onset and diagnosis. Among studies reporting this data, the mean ± standard deviation (SD) age for ALS disease onset was 61.8 ± 3.8 years (range 54–67); mean ± SD age for ALS diagnosis was 64.4 ± 2.9 years (range 58–68). The mean ± SD diagnostic delay was 12.6 ± 2.6 months (range 8.6–16.8). In Logroscino et al. [49], the median (IQR) age at diagnosis was 65.2 (56.0, 72.2) years. The mean ± SD time from symptom onset to diagnosis was 371 ± 372 days (12.2 ± 12.2 months) [49].
Of the 37 included studies, 35 (95%) reported incidence rates. In Europe, crude incidence estimates ranged from 0.5/100,000 population in Belgrade, Yugoslavia (Serbia since 1991) [31] to 3.6/100,000 individuals in the Faroe Islands [22]. Incidence rates in Canada [15] and the United States [25, 46] were similar; rates in China [36, 37] were lower than those in Japan [29, 43] or New Zealand [26] (fig. 2). Table S2 shows incidence rates and 95% confidence intervals (CIs) for those studies reporting it. The study by Logroscino et al. [49] is not included in this figure (and other calculations); however, as mentioned above, the incidence rate was 2.7/100,000 person-years. In general, incidence rates were higher for the prospective (median [IQR], 2.39 [2.15, 2.68]) versus retrospective (median [IQR], 1.52 [1.22, 2.04]) studies.
Fig. 2.

Country-specific crude incidence rate of ALS. *Logroscino et al. [49] not included in this figure (latest year of data collection during study); †Rates are adjusted.
Twenty of the 37 (54%) included studies reported prevalence. In Europe, crude prevalence ranged from 1.1/100,000 population in Yugoslavia [31] to 8.2/100,000 individuals in the Faroe Islands [22], although values for the Netherlands [21] and 2 regions in Italy [16, 20] were also among the highest in Europe, at ~8.0/100,000 population each (fig. 3). Table S2 shows prevalence values and 95% CIs for those studies reporting it. In other regions, prevalence ranged from 1.0/100,000 in China [37] to 11.3/100,000 population in Japan [43]. In general, and excluding the Japanese study as an outlier [43], prevalence estimates from prospective studies were higher (median [IQR], 7.89 [6.25, 7.98]) than estimates from retrospective studies (median [IQR], 4.04 [3.92, 4.70]). For both incidence and prevalence, there did not appear to be a strong relationship between reported rates and the data collection method or number of contributing data sources.
Fig. 3.

Country-specific crude prevalence rate of ALS. Text inside bars represents point prevalence estimate date.
We used crude incidence and prevalence to calculate the medians and IQRs for all of the studies combined and for different regions, including Europe, North America (i.e. United States and Canada), and Asia. In fig. 4, summary statistics for incidence and prevalence are shown for all of the studies combined, the European studies, and the Italian studies, as well as by prospective or retrospective design. For all studies combined (n = 34), the median (IQR) incidence rate was 1.90 (1.37, 2.40); values were higher in the prospective (2.26 [1.80, 2.60]) versus retrospective (1.50 [0.92, 2.00]) studies. Owing to the large number of European studies (n = 24), the summary statistics for this region were comparable to those calculated for all included studies (fig. 4). The subset of Italian studies (n = 10) showed the least inter-study variability. For prevalence, the overall median (IQR) was 4.48 (3.03, 6.70) for 20 combined studies. As with incidence, values were also higher among the prospective studies (7.10 [5.88, 7.97]) versus retrospective studies (3.89 [2.58, 4.27]); however, the inter-study variability was larger (fig. 4), which is likely due, at least in part, to the higher prevalence in the study from Japan [43]. Indeed, much of this variability was removed in the subset of European studies (n = 13), and even more so in the Italian studies (n = 4).
Fig. 4.

Summary statistics for all studies and by region. *Lighter shaded areas indicate the lowest and highest reported rate; darker shaded area represent the inter-quartile (IQ) range.
Using these statistics, we estimated the number of ALS cases in these regions by applying their respective population estimates for 2010 as the multiplier. In Europe, the estimated number of incident cases was about 15,400; prevalent cases were about 40,000 (table 1). Case estimates were similar for Asia, where incident cases were estimated to be about 15,000 and prevalent cases totaled 45,000. By contrast, incident case estimates for the United States and Canada were lower (5,432 and 762, respectively), likely due to the small number of studies and smaller overall populations. Only 2 US studies evaluated prevalence, and the resulting prevalent cases estimate was about 10,500 (table 1).
Table 1.
ALS case estimates for incidence (I) and prevalence (P) in the total population using descriptive statistics from included studies by region
| EUROPE | Incidence (per 100,000) | Prevalence (per 100,000) | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Statistics based on search results | IQ-25 | Median | IQ-75 | IQ-25 | Median | IQ-75 |
|
|
|
|||||
| All European studies (n = 24-I/13-P) | 1.47 | 2.08 | 2.43 | 4.06 | 5.40 | 7.89 |
| Prospective (n = 12-I/7-P) | 2.15 | 2.39 | 2.68 | 6.25 | 7.89 | 7.98 |
| Retrospective (n = 12-I/6-P) | 1.22 | 1.52 | 2.04 | 3.92 | 4.04 | 4.70 |
| Estimated cases in Europe, n | ||||||
|
|
|
|||||
| All European studies (n = 24-I/13-P) | 10,852 | 15,355 | 17,938 | 29,971 | 39,863 | 58,244 |
| Prospective (n = 12-I/7-P) | 15,871 | 17,643 | 19,784 | 46,137 | 58,244 | 58,908 |
| Retrospective (n = 12-I/6-P) | 9,006 | 11,221 | 15,059 | 28,937 | 29,823 | 34,695 |
| NORTH AMERICA | Incidence (per 100,000) | Prevalence (per 100,000) | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Statistics based on search results | IQ-25 | Median | IQ-75 | IQ-25 | Median | IQ-75 |
|
|
|
|||||
| All North American studies (n = 3-I/2-P) | 1.75 | 1.80 | 2.02 | – | – | – |
| US studies (n = 2-I/2-P) | 1.73 | 1.75 | 1.78 | 3.15 | 3.40 | 3.65 |
| Canadian study (n = 1-I/0-P) | – | 2.24 | – | – | – | – |
| Estimated cases in North America, n | ||||||
|
|
|
|||||
| All North American studies (n = 3-I/2-P) | 6,027 | 6,199 | 6,957 | – | – | – |
| US studies (n=2-I/2-P) | 5,370 | 5,432 | 5,525 | 9,777 | 10,553 | 11,329 |
| Canadian study (n=1-I/0-P) | – | 762 | – | – | – | – |
| China and Japan | Incidence (per 100,000) | Prevalence (per 100,000) | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Statistics based on search results | IQ-25 | Median | IQ-75 | IQ-25 | Median | IQ-75 |
|
|
|
|||||
| All Asian studies (n = 5-I/4-P) | 0..40 | 0.60 | 1.43 | 1.44 | 2.34 | 5.13 |
| Chinese studies (n = 2-I/2-P) | 0.38 | 0.46 | 0.53 | 1.48 | 2.01 | 2.54 |
| Japanese studies (n = 2-I/1-P) | 1.70 | 1.97 | 2.23 | – | 11.3 | – |
| Estimated cases in Asia, n | ||||||
|
|
|
|||||
| All Asian studies (n = 5-I/4-P) | 6,167 | 9,251 | 22,048 | 22,203 | 36,079 | 79,097 |
| Chinese studies (n = 2-I/2-P) | 5,097 | 6,170 | 7,109 | 19,852 | 26,961 | 34,070 |
| Japanese studies (n = 2-I/1-P) | 2,151 | 2,493 | 2,822 | 14,299 | ||
Total European population (2010): 738,199,000*
Total US and Canada population (2010): 344,401,000; US population (2010): 310,384,000; Canada population (2010): 34,017,000*
Total China, Japan, and Iran population (2010): 1,541,845,000; China population (2010): 1,341,335,000; Japan population (2010): 126,536,000; Iran population (2010): 73,974,000*
Twenty-one of the 35 studies that evaluated incidence also described changes in incidence during the course of the study. Study durations ranged from 3 years [27] to 74 years [46]. As the duration of the study increased, the number and size of the time intervals varied substantially. In most of the longer-term studies, the entire study duration was divided into 2 periods covering longer intervals. Overall, 12 of 21 (57%) studies reported stable incidence rates over time [14, 16, 19, 22, 27, 32, 34, 35, 39, 41, 44, 46]; 5 reported increases in rates [20, 26, 33, 38, 40], and 4 reported highly variable rates over time [13, 24, 29, 42]. Two studies evaluated changes in incidence over time but compared an updated 1-year [15] or 5-year analysis [43] with previous publications excluded from the review (as older, related studies).
When incidence and prevalence were plotted as a function of the study duration (range 1–22 years), a clear trend was not apparent (fig. 5, top panel). Two temporal outliers, a 34-year study [38] and a 74-year study [46], were not included in fig. 5; however, incidence rates (1.6 and 1.7/100,000 population, respectively) were comparable with other charted values. Overall, the inter-study variation was notably smaller for incidence than for prevalence. When incidence and prevalence rates were plotted as a function of the latest year of data collection, there was a trend toward a slight increase in incidence and prevalence during the 20-year period between 1989 and 2009 (fig. 5, bottom panel).
Fig. 5.

Temporal trends in incidence and prevalence. Panel A does not include 2 study outliers: Govoni et al. (1964–1998; 34 years) [38] and Sorenson et al. (1925–1998; 74 years) [46].
Temporal trends were also evaluated by looking specifically at the relationship between study date(s) and publication of the El Escorial criteria (EEC) for the diagnosis of ALS, which were used by the majority of the studies to identify ALS cases from the data sources. The EEC were introduced in 1994 [50] and revised in 2000 [51]. For the 6 studies conducted before 1994, the median (IQR) incidence was 1.2 (0.6, 1.4); the corresponding value for prevalence was 2.5 (1.0, 4.3). For the 12 studies that spanned the publication dates, median (IQR) incidence was 2.0 (1.6, 2.5) and prevalence was 4.1 (4.0, 6.1). For the 11 studies conducted after the publication dates, the median (IQR) incidence was 2.2 (1.5, 2.5) and prevalence was 4.9 (3.1, 7.9). In the Logroscino et al. study [49], which was conducted after the first [50] but before the second EEC publication [51], the mean annual crude incidence rate was 2.7/100,000 person-years (95% CI: 2.8–3.3).
When incidence and prevalence were plotted as a function of the median age of the population, there was a trend toward increased incidence and prevalence with advancing age (fig. 6). Although Iran [45] was an outlier with a median age of 24 years, incidence (0.4/100,000) and prevalence (1.6/100,000) were consistent with the trend (fig. 6). Nevertheless, the inter-country variability in incidence and prevalence was still large for both the temporal and age-related comparisons. Finally, there did not appear to be a relationship between incidence and prevalence and the populations used for their calculations (fig. S1). Twenty-two studies used the total population [14, 16, 19, 20, 24, 26, 28, 31–40, 42–44, 46, 48], which included persons of any age. Fifteen studies used only the adult population, defined as patients aged ≥15 years [17, 18, 21, 27, 30, 45], ≥18 years [13, 15, 22, 23, 25, 41, 47, 49], or >20 years [29]. In these studies, the adult population was selected with the rationale that it better represented the population at risk for ALS. Among the 35 studies reporting incidence, 26 (74%) also reported incidence by age cohorts; however, the number of intervals and bin widths for the distributions were varied. Fig. 7 is a schematic representation of the age cohorts for which ALS incidence rates peaked during the study, showing the age ranges and the number of studies reporting for each range. For 77% of the studies, ALS incidence peaked between the ages of 60 and 75 years.
Fig. 6.
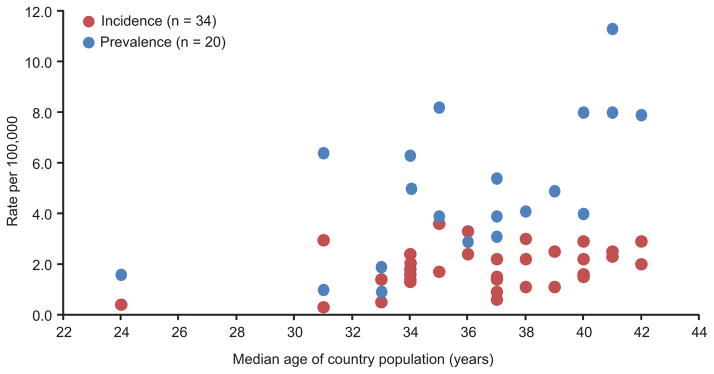
Age-related trends in incidence and prevalence.
Fig. 7.

Distribution of age cohorts evaluated for peaked ALS incidence rates. n = number of studies reporting for the cohort.
Incidence by geographic location (i.e. latitude) is presented in fig. S2 for Europe and Italy (top panel) and for the other countries in North America, South America (i.e. Uruguay), Asia, and the Pacific (i.e. New Zealand; bottom panel). With the exception of New Zealand, there appeared to be a trend for ALS incidence rates to increase in countries with higher latitudes.
Discussion
In this systematic review, we conducted a comprehensive search of the biomedical literature to identify observational studies on the epidemiology of ALS published between 1995 and 2011, particularly those reporting data for incidence and prevalence. More than 1,700 articles were identified; we evaluated 37 studies that met the a priori inclusion/exclusion criteria. Overall, reported incidence and prevalence varied widely. Some studies reported outliers in incidence rates. Low incidence rates reported for Iran [45] and China [36, 37] could be related to the retrospective design and the use of hospital records as the only source of data. By contrast, high incidence rate reported for the Faroe Islands [22] likely reflects the small population – as reflected by the large confidence interval – and the excellent identification of cases by a single investigator. The higher incidence rate in the Faroe Islands may also be due to a genetic founder effect based on the proximity of the islands to Scandinavia and the history of settlement by the Vikings.
As there were no exclusion criteria relating to country or continent, the variations in rates may be due to true demographic and/or geographic differences as well as other factors not previously considered. Under-ascertainment of cases is another possible explanation, largely related to different sources of data utilized in various studies. Most of the studies were conducted in Europe. Italy was well represented with 11 studies. Two studies were conducted in the Southern Hemisphere — 1 in New Zealand [26] and 1 in Uruguay [28]. The incidence rate in Iran [45], located in western Asia, was comparable with rates for China [36, 37], but not Japan [29, 43].
Prospective and retrospective study designs were comparably represented; incidence and prevalence were generally higher in prospective versus retrospective studies. Data collected prospectively are more accurate than are data collected retrospectively in terms of identification of new cases [52, 53]. Moreover, a prospective design allows for the continuous, long-term collection of data to characterize disease progression [13]. By definition, retrospective studies do not have time constraints; thus, numerous data sources can be accessed. The disadvantage is that most retrospective studies are limited by inadequate recorded information [44]. Nevertheless, while higher incidence rates can serve as a proxy for better case ascertainment (i.e. via prospective data collection), low incidence rates may reflect true population differences and are not always indicative of a poorly designed study.
A variety of contributing data sources were used, and most studies used multiple sources, primarily HDD, hospital and/or ED records, neurology departments, and death certificates. In an early systematic review, the types of sources used were far fewer, but the vast majority of ALS cases were identified from neurology departments, hospitals, and mortality data [54]. There did not appear to be a relationship between incidence and/or prevalence and the number of data sources used to identify ALS cases. By contrast, when Chancellor and Warlow [10] separated their studies according to the quality of the methodology used, crude incidence rates reflected the likelihood of complete case ascertainment. That is, with a few exceptions, rates were higher in studies that used multiple sources, particularly those that were computerized or based on national statistics. The authors suggested that the best incidence studies are those that use multiple data sources and/or systematic medical documentation systems [10].
Case ascertainment methods included delineating the catchment area, identifying the population of interest, and defining the ALS diagnostic criteria. If the methods are accurate, sensitivity and/or specificity will be increased. Some of the studies assessed incidence and prevalence for an extremely large catchment area (e.g. country level) [35, 37] or multiple catchment areas (e.g. European countries) [49]; other studies used smaller regions, such as the provinces of Italy [14, 32, 33, 44] and North American states [47] and counties [25, 46, 48]. Compared with the other studies, catchment areas in the 4 US-based studies were small. Both of the incidence studies [25, 46] and 1 prevalence study [48] were conducted at the county level. One prevalence study evaluated ALS cases at the state level (South Carolina) [47]. In general, smaller catchment areas correlate with better case ascertainment [53]; however, if the area is too small, there is a risk for under- or over-estimating incidence owing to the relative rarity of ALS. Overall, the use of different methodologies for case ascertainment, including retrospective versus prospective study design, fewer data sources, and smaller catchment area size, may produce under-ascertainment of cases with consequent lower incidence and prevalence rates.
Collectively, data from the included studies showed positive temporal trends with the latest year of data collection; however, no formal correlational analyses were performed. Most of the studies analyzed temporal changes in ALS incidence rates, and the results varied. Nearly 60% found ALS incidence rates to be stable over time; some studies showed increased rates and others showed highly variable rates during the course of the study. Across studies, there was a trend for a gradual increase in incidence and prevalence with the latest year of data collection. In another systematic review, Worms [55] reported a slight increase in ALS incidence from 1.3/100,000 patient-years in the 1960s–1970s to 1.9/100,000 patient-years in the 1990s, but also noted broad regional differences in temporal changes for both incidence and prevalence.
The majority of studies showed an increase in incidence with age. Some investigators suggest that the aging of the global population contributes to this finding [34, 38]; others attribute the age-associated increase in incidence rates to other factors [22, 35]. At least 1 study used direct methods to standardize the population to a uniform age distribution, which controls for the potential effects of aging [35]. Most of the included studies reported that ALS incidence rate peaked between the ages of 60 and 75 years, which is consistent with other systematic reviews [10, 11, 55]. In the Cima study [34], the annual incidence of ALS peaked later (ages 65–74 years) than it did during a previous study of the same area (ages 44–64 years). In a series of studies in Nova Scotia, Canada, conducted between 1984 and 2003, the age cohort for which ALS incidence rate peaked was the same in each study (i.e. 70–79 years); however, crude rates decreased from ~13.0/100,000 in 1984 to ~10.0/100,000 in 1995 to ~7.0/100,000 in 2003 [15].
Among the European studies included in this analysis, there appeared to be a trend for ALS incidence rates to increase in countries with higher latitudes. Although the number of studies is small, the finding is consistent with genetic evidence suggesting that the risk for ALS is highest in Scandinavia and follows a north-to-south gradient [56–58]. Immigration and migration were also cited as contributing factors to changing incidence rates [29, 33, 43]. In a follow-up study to Abhinav [30], Scott et al. [59] found geographic clustering of ALS in southeast England, with larger than expected numbers of cases in the Greater London area. It appears that the relative risk for ALS is higher in areas of higher population density [60]. In a study conducted 2 decades earlier, Leone et al. [61] reported that population density was also related to a non-random distribution of ALS cases in Turin, Italy.
Overall, our results with regard to age- and temporal-related trends in incidence and prevalence are consistent with the literature published during the past 50 years. Furthermore, as discussed previous systematic reviews [9–11, 55], it is likely that any true intercontinental and inter-country differences in incidence and prevalence are driven primarily by the different demographic compositions of the populations under study. To understand these differences, researchers are using genomic epidemiology with increasing frequency [62], and several genome-wide association studies (GWAS) of patients with ALS have been published. In isolated and genetically homogeneous populations such as the Finnish [63] and Sardinians [64], genetics plays a major role in the development of ALS.
Recent GWAS of Finnish patients with ALS identified a locus on chromosome 9p21 that accounted for more than 40% of familial ALS cases and nearly 25% of all ALS cases [58, 63]. Subsequent studies identified Finnish haplotype in other European populations [57, 58, 63] and determined that it arose from a single founder mutation [63]. The cause of the chromosome 9p21 mutation was identified as a large, hexanecleotide repeat expansion located within the non-coding portion of C9orf72 [65, 66]. In a follow-up study, the frequency of the expansion was evaluated in 17 regions worldwide, including countries in Europe, ethnic populations in the United States, and countries in Asia, Australia, the Middle East, and Pacific lslands [56]. Every patient with the expansion also had the Finnish founder haplotype, indicating that the mutation occurred in 1 individual and was disseminated throughout these populations [56]. The mutation frequency was highest in Finland (21%) and followed a north-to-south gradient. The estimated age of the mutation was about 1,500 years, corresponding to 100 generations in Finland, about 80 in Germany, and approximately 60 in Italy [56]. It is possible that genetics plays a part in other, more diverse populations that have yet to be investigated using GWAS.
Although almost every study analyzed incidence or prevalence by different geographic (e.g. country, province, state, county, city), demographic (e.g. age, gender), and clinical (e.g. site of onset, clinical type) characteristics, only a few studies reported rates for race and/or ethnicity. None of the European studies reported incidence or prevalence by race or ethnicity. Murphy et al. [26] briefly mentioned that the predominant ethnicity of ALS cases was representative of the Canterbury, New Zealand population. For North America, likely the most ethnically diverse country evaluated, only 2 studies mentioned ethnicity. In a study by Turabelidze et al. [48], all of the ALS cases identified in Jefferson county, Missouri (n = 36) were non-Hispanic whites. McGuire [25] reported age-adjusted incidence rates for nonwhite men (0.74/100,000/y [95% CI, 0.00–1.96]) and nonwhite women (0.53/100,000/y [95% CI 0.00–1.91]) in western Washington. These rates were notably lower than the age-adjusted rates among white men (2.1/100,000/year [95% CI 1.3–2.9]) and women (1.9/100,000/year [95% CI 1.1–2.7]) [25].
In a systematic review of the ALS literature between 1966 and 2006, 6 of 61 (10%) evaluable studies, mainly from the United States, evaluated incidence rates stratified by ethnicity [9]. In addition to the McGuire study [25], Annegers et al. [67] observed significantly lower MND incidence rates among those of Hispanic ethnicity compared with affected African American and non-Hispanic whites in Harris county, Texas. In 3 other US-based studies, MND mortality rates were significantly lower among nonwhite versus white populations [9]. ALS mortality rates appear to be lowest in highly ethnically “mixed” populations [68]. Thus, further studies, using sound methodologies, are warranted in countries with different ethnic populations, including Africa and South America.
Since the literature search was conducted in November 2011, a number of other studies on ALS epidemiology have been published, including a number of reviews on the epidemiology of long-term neurologic conditions in the United Kingdom [69], the utility of mortality data to estimate the incidence of ALS [70], and a paper focused on the heterogeneity of the disease and an appraisal of clinical trials [71]. Additional prospective, population-based studies have come out of Italy, including reports for ALS epidemiology in Liguria [72], Sardinia [73, 74], and Sicily [75]. Incidence rates (95% CI) ranged from 1.33/100,000 (not reported) [73] to 3.22/100,000 (2.66–3.90) [72], which are consistent with the rates published in the Italian reports included in this review. Kihira [76] published a 5-decade review of the changes in ALS incidence on the Kii Peninsula of Japan. Two studies were conducted in the United States [77, 78], and Rabkin et al. [79] compared the use of tracheostomy with invasive ventilation in the United States and Japan.
Despite the large and growing volume of literature on ALS epidemiology, additional large-scale, prospective, population-based studies using standardized diagnostic criteria are warranted. Given the increasing number of ALS registries across Europe, multi-country studies similar to that conducted by Logroscino and colleagues [49] are certainly feasible. Moreover, there are many European countries for which the epidemiology of ALS has not yet been investigated or are in need of updated estimates, including Spain, Germany, and Russia, to name a few. Outside Europe, there is a need for more studies in Asia, as those conducted to date involved relatively small catchment areas. Other important continents and countries include Africa, the Middle East, Australia, Greenland, and Iceland. In North America, the epidemiology of ALS in Canada is well described; however, for the United States, there have been only a few studies involving small catchment areas. A nationwide study is clearly warranted.
In 2008, the ALS Registry Act was passed into law, and in 2009 the Agency for Toxic Substance and Disease Registry (ATSDR) implemented the National ALS Registry using a 2-pronged approach: (1) use existing national administrative databases (e.g. Medicare, Medicaid) to identify prevalent cases, and (2) use a secure Web portal (www.cdc.gov/als) to capture ALS cases not included in the administrative databases [80]. At this Web site, ALS patients can self-identify and enroll in the ALS registry and participate in various surveys. ATSDR has other surveillance activities underway in a number of smaller geographic areas (i.e. state and metropolitan levels) to obtain timely, population-based estimates of ALS across the United States [80].
Conclusions
The body of literature on ALS epidemiology is large but limited geographically. Most research has been conducted in Europe. There is wide variability in incidence and prevalence, which is likely due, at least in part, to differences in study design. In some larger regions, including North America, there is a paucity of data from which to derive current case estimates. Additional epidemiologic studies that optimize case ascertainment and cover larger catchment areas will help improve our understanding of the global population burden of ALS. Clearly, population demographics contribute to the diversity in ALS epidemiology. Across the globe, the incidence of ALS increases with age, and it is likely that the overall incidence of ALS will increase as the world population ages. Overall, there are no clear indications of a change in the incidence of ALS over time, but long-term studies in the same population and using the same case ascertainment methodology are still lacking. Geographic differences and environmental risk factors have been the subject of study for decades, whereas recent advances in genomic epidemiology will continue to provide a better understanding of genetic risk and susceptibility for this devastating disease.
Supplementary Material
Acknowledgments
This report and medical writing assistance was funded by Biogen Idec. The research leading to these results has received funding from the European Community’s Health Seventh Framework Programme (FP7/2007–2013; grant agreements no. 259867 and 278611) (AC).
Footnotes
Disclosure Statement
A Chiò, G Logroscino, and B Traynor report no conflicts of interest. J Collins and J Simeone are employed by United BioSource Corporation (UBC), Epidemiology & Database Analytics. LA Goldstein is employed by UBC, Scientific Solutions. Leigh Ann White is employed by and owns stock in Biogen Idec.
References
- 1.Rowland LP. Amyotrophic lateral sclerosis. Curr Opin Neurol. 1994;7:310–315. doi: 10.1097/00019052-199408000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Sejvar JJ, Holman RC, Bresee JS, Kochanek KD, Schonberger LB. Amyotrophic lateral sclerosis mortality in the United States, 1979–2001. Neuroepidemiology. 2005;25:144–152. doi: 10.1159/000086679. [DOI] [PubMed] [Google Scholar]
- 3.Ng L, Talman P, Khan F. Motor neurone disease: disability profile and service needs in an Australian cohort. Int J Rehabil Res. 2011;34:151–159. doi: 10.1097/MRR.0b013e328344ae1f. [DOI] [PubMed] [Google Scholar]
- 4.Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–323. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.del Aguila MA, Longstreth WT, Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–819. doi: 10.1212/01.wnl.0000049472.47709.3b. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Bastida J, Perestelo-Perez L, Monton-Alvarez F, Serrano-Aguilar P, Alfonso-Sanchez JL. Social economic costs and health-related quality of life in patients with amyotrophic lateral sclerosis in Spain. Amyotroph Lateral Scler. 2009;10:237–243. doi: 10.1080/17482960802430781. [DOI] [PubMed] [Google Scholar]
- 7.Schepelmann K, Winter Y, Spottke AE, Claus D, Grothe C, Schroder R, Heuss D, Vielhaber S, Mylius V, Kiefer R, Schrank B, Oertel WH, Dodel R. Socioeconomic burden of amyotrophic lateral sclerosis, myasthenia gravis and facioscapulohumeral muscular dystrophy. Journal of neurology. 2010;257:15–23. doi: 10.1007/s00415-009-5256-6. [DOI] [PubMed] [Google Scholar]
- 8.Wolfson C, Kilborn S, Oskoui M, Genge A. Incidence and prevalence of amyotrophic lateral sclerosis in Canada: a systematic review of the literature. Neuroepidemiology. 2009;33:79–88. doi: 10.1159/000222089. [DOI] [PubMed] [Google Scholar]
- 9.Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007;68:1002–1007. doi: 10.1212/01.wnl.0000258551.96893.6f. [DOI] [PubMed] [Google Scholar]
- 10.Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality, incidence and distribution since 1950. Journal of neurology, neurosurgery, and psychiatry. 1992;55:1106–1115. doi: 10.1136/jnnp.55.12.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Román GC. Neuroepidemiology of amyotrophic lateral sclerosis: clues to aetiology and pathogenesis. Journal of neurology, neurosurgery, and psychiatry. 1996;61:131–137. doi: 10.1136/jnnp.61.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brooke E. The current and future use of registers in health information systems [Google Scholar]
- 13.Argyriou AA, Polychronopoulos P, Papapetropoulos S, Ellul J, Andriopoulos I, Katsoulas G, Salakou S, Chroni E. Clinical and epidemiological features of motor neuron disease in south-western Greece. Acta neurologica Scandinavica. 2005;111:108–113. doi: 10.1111/j.1600-0404.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 14.Beghi E, Millul A, Micheli A, Vitelli E, Logroscino G. Incidence of ALS in Lombardy, Italy. Neurology. 2007;68:141–145. doi: 10.1212/01.wnl.0000250339.14392.bb. [DOI] [PubMed] [Google Scholar]
- 15.Bonaparte JP, Grant IA, Benstead TJ, Murray TJ, Smith M. ALS incidence in Nova Scotia over a 20-year-period: a prospective study. The Canadian journal of neurological sciences. 2007;34:69–73. doi: 10.1017/s0317167100005813. [DOI] [PubMed] [Google Scholar]
- 16.Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. 2009;72:725–731. doi: 10.1212/01.wnl.0000343008.26874.d1. [DOI] [PubMed] [Google Scholar]
- 17.Donaghy C, Clarke J, Patterson C, Kee F, Hardiman O, Patterson V. The epidemiology of motor neuron disease in Northern Ireland using capture-recapture methodology. Amyotroph Lateral Scler. 2010;11:374–378. doi: 10.3109/17482960903329569. [DOI] [PubMed] [Google Scholar]
- 18.Donaghy C, O’Toole O, Sheehan C, Kee F, Hardiman O, Patterson V. An all-Ireland epidemiological study of MND, 2004–2005. Eur J Neurol. 2009;16:148–153. doi: 10.1111/j.1468-1331.2008.02361.x. [DOI] [PubMed] [Google Scholar]
- 19.Forbes RB, Colville S, Parratt J, Swingler RJ. The incidence of motor neuron disease in Scotland. Journal of neurology. 2007;254:866–869. doi: 10.1007/s00415-006-0454-y. [DOI] [PubMed] [Google Scholar]
- 20.Georgoulopoulou E, Vinceti M, Bonvicini F, Sola P, Goldoni CA, De Girolamo G, Ferraro D, Nichelli P, Mandrioli J. Changing incidence and subtypes of ALS in Modena, Italy: A 10-years prospective study. Amyotroph Lateral Scler. 2011;12:451–457. doi: 10.3109/17482968.2011.593037. [DOI] [PubMed] [Google Scholar]
- 21.Huisman MH, de Jong SW, van Doormaal PT, Weinreich SS, Schelhaas HJ, van der Kooi AJ, de Visser M, Veldink JH, van den Berg LH. Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. Journal of neurology, neurosurgery, and psychiatry. 2011;82:1165–1170. doi: 10.1136/jnnp.2011.244939. [DOI] [PubMed] [Google Scholar]
- 22.Joensen P. Incidence of amyotrophic lateral sclerosis in the Faroe Islands. Acta neurologica Scandinavica. 2011 doi: 10.1111/j.1600-0404.2011.01611.x. [DOI] [PubMed] [Google Scholar]
- 23.Logroscino G, Beghi E, Zoccolella S, Palagano R, Fraddosio A, Simone IL, Lamberti P, Lepore V, Serlenga L. Incidence of amyotrophic lateral sclerosis in southern Italy: a population based study. Journal of neurology, neurosurgery, and psychiatry. 2005;76:1094–1098. doi: 10.1136/jnnp.2004.039180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marin B, Gil J, Preux PM, Funalot B, Couratier P. Incidence of amyotrophic lateral sclerosis in the Limousin region of France, 1997–2007. Amyotroph Lateral Scler. 2009;10:216–220. doi: 10.1080/17482960902721626. [DOI] [PubMed] [Google Scholar]
- 25.McGuire V, Longstreth WT, Jr, Koepsell TD, van Belle G. Incidence of amyotrophic lateral sclerosis in three counties in western Washington state. Neurology. 1996;47:571–573. doi: 10.1212/wnl.47.2.571. [DOI] [PubMed] [Google Scholar]
- 26.Murphy M, Quinn S, Young J, Parkin P, Taylor B. Increasing incidence of ALS in Canterbury, New Zealand: a 22-year study. Neurology. 2008;71:1889–1895. doi: 10.1212/01.wnl.0000336653.65605.ac. [DOI] [PubMed] [Google Scholar]
- 27.O’Toole O, Traynor BJ, Brennan P, Sheehan C, Frost E, Corr B, Hardiman O. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. Journal of neurology, neurosurgery, and psychiatry. 2008;79:30–32. doi: 10.1136/jnnp.2007.117788. [DOI] [PubMed] [Google Scholar]
- 28.Vazquez MC, Ketzoian C, Legnani C, Rega I, Sanchez N, Perna A, Penela M, Aguirrezabal X, Druet-Cabanac M, Medici M. Incidence and prevalence of amyotrophic lateral sclerosis in Uruguay: a population-based study. Neuroepidemiology. 2008;30:105–111. doi: 10.1159/000120023. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida S, Uebayashi Y, Kihira T, Kohmoto J, Wakayama I, Taguchi S, Yase Y. Epidemiology of motor neuron disease in the Kii Peninsula of Japan, 1989–1993: active or disappearing focus? Journal of the neurological sciences. 1998;155:146–155. doi: 10.1016/s0022-510x(97)00300-6. [DOI] [PubMed] [Google Scholar]
- 30.Abhinav K, Stanton B, Johnston C, Hardstaff J, Orrell RW, Howard R, Clarke J, Sakel M, Ampong MA, Shaw CE, Leigh PN, Al-Chalabi A. Amyotrophic lateral sclerosis in South-East England: a population-based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry) Neuroepidemiology. 2007;29:44–48. doi: 10.1159/000108917. [DOI] [PubMed] [Google Scholar]
- 31.Alcaz S, Jarebinski M, Pekmezovic T, Stevic-Marinkovic Z, Pavlovic S, Apostolski S. Epidemiological and clinical characteristics of ALS in Belgrade, Yugoslavia. Acta neurologica Scandinavica. 1996;94:264–268. doi: 10.1111/j.1600-0404.1996.tb07063.x. [DOI] [PubMed] [Google Scholar]
- 32.Bonvicini F, Vinceti M, Marcello N, Rodolfi R, Rinaldi M. The epidemiology of amyotrophic lateral sclerosis in Reggio Emilia, Italy. Amyotroph Lateral Scler. 2008;9:350–353. doi: 10.1080/17482960802196150. [DOI] [PubMed] [Google Scholar]
- 33.Chio A, Cucatto A, Calvo A, Terreni AA, Magnani C, Schiffer D. Amyotrophic lateral sclerosis among the migrant population to Piemonte, northwestern Italy. Journal of neurology. 1999;246:175–180. doi: 10.1007/s004150050330. [DOI] [PubMed] [Google Scholar]
- 34.Cima V, Logroscino G, D’Ascenzo C, Palmieri A, Volpe M, Briani C, Pegoraro E, Angelini C, Soraru G. Epidemiology of ALS in Padova district, Italy, from 1992 to 2005. Eur J Neurol. 2009;16:920–924. doi: 10.1111/j.1468-1331.2009.02623.x. [DOI] [PubMed] [Google Scholar]
- 35.Fang F, Valdimarsdottir U, Bellocco R, Ronnevi LO, Sparen P, Fall K, Ye W. Amyotrophic lateral sclerosis in Sweden, 1991–2005. Archives of neurology. 2009;66:515–519. doi: 10.1001/archneurol.2009.13. [DOI] [PubMed] [Google Scholar]
- 36.Fong GC, Cheng TS, Lam K, Cheng WK, Mok KY, Cheung CM, Chim CS, Mak W, Chan KH, Tsang KL, Kwan MC, Tsoi TH, Cheung RT, Ho SL. An epidemiological study of motor neuron disease in Hong Kong. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:164–168. doi: 10.1080/14660820510028412a. [DOI] [PubMed] [Google Scholar]
- 37.Fong KY, Yu YL, Chan YW, Kay R, Chan J, Yang Z, Kwan MC, Leung KP, Li PC, Lam TH, Cheung RT. Motor neuron disease in Hong Kong Chinese: epidemiology and clinical picture. Neuroepidemiology. 1996;15:239–245. doi: 10.1159/000109913. [DOI] [PubMed] [Google Scholar]
- 38.Govoni V, Granieri E, Capone J, Manconi M, Casetta I. Incidence of amyotrophic lateral sclerosis in the local health district of Ferrara, Italy, 1964–1998. Neuroepidemiology. 2003;22:229–234. doi: 10.1159/000070563. [DOI] [PubMed] [Google Scholar]
- 39.Gross-Paju K, Oopik M, Luus SM, Kalbe I, Puksa L, Lepik T, Kaasik AE. Motor neurone disease in South Estonia. Diagnosis and incidence rate. Acta neurologica Scandinavica. 1998;98:22–28. doi: 10.1111/j.1600-0404.1998.tb07373.x. [DOI] [PubMed] [Google Scholar]
- 40.Guidetti D, Bondavalli M, Sabadini R, Marcello N, Vinceti M, Cavalletti S, Marbini A, Gemignani F, Colombo A, Ferrari A, Vivoli G, Solime F. Epidemiological survey of amyotrophic lateral sclerosis in the province of Reggio Emilia, Italy: influence of environmental exposure to lead. Neuroepidemiology. 1996;15:301–312. doi: 10.1159/000109920. [DOI] [PubMed] [Google Scholar]
- 41.Gundersen MD, Yaseen R, Midgard R. Incidence and clinical features of amyotrophic lateral sclerosis in More and Romsdal County, Norway. Neuroepidemiology. 2011;37:58–63. doi: 10.1159/000329523. [DOI] [PubMed] [Google Scholar]
- 42.Huber S, Henn V. Unchanged incidence and prevalence of amyotrophic lateral sclerosis in the canton of Zurich. Schweiz Arch Neurol Psychiatr. 1995;146:52–54. [PubMed] [Google Scholar]
- 43.Kihira T, Yoshida S, Hironishi M, Miwa H, Okamato K, Kondo T. Changes in the incidence of amyotrophic lateral sclerosis in Wakayama, Japan. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:155–163. doi: 10.1080/14660820510030031. [DOI] [PubMed] [Google Scholar]
- 44.Mandrioli J, Faglioni P, Merelli E, Sola P. The epidemiology of ALS in Modena, Italy. Neurology. 2003;60:683–689. doi: 10.1212/01.wnl.0000048208.54755.78. [DOI] [PubMed] [Google Scholar]
- 45.Sajjadi M, Etemadifar M, Nemati A, Ghazavi H, Basiri K, Khoundabi B, Mousavi SA, Kabiri P, Maghzi AH. Epidemiology of amyotrophic lateral sclerosis in Isfahan, Iran. Eur J Neurol. 2010;17:984–989. doi: 10.1111/j.1468-1331.2010.02972.x. [DOI] [PubMed] [Google Scholar]
- 46.Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology. 2002;59:280–282. doi: 10.1212/wnl.59.2.280. [DOI] [PubMed] [Google Scholar]
- 47.Stickler DE, Royer JA, Hardin JW. Validity of hospital discharge data for identifying cases of amyotrophic lateral sclerosis. Muscle & nerve. 2011;44:814–816. doi: 10.1002/mus.22195. [DOI] [PubMed] [Google Scholar]
- 48.Turabelidze G, Zhu BP, Schootman M, Malone JL, Horowitz S, Weidinger J, Williamson D, Simoes E. An epidemiologic investigation of amyotrophic lateral sclerosis in Jefferson County, Missouri, 1998–2002. Neurotoxicology. 2008;29:81–86. doi: 10.1016/j.neuro.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 49.Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E. Incidence of amyotrophic lateral sclerosis in Europe. Journal of neurology, neurosurgery, and psychiatry. 2010;81:385–390. doi: 10.1136/jnnp.2009.183525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. Journal of the neurological sciences. 1994;124 (Suppl):96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 51.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 52.Beghi E, Logroscino G, Chio A, Hardiman O, Mitchell D, Swingler R, Traynor BJ. The epidemiology of ALS and the role of population-based registries. Biochimica et biophysica acta. 2006;1762:1150–1157. doi: 10.1016/j.bbadis.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 53.Logroscino G, Traynor BJ, Hardiman O, Chio A, Couratier P, Mitchell JD, Swingler RJ, Beghi E. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. Journal of neurology, neurosurgery, and psychiatry. 2008;79:6–11. doi: 10.1136/jnnp.2006.104828. [DOI] [PubMed] [Google Scholar]
- 54.Pedro-Cuesta J, Litvan I. Epidemiology of motor neuron disease. In: Anderson DW, Schoenberg DG, editors. Neuroepidemiology A Tribute to Bruce. Schoenberg: CRC Press; 1991. pp. 265–296. [Google Scholar]
- 55.Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. Journal of the neurological sciences. 2001;191:3–9. doi: 10.1016/s0022-510x(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 56.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet neurology. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mok K, Traynor BJ, Schymick J, Tienari PJ, Laaksovirta H, Peuralinna T, Myllykangas L, Chio A, Shatunov A, Boeve BF, Boxer AL, DeJesus-Hernandez M, Mackenzie IR, Waite A, Williams N, Morris HR, Simon-Sanchez J, van Swieten JC, Heutink P, Restagno G, Mora G, Morrison KE, Shaw PJ, Rollinson PS, Al-Chalabi A, Rademakers R, Pickering-Brown S, Orrell RW, Nalls MA, Hardy J. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiology of aging. 2012;33:209, e203–208. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, van der Kooi AJ, de Visser M, Dahlberg C, Estrada K, Rivadeneira F, Hofman A, Zwarts MJ, van Doormaal PT, Rujescu D, Strengman E, Giegling I, Muglia P, Tomik B, Slowik A, Uitterlinden AG, Hendrich C, Waibel S, Meyer T, Ludolph AC, Glass JD, Purcell S, Cichon S, Nothen MM, Wichmann HE, Schreiber S, Vermeulen SH, Kiemeney LA, Wokke JH, Cronin S, McLaughlin RL, Hardiman O, Fumoto K, Pasterkamp RJ, Meininger V, Melki J, Leigh PN, Shaw CE, Landers JE, Al-Chalabi A, Brown RH, Jr, Robberecht W, Andersen PM, Ophoff RA, van den Berg LH. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21. 2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nature genetics. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 59.Scott KM, Abhinav K, Stanton BR, Johnston C, Turner MR, Ampong MA, Sakel M, Orrell RW, Howard R, Shaw CE, Leigh PN, Al-Chalabi A. Geographical clustering of amyotrophic lateral sclerosis in South-East England: a population study. Neuroepidemiology. 2009;32:81–88. doi: 10.1159/000177032. [DOI] [PubMed] [Google Scholar]
- 60.Scott KM, Abhinav K, Wijesekera L, Ganesalingam J, Goldstein LH, Janssen A, Dougherty A, Willey E, Stanton BR, Turner MR, Ampong MA, Sakel M, Orrell R, Howard R, Shaw CE, Nigel Leigh P, Al-Chalabi A. The association between ALS and population density: A population based study. Amyotroph Lateral Scler. 2010;11:435–438. doi: 10.3109/17482961003754552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leone M, Chio A, Mortara P, Rosso MG, Schiffer D. Motor neuron disease in the Province of Turin, Italy, 1971–1980. Acta neurologica Scandinavica. 1983;68:316–327. doi: 10.1111/j.1600-0404.1983.tb04839.x. [DOI] [PubMed] [Google Scholar]
- 62.Traynor BJ. The era of genomic epidemiology. Neuroepidemiology. 2009;33:276–279. doi: 10.1159/000235639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai SL, Myllykangas L, Sulkava R, Jansson L, Hernandez DG, Gibbs JR, Nalls MA, Heckerman D, Tienari PJ, Traynor BJ. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet neurology. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chio A, Borghero G, Pugliatti M, Ticca A, Calvo A, Moglia C, Mutani R, Brunetti M, Ossola I, Marrosu MG, Murru MR, Floris G, Cannas A, Parish LD, Cossu P, Abramzon Y, Johnson JO, Nalls MA, Arepalli S, Chong S, Hernandez DG, Traynor BJ, Restagno G. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Archives of neurology. 2011;68:594–598. doi: 10.1001/archneurol.2010.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Annegers JF, Appel S, Lee JR, Perkins P. Incidence and prevalence of amyotrophic lateral sclerosis in Harris County, Texas, 1985–1988. Archives of neurology. 1991;48:589–593. doi: 10.1001/archneur.1991.00530180041015. [DOI] [PubMed] [Google Scholar]
- 68.Zaldivar T, Gutierrez J, Lara G, Carbonara M, Logroscino G, Hardiman O. Reduced frequency of ALS in an ethnically mixed population: a population-based mortality study. Neurology. 2009;72:1640–1645. doi: 10.1212/WNL.0b013e3181a55f7b. [DOI] [PubMed] [Google Scholar]
- 69.Hoppitt T, Pall H, Calvert M, Gill P, Yao G, Ramsay J, James G, Conduit J, Sackley C. A systematic review of the incidence and prevalence of long-term neurological conditions in the UK. Neuroepidemiology. 2011;36:19–28. doi: 10.1159/000321712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marin B, Couratier P, Preux PM, Logroscino G. Can mortality data be used to estimate amyotrophic lateral sclerosis incidence? Neuroepidemiology. 2011;36:29–38. doi: 10.1159/000321930. [DOI] [PubMed] [Google Scholar]
- 71.Beghi E, Chio A, Couratier P, Esteban J, Hardiman O, Logroscino G, Millul A, Mitchell D, Preux PM, Pupillo E, Stevic Z, Swingler R, Traynor BJ, Van den Berg LH, Veldink JH, Zoccolella S. The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Lateral Scler. 2011;12:1–10. doi: 10.3109/17482968.2010.502940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.di Poggio MB, Sormani MP, Truffelli R, Mandich P, Origone P, Verdiani S, Mantero V, Scialo C, Schenone A, Mancardi GL, Caponnetto C. Clinical epidemiology of ALS in Liguria, Italy. Amyotroph Lateral Scler. 2012 doi: 10.3109/17482968.2012.729062. [DOI] [PubMed] [Google Scholar]
- 73.Giagheddu M, Puggioni G, Tacconi P, Pirastru MI, Cannas A, Tamburini G, Congia S. Amyotrophic lateral sclerosis in Sardinia (Italy): epidemiologic features from 1957 to 2000. Acta neurologica Scandinavica. 2012 doi: 10.1111/j.1600-0404.2012.01705.x. [DOI] [PubMed] [Google Scholar]
- 74.Pugliatti M, Parish LD, Cossu P, Leoni S, Ticca A, Saddi MV, Ortu E, Traccis S, Borghero G, Puddu R, Chio A, Pirina P. Amyotrophic lateral sclerosis in Sardinia, insular Italy, 1995–2009. Journal of neurology. 2012 doi: 10.1007/s00415-012-6681-5. [DOI] [PubMed] [Google Scholar]
- 75.Ragonese P, Cellura E, Aridon P, D’Amelio M, Spataro R, Taiello AC, Maimone D, La Bella V, Savettieri G. Incidence of amyotrophic lateral sclerosis in Sicily: A population based study. Amyotroph Lateral Scler. 2012;13:284–287. doi: 10.3109/17482968.2012.662689. [DOI] [PubMed] [Google Scholar]
- 76.Kihira T, Yoshida S, Kondo T, Iwai K, Wada S, Morinaga S, Kazimoto Y, Kondo T, Okamoto K, Kokubo Y, Kuzuhara S. An increase in ALS incidence on the Kii Peninsula, 1960–2009: a possible link to change in drinking water source. Amyotroph Lateral Scler. 2012;13:347–350. doi: 10.3109/17482968.2012.674140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wagner L, Archer NP, Williamson DM, Henry JP, Schiffer R, Jackson CE. Prevalence of amyotrophic lateral sclerosis in Texas, 1998–2003. Texas medicine. 2012;108:e1. [PubMed] [Google Scholar]
- 78.Wittie M, Nelson LM, Usher S, Ward K, Benatar M. Utility of Capture-Recapture Methodology to Assess Completeness of Amyotrophic Lateral Sclerosis Case Ascertainment. Neuroepidemiology. 2013;40:133–141. doi: 10.1159/000342156. [DOI] [PubMed] [Google Scholar]
- 79.Rabkin J, Ogino M, Goetz R, McElhiney M, Marziliano A, Imai T, Atsuta N, Morita M, Tateishi T, Matsumura T, Mitsumoto H. Tracheostomy with invasive ventilation for ALS patients. Neurologists’ roles in the US and Japan. Amyotroph Lateral Scler. 2012 doi: 10.3109/17482968.2012.726226. [DOI] [PubMed] [Google Scholar]
- 80.Antao VC, Horton DK. The National Amyotrophic Lateral Sclerosis (ALS) Registry. Journal of environmental health. 2012;75:28–30. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.