

Abstract
A substantial obstacle to the existing treatment of bacterial diseases is the lack of specific probes that can be used to diagnose and treat pathogenic bacteria in a selective manner while leaving the microbiome largely intact. To tackle this problem, there is an urgent need to develop pathogen-specific therapeutics and diagnostics. Here, we describe recent evidence that indicates distinctive glycans found exclusively on pathogenic bacteria could form the basis of targeted therapeutic and diagnostic strategies. In particular, we highlight the use of metabolic oligosaccharide engineering to covalently deliver therapeutics and imaging agents to bacterial glycans.
1. Introduction
The introduction of small molecule antibiotics revolutionized medicine. Prior to the discovery of antibiotics like penicillin, bacterial diseases ravaged mankind. Since that time, antibiotics have saved millions of lives. However, despite the undeniable impact antibiotics have had on curing bacterial diseases, existing antibiotics suffer from a number of drawbacks that must be overcome.
One widespread challenge in the health care industry is the emergence and spread of pathogenic bacterial strains that are resistant to existing antibiotics. In hospitals, 50% of Staphylococcus aureus isolates are methicillin resistant1, and vancomycin, a drug once known as the “antibiotic of last resort,” is no longer effective for some infected individuals.2 To address this challenge, chemists have employed three main approaches. One common method focuses on altering first-generation antibiotics to create variants that circumvent antibiotic resistance mechanisms.3 Another successful tactic emphasizes the discovery or development of novel classes of antibiotics.3 The third course of action employs combination therapies that inactivate resistance mechanisms and thus restore the efficacy of the initial antibiotic.4 However, the presence of sustained selective pressure combined with the mutability of bacteria produces drug resistant strains that continue to challenge chemists. The dwindling antibiotic pipeline has further eroded our ability to combat infection.5
Another challenge is the crucial role that microorganisms play in human health, and the unintended consequences antibiotics can have on our beneficial flora.6 In the human body, microbial cells outnumber human cells by a factor of ten.7 Disrupting the microbiome with a course of broad-spectrum antibiotics can alter the composition of gut bacteria for years, resulting in deleterious consequences on human health.6 Indeed, patients suffering from stomach ulcers who were treated with antibiotics for one week had a shift in their gut microbiome that lasted for up to four years post-antibiotic treatment.8 Such disturbances to the normal gut microbiota have been associated with obesity, autoimmune disorders, allergies, and malnutrition.9, 10 Moreover, interference of the microbiota with oral antibiotics enables pathogens to gain a foothold in the gut.11 Given the vital role of beneficial flora in human health, we need to establish new narrow spectrum therapeutics that do not disturb symbiotic bacteria.
The use of microorganism-specific antibiotics rather than broad-spectrum antibiotics slows the evolution and spread of antibiotic resistance12 by minimizing the likelihood of resistance gene transfer across bacterial species. In addition, microorganism-specific antibiotics treat pathogenic bacteria in a discriminating manner while leaving the host microbiome largely intact. Therefore, this method mitigates immediate health problems and minimizes long-term deleterious effects on beneficial bacteria. The practical deployment of microorganism-specific therapeutics requires both narrow spectrum antibiotics and rapid diagnostic tests that pinpoint the organism responsible for a patient’s infection. Therefore, there is an urgent need to develop novel antibiotics and diagnostics aimed at specific bacterial populations.
Bacterial glycans represent intriguing targets of therapeutics and diagnostics. They are linked to pathogenesis, have distinctive structures, and, in some cases, are present on only a small number of pathogenic bacteria.13 Here we provide an overview of bacterial glycans and how they can be harnessed to diagnose and treat bacterial diseases in a discriminating manner. We begin with a brief overview of bacterial glycan structures and their links to pathogenesis. We then highlight approaches to metabolically label these glycans with chemical reporters. Finally, we describe approaches to covalently target bacterial glycans with therapeutics or imaging agents.
1.1 Bacterial glycans are attractive pathogen-specific targets
Bacterial cells are coated with an impressive array of glycan structures that comprise their cell wall. The cell wall forms a suit of armour that protect the cell from its environment and osmotic lysis. Due to its critical importance in bacterial survival and its surface accessibility, the cell wall is a common target of antibiotics.14 Blockbuster antibiotics such as penicillin15, vancomycin16, and bacitracin17 all interfere with bacterial cell wall biosynthesis, which testifies to the cell wall’s attractiveness as a drug target for novel therapeutics. The bacterial cell wall remains an exciting target, as it is covered with distinctive surface accessible structures that are linked to pathogenesis.
Most bacteria can be grouped into one of three categories depending on their cell wall architecture: Gram-negative bacteria, Gram-positive bacteria, and mycobacteria (Fig. 1).18 Gram-negative bacteria have inner and outer cell membranes, with peptidoglycan in the intervening periplasmic space and lipopolysaccharide (LPS) and capsular polysaccharide (CPS) associated with the outer membrane (Fig. 1a).19 Like Gram-negative cells, Gram-positive cells have a dense peptidoglycan layer and capsular polysaccharide on their cell surface (Fig. 1b). In contrast, Gram-positive bacteria contain only one cell membrane and produce teichoic acids on their surfaces rather than LPS.19 Teichoic acids are either covalently attached to the peptidoglycan or anchored in the cell membrane via a lipid tail (Fig. 1b).20 Finally, mycobacterial species, including the widespread human pathogen Mycobacterium tuberculosis, have such idiosyncratic cell walls that, although technically Gram-positive bacteria, they can be placed in a category of their own (Fig. 1c). The mycobacterial cell membrane is encased by a thick layer of peptidoglycan and lipoarabinomannan, which are further elaborated with arabinogalactan and mycolic acids, and finally capped with trehalose-linked lipids.21 This exceptional cell wall has permeability characteristics that enable mycobacteria to evade antibiotics that target cell wall biosynthesis in Gram-negative and Gram-positive bacteria.22 An additional differentiating feature is that some but not all strains of Gram-positive, Gram-negative, and mycobacteria also synthesize glycosylated proteins and present these glycoproteins on their cell surfaces.13, 23 Regardless of bacterial sub-class, bacterial cells are covered with unusual glycans that are absent from human cells and have the potential to underpin novel therapeutic and diagnostic strategies.
Fig. 1.

Bacterial cell walls are coated with a diverse array of glycan structures. (a) Gram-negative bacteria contain both an inner membrane (IM) and an outer membrane (OM). The space between the membranes is rigidified by the peptidoglycan. Embedded in the outer membrane are lipopolysaccharide (LPS), capsular polysaccharide (CPS), and glycoproteins. (b) Gram-positive bacteria contain only one membrane (M), which is reinforced with a thick coating of peptidoglycan. Capsular polysaccharides, teichoic acids, and glycoproteins are found on the periphery of the cell. (c) Mycobacteria are Gram-positive bacteria that contain distinctive glycans on their cells, including lipoarabinomannan, arabinogala ctan, mycolic acid, and trehalose-linked lipids. Image is not to scale.
An analysis of the structures of glycans found on bacterial cells reveals exclusively bacterial monosaccharide building blocks (Fig. 2). Some of these monosaccharides are widely prevalent in bacteria, while others are limited to a small number of bacterial pathogens.13 For example, all bacterial cells are coated with peptidoglycan, a network of repeating units of β-1,4-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) crosslinked by short peptides (Fig. 2). Though GlcNAc is used broadly in prokaryotic and eukaryotic cells, MurNAc is a uniquely bacterial glycan. Thus, a therapeutic or imaging agent that targets MurNAc or peptidoglycan would influence all bacteria. In contrast, only Gram-negative bacteria synthesize LPS, which contains the distinctive monosaccharides 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) and L-glycero-D-mannoheptose (heptose; Fig. 2).24 Consequently, an antibiotic or diagnostic that targets these structures would only impact Gram-negative bacteria. Similarly, Gram-positive cells are characterized by the presence of teichoic acids.20 Hence, interfering with teichoic acid biosynthesis would impact only Gram-positive cells. Mycobacteria, in contrast, could be detected or targeted by their trehalose-linked lipids or arabinogalactan, for example (Fig. 2). These structures could form the basis of a strategy to diagnose or destroy each of the major classes of bacteria in a broad-spectrum manner.
Fig. 2.

Structures of representative glycans found on bacterial cells, with exclusively bacterial monosaccharides highlighted in color. Structures are grouped according to type of glycoconjugate, including peptidoglycan, glycolipids, polysaccharides, and glycoproteins, with known distribution indicated in pa rentheses. Abbreviations: MurNAc = muramic acid; KDO = 3-deoxy-D-manno-oct-2-ulosonic acid; heptose = L-glycero-D-mannoheptose; galf = galactofuranose; AAT = 2-acetamido-4-amino-2,4,6-trideoxyhexose; Pse = pseudaminic acid; Leg = legionaminic acid; DATDH = 2,4-diacetamido-2,4,6-trideoxyhexose; FucNAc = N-acetylfucosamine; Bac = bacillosamine
Potentially attractive targets are rare carbohydrates, which are present on select bacteria and thus could form the foundation of narrow-spectrum therapeutics and diagnostics. For example, Neisseria meningitides utilizes 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH)25, Pseudomonas aeruginosa installs FucNAc residues26, and Bacteroides fragilis appends 2-acetamido-4-amino-2,4,6-trideoxy-galactose (AAT)27 into its cell surface polysaccharides (Fig. 2). These distinctive building blocks are exclusively present on a small number of bacterial species.13 Similarly, Campylobacter jejuni’s and Helicobacter pylori’s glycoproteins contain the amino- and deoxy-monosaccharides pseudaminic acid, legionaminic acid, and bacillosamine28, 29 (see Fig. 2). These sugars have limited expression on pathogenic bacteria,28, 30, 31 and there are no reports of these monosaccharides in commensal bacterial that dominate the human gut microbiome9 (e.g. Bacteroides sp., Prevotella sp.). These insights indicate that certain bacterial sugars are not widely used and are thus attractive targets of selective interference and examination.
Bacterial glycans are great targets because they are often linked to bacterial fitness and pathogenesis. Indeed, bacterial cells with improperly formed peptidoglycan undergo osmotic lysis32, those with altered LPS are destroyed by the host’s immune system32, and bacteria that lack teichoic acids are attenuated in host colonization and infection.33 Similarly, trehalose-deficient mycobacteria are not viable34 and bacterial strains with inactivated protein glycosylation genes exhibit reduced host cell binding and colonization defects28, 35-37. These observations reveal that bacterial glycans perform crucial functions and are, in several cases, linked to pathogenicity. The ability to target these structures and visualize them could address the pressing needs to develop new antibiotics and diagnostics for bacterial diseases.
1.2 Traditional approaches to targeting bacterial glycans are powerful yet have limitations
Traditional approaches to targeting bacterial glycans for prophylactic or therapeutic purposes have had remarkable successes in the clinic.13 For example, vaccines based on bacterial carbohydrate epitopes are at the forefront of preventative medicine and are used to immunize children against bacterial pathogens such as Neisseria meningitides (e.g. Menactra)38, Streptococcus pneumoniae (e.g. Prevnar)39, and Haemophilus influenza (Act-HIB)40. Continued efforts to develop a more extensive collection of anti-bacterial vaccines are underway. Though vaccination is a successful preventative measure, it does not help individuals who are already suffering from a bacterial infection. Therefore, antibiotics are critical for battling active infections. Our antibiotic arsenal contains numerous small molecule inhibitors of enzymes involved in bacterial glycoconjugate biosynthesis such as penicillin15, vancomycin16, and bacitracin17 which interfere with peptidoglycan biosynthesis. Recent developments have enabled the elucidation of peptidoglycan biosynthesis41, bacterial O-linked glycan biosynthesis25, and rare monosaccharide biosynthetic pathways42, thereby facilitating high-throughput screening of inhibitors of these pathways42. Despite their successes, traditional approaches that target bacteria by inhibiting cell wall assembly with therapeutics have limitations. Traditional antibiotics target molecules that promote growth. As such, they miss a subset of bacteria that are not actively growing, in particular, dormant and persistent bacteria.43 Additionally, some bacteria evade antibiotics due to their location within the host. Finally, the majority of antibiotics are broad-spectrum which endangers our beneficial flora. Thus, though traditional approaches to targeting bacterial glycans have much to offer to prevent and treat bacterial disease, complementary approaches should be explored.
In contrast, bacterial glycans have played a minimal role in diagnosing and tracking infection. The notable exception is with patient antisera, which often contains antibodies that bind to bacterial cell wall structures and can be used to establish whether a patient has had a particular bacterial infection.44 Rather than using an indirect immune response in an ex vivo assay, efforts to image bacterial glycans in animal based models could lead to improved diagnostic capabilities.
New approaches that selectively target membrane glycans on both growing and persistent bacteria with therapeutics or imaging agents have the potential to address unmet needs. In this review, we focus on the potential benefits of covalent targeting of bacterial glycans with small molecule therapeutics and diagnostics. To ramp up the armamentarium against bacteria, we discuss a novel and highly modular approach to interfering with bacteria and imaging bacterial glycans based on metabolic labelling with bioorthogonal chemistries.
2. Metabolic labelling of bacterial glycans with chemical reporters
Metabolic oligosaccharide engineering (MOE)45, 46 and bioorthogonal chemistry47, 48 have the potential to transform the way bacterial diseases are treated and bacterial infections are visualized. MOE is a two-step chemical approach that was pioneered by Bertozzi49, 50, Reutter51, 52, and colleagues to study and target eukaryotic glycans and has been recently extended to bacterial glycans. In the first step of MOE, an unnatural sugar containing a bioorthogonal chemical reporter is taken up and processed by permissive carbohydrate biosynthetic enzymes in living cells, ultimately leading to the metabolic replacement of endogenous sugars with unnatural variants (Fig. 3). In the second step, cells that are covered with chemical reporters undergo a reaction with exquisitely selective reactive partners to yield covalent adducts (Fig. 3). Reactive partners can be conjugated to therapeutics or diagnostics to enable the covalent delivery of drugs for targeted bacterial killing or diagnostic imaging.
Figure 3.
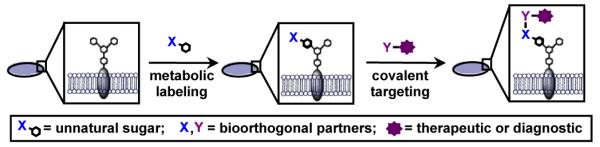
Metabolic oligosaccharide engineering can be employed to covalently deliver therapeutics or diagnostics to bacterial glycans. First, bacterial cells metabolically process an unnatural sugar that contains a bioorthogonal chemical reporter (X) into cellular glycans. In a second step, the chemical reporter undergoes covalent elaboration with a reactive partner (Y) conjugated to a therapeutic or diagnostic to fo6rm a covalent adduct.
This versatile covalent targeting strategy leaves room for creativity and permits the design of a large variety of both broad- and narrow-spectrum therapeutics and diagnostics. Essentially, by treating a patient with a carefully chosen sugar pill followed by a therapeutic or diagnostic cocktail, bacterial cells could be targeted or visualized by virtue of their glycan coat. In this section we will discuss ways to make this strategy highly selective for certain populations of bacteria by modulating (1) the unnatural sugar substrate chosen for metabolic labelling and (2) the bioorthogonal chemistry used for glycan targeting. In the final section, we will discuss a plethora of options for covalent targeting of bacterial glycans with therapeutics or imaging agents.
2.1 Unnatural substrates for metabolic labelling of bacterial glycans
The first step of MOE requires an unnatural sugar that is taken up by bacteria and processed by the cell’s metabolic pathways.45, 46 Uptake is aided by transporters in the bacterial membrane53 or by passive diffusion across the lipid bilayer, which can be facilitated by the temporary masking of hydrophilic hydroxyl groups with hydrophobic acetyl groups.54 Once the unnatural sugars enter cells, non-specific esterases remove temporary protecting groups to yield free hydroxyls.55 Permissive carbohydrate biosynthetic enzymes then process the unnatural sugars and incorporate them into surface glycans in place of natural sugars. As detailed below, several classes of bacterial glycans have been labelled with ketone (−COCH3), azide (−N3), or alkyne (−CCH) chemical reporters to date (Fig. 4). These examples illustrate that subtle structural perturbations are well tolerated by a variety of carbohydrate biosynthetic enzymes in bacteria, setting the stage to choose an appropriate metabolic precursor to label and target select bacterial glycans.
Fig. 4.

Literature examples of unnatural metabolic precursors that are processed and incorporated into bacterial glycans. (a) Sadamoto et al. demonstrated that a ketone-bearing analog of the peptidoglycan precursor UDP-MurNAc-pentapeptide is metabolically incorporated into peptidoglycan on Gram-positive and Gram-negative bacteria.56 (b) VanNieuwenhze, Bertozzi and colleagues found that azide- and alkyne-bearing analogs of D-amino acids are metabolically incorporated into peptidoglycan in myriad bacteria.58,59 (c) Dumont et al. reported that an alkyne-containing variant of Kdo is incorporated into LPS of Gram-negative bacteria.60 (d) Swarts et al. revealed that azide-containing trehalose derivatives are processed by Mycobacteria and incorporated into their glycolipids.62 (e) Liu et al. found that 6-deoxy-AltNAc4NAz is processed by C. jejuni, converted to azido-pseudaminic acid, and displayed on flagellin glycoproteins.63 (f) Champasa et al. established that GlcNAz is metabolically processed into H. pylori’s glycoproteins.64 Further, Memmel et al. found that GlcNAz is incorporated into S. aureus’ surface glycans.66 (g) Finally, Wu and coworkers demonstrated that both alkynyl and azide-containing fucose derivatives are tolerated by carbohydrate processing enzymes in Parabacteroides sp.67 Abbreviations: MurNAc = muramic acid; D-Ala = D-alanine; Kdo = 3-deoxy-D-manno-oct-2-ulosonic acid; Pse = pseudaminic acid.
The pioneering report of metabolic labelling of bacterial glycans with chemical reporters employed a ketone-modified glycopeptide analogue of a naturally occurring peptidoglycan precursor, uridine diphosphate (UDP)-MurNAc pentapeptide (Fig. 4a).56 In this study, Sadamoto and coworkers metabolically introduced ketones into the peptidoglycan of Gram-negative (Escherichia coli) and Gram-positive bacteria (Lactobacillus species) by supplementing the bacteria with a derivative of the precursor bearing a ketone modification at the peptide side chain (Fig. 4a). The cell wall precursor is a substrate of bacterial but not human cells and therefore should enable selective labelling of bacteria with ketones. Using a hydrazide-based probe to target the ketone moiety on the cell walls, these authors were able to modulate levels of bacterial adhesion, a key property that is essential for colonization and persistence.57 As an alternative to labelling peptidoglycan with a glycan-containing metabolic precursor, supplementing bacteria with azide- and alkyne-bearing D-amino acids results in labelling of newly synthesized peptidoglycan (Fig. 4b), yet no labelling of proteins or teichoic acids.58, 59 Notably, labelling with these peptidoglycan precursors occurs in Gram-positive bacteria, Gram-negative bacteria, and mycobacteria and thus could form the basis of a broad-spectrum antibiotic or bacterial imaging strategy.
Though peptidoglycan-labelling studies have involved chemically modifying the peptide moiety in their chosen metabolic precursors, subsequent metabolic labelling experiments with bacteria have primarily modified monosaccharide substrates. For example, Dumont et al. synthesized an alkyne-containing variant of Kdo (Fig. 4c), a characteristic monosaccharide present in the inner core of LPS, and reported that alkyne-modified Kdo is incorporated into Gram-negative bacteria (e.g. E. coli, Salmonella typhimurium, Legionella pneumophila) but not Gram-positive bacteria (e.g. S. aureus and Bacillus subtilis).60 This observed labelling pattern is consistent with the presence of LPS on Gram-negative but not Gram-positive bacterial cells. Hence, antibiotics or diagnostics that target alkyne-Kdo would only affect Gram-negative bacteria.
The development of alternative therapeutic approaches is particularly urgent for multidrug resistant bacteria such as Mycobacterium tuberculosis. Recent investigations into the unique outer membrane of mycobacteria reveals structures, such as trehalose monomycolate and trehalose dimycolate, that are essential for cell wall biosynthesis and disease progression.61 By taking advantage of the natural trehalose recycling pathway, Bertozzi, Swarts, and coworkers selectively labelled glycolipids on Mycobacterium smegmatis cells with four distinct azido-trehalose (TreAz) derivatives (Fig. 4d).62 TreAz containing glycolipids were successfully installed in the mycobacterium cell surfaces, which allowed for targeted delivery of azide-specific cyclooctyne probes.62 Given the narrow expression of trehalose on mycobacteria and its absence from Gram-negative and Gram-positive bacteria, metabolic labelling with TreAz derivatives has the potential to yield selective labelling of mycobacterial cells with azides and therefore could form the basis of mycobacteria-specific therapeutics and diagnostics.
Narrow-spectrum therapeutics and diagnostics could be developed based on the expression of certain rare monosaccharides on bacteria. For example, pseudaminic acid is present on only a small number of pathogenic bacteria, while neither pseudaminic acid nor any enzyme in its biosynthetic pathway are found in mammalian cells.28, 42 Intrigued by this knowledge, Liu et al. designed and synthesized an azide-containing variant of 6-deoxy-AltdiNAc, a dedicated precursor to pseudaminic acid.63 The authors demonstrated that C. jejuni’s pseudaminic acid biosynthetic pathway is permissive of the azide-containing substrate 6-deoxy-AltNAc-4-NAz and that this substrate has only one fate in C. jejuni – conversion into azido-pseudaminic acid and incorporation into flagellin glycoproteins (Fig. 4e).63 Thus, this work indicates that pseudaminic acid biosynthesis could be exploited to incorporate azides selectively onto a handful of pathogenic bacterial cells for therapeutic or diagnostic purposes.
The discovery and access to unique bacterial sugars for targeting specific bacterial populations remains the chief bottleneck in MOE. Current efforts to profile bacterial glycan structures could yield additional unique glycan structures to augment this targeting approach. Indeed, several rare hexosamine bacterial monosaccharides such as FucNAc, Bac, AAT, and DATDH have been identified. Synthetic methods developed by Kulkarni and colleagues have permitted unprecedented and expedited access to these rare sugars.64, 65 Synthetic access to these monosaccharides opens the door to the creation of chemical reporter-containing variants of these sugars and thus expands the possibilities for metabolic labelling of unique bacterial glycans.64, 65
In addition to uniquely bacterial glycans, sugars common to both bacterial and mammalian cells can also be utilized in this approach. Selective targeting of bacteria with common sugars is possible if the sugars are differentially incorporated onto bacterial cells compared to mammalian cells. For example, GlcNAc is a common building block present in N-linked glycoproteins on the surface of mammalian cells. Surprisingly, metabolic labelling of H. pylori and mammalian cells with peracetylated N-azidoacetylglucosamine (GlcNAz) led to appreciable levels of azide-labelled glycans on the surface of H. pylori66, yet not on the surface of kidney epithelial cells67 (Fig. 4f). The selective incorporation of the common metabolic precursor GlcNAz on bacterial cell surfaces provides an intriguing area of pursuit for preferentially tagging bacteria in the context of mammalian cells. Recent work by Memmel et al. revealed that S. aureus also metabolically incorporate GlcNAz into surface glycans.68 Intriguingly, azide-functionalized S. aureus covalently targeted with alkyne dyes display reduced adherence to host cells. These exciting studies indicate that even broad metabolic precursors could enable selective targeting of bacteria. GlcNAz may represent a special case, however, as azide and alkyne derivatives of the widespread sugar L-fucose are incorporated into fucosylated glycans on both bacterial (e.g. Parabacteroides sp. and engineered E. coli)69 and mammalian cell’s surfaces70, 71 (Fig. 4g). Nevertheless, the GlcNAz results suggests that the selective labelling of bacteria with unnatural derivatives of common metabolic sugars merits investigation.
The above-described studies establish that distinctive glycans found in the cell wall of bacteria can be metabolically labelled with dedicated precursors containing chemical reporters. Labelled bacteria are poised for targeting with reactive partners for imaging or selective damage. The breadth of these examples underscores the impact of metabolic labelling for potential therapeutic and diagnostic applications. We are now poised to develop new unnatural sugars to specifically target a narrow range of bacteria using myriad biocompatible chemistries discussed in the next section.
2.2 Bioorthogonal chemistries for glycan targeting
Covalent targeting of bacterial glycans requires chemistries that are compatible with living systems. Ideally, the chemistries chosen should employ chemical reporters that are normally absent from biological systems, stable in water, nonreactive with biological functional groups, and capable of undergoing covalent elaboration with exquisitely selective reactive partners under physiological conditions.48 Several chemistries have been developed that meet these demands; these “bioorthogonal” chemistries are discussed in depth in recent reviews.72-74 In this section, we highlight select bioorthogonal chemistries that demonstrate the potential to covalently target bacterial glycans in living systems (Fig. 5). Crucially, these chemistries use small chemical reporters that are likely to be tolerated by permissive carbohydrate biosynthetic pathways and unlikely to perturb the function of labelled glycans. Moreover, the chemistries we discuss have the potential to proceed in vivo at appreciable rates, either on their own or with the aid of a biocompatible catalyst. We will briefly enumerate the application of these chemistries in the context of glycans and highlight their relative advantages and disadvantages in cell and animal based applications.
Fig. 5.

Reported bioorthogonal chemistries with promise for use in covalent targeting of bacterial glycans. The highlighted chemistries entail small chemical reporters that are distinctive in biological systems, bioinert, and undergo exquisitely selective covalent elaboration with the indicat ed reactive partners. The second order rate constants were reported in the following references: (a)75, (b)76, (c)83, (d)86, (e)73, (f)98, (g)86, (h)110, (i)113,116, (j)114, (k)115, (l)118, (m)119.
As discussed in the previous section, ketones, azides and alkynes are chemical reporters that have been used to label bacterial glycans. Originally, ketones were explored as chemical reporters because they are virtually absent from cell surfaces and are not found in naturally occurring amino acids, glycoconjugates, or lipids.49 Ketones undergo covalent elaboration with hydrazide and amino-oxy probes in near neutral environments (pH 5-6) to form stable hydrazone or oxime products48, respectively (Fig. 5a, b).75, 76 Although the resulting oxime products are more stable to hydrolysis than hydrazones77, ketone ligation with hydrazide proceeds more favourably at biological pH.76 Remarkably, ketone-hydrazide chemistry is bioinert in the extracellular environment and has been previously accomplished in both Gram-negative and Gram-positive bacteria.49, 78 Furthermore, Rideout and coworkers demonstrated that this chemistry is robust even in the context of a living animal.79, 80 Thus, bacteria labelled with ketones are poised for safe and selective covalent targeting with therapeutics and imaging agents within a host.
The introduction of the azide as a reporter in biological systems represents a turning point in the development of bioorthogonal chemistries, as azides are truly absent from biological systems.47 Unlike their inorganic counterparts, alkyl azides are relatively nontoxic and are sufficiently tolerated by cells50 and whole organisms81, 82. Organic azides act as soft nucleophiles that react with select partners to form adducts. For example, azides react with triarylphosphines via Staudinger ligation to yield amides (Fig. 5c).83 This robust chemistry proceeds in mice without side reactions or apparent detriment to physiology.47 Therefore, the Staudinger ligation has properties that make it well suited to probe bacterial glycans in the context of infected individuals. Although the Staudinger ligation is highly selective, triarylphosphines are easily inactivated through oxidation and have slow reaction kinetics47, which could pose pharmacokinetic liabilities for targeting bacterial glycans in vivo.
Therefore, alternative azide-based bioorthogonal reactions have been explored. Meldal, Sharpless and coworkers demonstrated that Cu(I)-catalysed azide-alkyne cycloaddition, also known as “click chemistry,” is highly selective84, 85 and incredibly fast (k = 1-10 M−1s−1)86 (Fig. 5d). However, the initial application of click chemistry in vivo was fairly limited due to the high toxicity profile of copper87, 88 at the concentration required to enable catalysis.84 To enhance the biocompatibility of this chemistry, Bertozzi and coworkers developed the “strain promoted azide-alkyne cycloaddition” (SPAAC), a bioorthogonal reaction between azides and strained cyclooctynes to yield triazoles (Fig. 5e).89, 90 Alterations to the cyclooctyne scaffold have led to a 100-fold improvement in reaction rates relative to the Staudinger ligation.73, 91 Indeed, the rates of reaction for the most recently developed reagents for SPAAC are comparable to that of Cu(I)-catalysed click chemistry.73, 92 Further, SPAAC proceeds in living animals, including in zebrafish93-95, nematodes96, and mice97. The light activatable conversion of cyclopropenes to cyclooctynes enables temporal, light-dependent control over SPAAC (Fig. 5f).98 Taken together, SPAAC offers an attractive choice for targeting bacterial glycans. This chemistry is not without problems as some cyclooctynes react promiscuously with endogenous nucleophiles, such as free thiols99, in the serum and in intracellular spaces. Modifications to the cyclooctyne scaffold, however, have been able to fine-tune reactivity, minimizing side reactions while maximizing biocompatibility.100, 101
An alternative approach to applying click chemistry in live cell imaging has relied on the deployment of copper chelators that mitigate Cu(I) toxicity. In particular, Finn and others have developed a number of water-soluble ligands that stabilize copper, accelerate click chemistry, prevent the formation of undesirable side products and oxidative damage to biomolecules, and ultimately sequester copper ions to facilitate removal.102 These ligands enable click chemistry to proceed in cells with minimal loss of cellular viability.102-107 Building upon this approach, Ting, Zhu, and co-workers employed an exciting strategy in their ligand design, which uses an azide-containing copper chelating agent. Ultimately, azide-containing copper ligands increase the effective concentration of Cu(I) at the catalytic site while reducing the actual copper concentration to the low micromolar range, at which copper toxicity is minimal.108, 109 The use of copper chelators has vastly enhanced the viability of the alkyne as a chemical reporter in multicellular organisms.
Like azides, terminal alkynes are rarely seen in biological chemical space48 and are small enough to minimize functional and structural perturbations. As a chemical reporter, alkynes can react with exogenous azides via Cu(I)-catalysed click chemistry, as discussed above (Fig. 5g). Alternatively, alkynes can react with indophenols in the presence of catalytic Pd(NO3)2 via Sonogashira cross coupling (Fig. 5h).110 This reaction proceeds at a rate comparable to that of ligand-mediated Cu(I)-catalysed click chemistry, exhibits excellent compatibility with proteins, and proceeds to completion in E. coli, Shigella and Salmonella.110 Especially intriguing, palladium toxicity111 appears lower than copper toxicity112. Taken together, the palladium-catalysed Sonogashira cross coupling offers a feasible option for probing bacterial glycans and holds several exciting possibilities for future developments.
In addition to the well-established ketone, azide, and alkyne chemical reporters, other functional groups have arisen as promising chemical reporters. In particular, Prescher and coworkers have demonstrated that the cyclopropene is tolerated by permissive carbohydrate biosynthetic pathways in mammalian cells and undergoes covalent modification with tetrazine conjugates on live cells (Fig. 5i).113 This chemistry proceeds at a rate comparable to the Staudinger ligation and is compatible with SPAAC between azides and cyclooctynes.113 Isomeric 3,3-disubstituted cyclopropenes also have the potential to serve as orthogonal chemical reporters, as they do not react with tetrazine but instead selectively react with nitrile imines to form a cycloadduct upon UV light activation (Fig. 5j).114 Thus, cyclopropenes are a viable approach to probing multiple bacterial glycans in tandem.
Terminal alkenes have also been explored as chemical reporters. Although alkenes are abundant in membrane lipids, endogenous alkenes are not accessible to chemical reactions because they are tightly packed within the lipid bilayer.115 Thus, exposed terminal alkenes on the cell surface represent a distinctive reactive group that can be preferentially tagged with bioorthogonal chemistries. Wittman et al. reported that the sialic acid biosynthetic pathway is permissive of an unnatural substrate bearing an alkene, allowing terminal alkenes to be incorporated into mammalian glycans.116 Further, these authors demonstrated that alkene-covered cells undergo highly selective ligation with tetrazine via an inverse Diels Alder (Fig. 5i).116 Moreover, this chemistry proceeds selectively in the presence of azides.116 Though cyclizations of tetrazines with terminal alkenes are slower than with cyclopropenes due to the lack of ring strain, modulating the tetrazine scaffold achieves a rate of reaction similar to the Staudinger ligation116. Alternatively, terminal alkenes undergo reaction with diaryl tetrazole via a light-mediated reaction to produce nitrile imines in mammalian cells (Fig. 5k).115 The resulting product is a highly fluorescent pyrazoline cycloadduct, and the reaction proceeds without detriment to living cells when activated by a two photon 700 nm laser.115 Thus, this chemistry has the potential to enable tracking of glycans in real-time.115
Isonitriles also have demonstrated utility in live cells.117 This functional group are net neutral, small, stable at neutral pH, and display no appreciable toxicity in mammals. Isonitriles undergo [4+1] cycloaddition with tetrazines at a rate comparable to second-generation strain promoted cyclooadditions (Fig. 5l).117, 118 Metabolic labelling of mammalian glycans with isonitrile-modified sugar substrates proceeds without cytotoxic or cytostatic effects.117 Moreover, isonitrile-tetrazine chemistry proceeds on live cells and is orthogonal to azide/cyclooctyne chemistry.117 Thus, isonitriles are yet another viable glycan reporter.
Finally, vinyl thioethers have emerged as chemical reporters. Vinyl thioethers are small, chemically stable, and act as electron-rich dienophiles in a hetero-Diels-Alder cycloaddition with o-quinone-methides (Fig. 5m).119 The rate of this reaction is on par with the Staudinger ligation, and the reaction is selective in the presence of thiols.119 Although this chemistry has not yet been performed to label and track glycans, Li et al. reported that this cycloaddition proceeds in live mammalian cells and could be used to localize exogenous vinyl thioether conjugated taxol in mammalian systems.119 Thus, vinyl thioethers could underlie a chemical strategy to visualize and target bacterial glycans.
In sum, the recent explosion of bioorthogonal chemistries has expanded the repertoire of chemical reporters that could be used for probing bacterial glycans. Further, the diverse modes of reactivity exhibited by these chemical reporters sets the stage for simultaneously tracking multiple bacterial glycans in living systems. Progress in translating these new chemistries into disease models and applying them to study bacterial glycans will provide a foundation for enhancing our understanding of host-pathogen interactions and for increasing our antibiotic repertoire.
3. Covalent targeting of bacterial glycans with therapeutics or imaging agents
Once bacterial glycans are metabolically labelled with chemical reporters, they are poised for covalent elaboration with selective reactive partners. Reactive probes that are conjugated to a therapeutic agent could catalyse cellular damage. Alternatively, reactive partners that comprise an imaging agent could be used to monitor dynamic changes in the bacterial glycan coat and disease progression. Below, we discuss recent examples of covalent targeting of bacterial glycans with therapeutics or diagnostics and highlight possible future directions.
3.1 Therapeutics that catalyse cellular damage or render bacteria innocuous
Covalent targeting of distinctive bacterial glycans with therapeutics has the potential to incite selective cellular damage or render bacteria harmless. Only two examples of covalently targeting bacterial glycans with therapeutics have been published to date. In the first example, Kaewsapsak et al. were able to recruit immune effector cells to target H. pylori’s surface glycans.67 Briefly, azide-covered H. pylori cells were covalently targeted via Staudinger ligation with phosphines conjugated to the immune stimulant 2,4-dinitrophenol (DNP) (Fig. 6a).67 DNP-modified H. pylori were then exposed to anti-DNP antibodies and immune effector cells for targeted H. pylori killing (Fig. 6a).67 Given the occurrence of naturally circulating anti-DNP antibodies in a high percentage of the human population, this bacterial targeting strategy has the potential to proceed in animals.120 Indeed, the level of H. pylori killing observed was on par with the requirements for anti-DNP mediated cell killing of cancer cells targeted with DNP-conjugates in vivo.121 This work establishes an important precedent for using immune stimulants for targeting bacterial glycans and could be extended to targeting diverse bacterial glycans with assorted immune stimulants, such as fluorescein, alpha-Gal, or 1,3 diketones.122
Fig. 6.
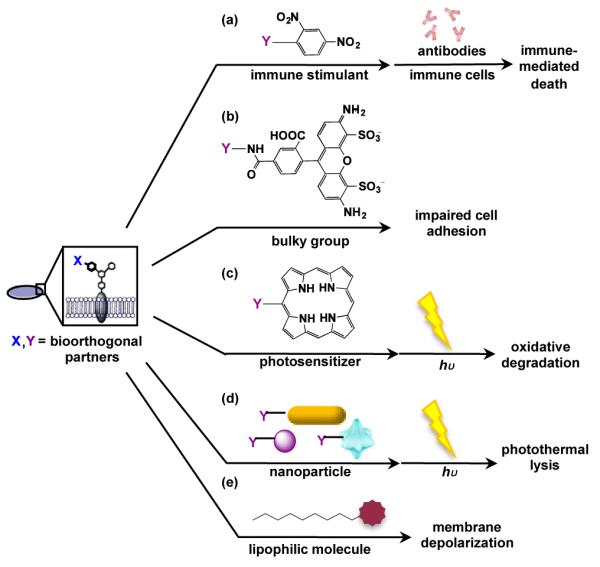
Several classes of therapeutics, including (a) immune stimulants, (b) bulky groups, (c) photosensitizers, (d) nanoparticles, and (e) lipophilic molecules, have the potential to catalyse cellular damage or render bacteria innocuous if they are covalently delivered to chemical reporter-labelled glycans on bacterial cells.
Rather than catalysing cell killing, an alternative therapeutic approach involves rendering bacteria innocuous within a host. Memmel et al. published a beautiful demonstration of this aim, in which they interfered with bacterial adhesion to host cells.68 In essence, they demonstrated that covalent delivery of a bulky fluorophore, alkyne-Alexa-488, to GlcNAz-covered S. aureus via Cu(I)-catalysed click chemistry led to reduced adhesion to host cells relative to controls (Fig. 6b).68 Given the crucial role of bacterial adhesion in infection123 and bacterial persistence124 in a host, inhibiting this process is an effective mechanism for drug therapy. The use of a fluorescent group provides the added benefit for tracking the allocation of the therapeutic in patients. This strategy has the potential to be broadened to an array of therapeutics that interfere with bacterial glycan function.
Although there are only two precedented examples of covalent targeting of bacterial glycans with therapeutics, we can take cues from the area of cancer research for inspiration on alternative therapeutics to target bacterial glycans. Here we discuss novel classes of therapeutics such as photosensitizers125 and nanoparticles126, and the use of toxins127 to catalyse damage to bacteria.
In photodynamic therapy, photosensitizers generate toxic reactive oxygen species when excited by light (Fig. 6c).128 The generated species then oxidize essential biological molecules and cause cell death (Fig. 6c).128 Since singlet oxygen species have an incredibly short life span in water, their toxicity is localized in the immediate vicinity where they are produced; thus, targeted cells are killed with selectivity.129 Several porphyrin-based photosensitizers are clinically approved for photodynamic eradication of cancer cells in the skin, head, neck, esophagus, lungs, and stomach.128 In addition, photodynamic therapy has been applied for the treatment of bacterial infection.130-132 Therefore, photosensitizer-containing reactive partners hold great potential for covalently targeting bacteria based on their distinctive glycans.
Antibiotic development has the potential to benefit from the burgeoning field of nanomedicine. Although nanoparticles are in their infancy and are just entering clinical trials, they display unique optical properties that could be harnessed for imaging and therapeutic purposes. In particular, nanoparticles can be tuned to absorb light in the NIR region and subsequently dissipate heat (Fig. 6d).133 Localized heating can disrupt cellular function or induce cellular lysis, ultimately leading to cellular destruction (Fig. 6d).133 Gold nanorods remain one of most popular agents for photothermal therapy as gold is bioinert. However, other nanostructures have been identified as great photothermal agents.133 One of the advantages of nanoparticles is that they can be tuned to absorb light at wavelengths that optimize depth of tissue penetration and targeted cell damage.134 In addition, their surface chemistries can be modified to promote solubility and biocompatibility. The light dependent cytotoxicity of nanoparticles makes these particles optimal for antimicrobial therapeutics. Indeed, Norman et al. harnessed the power of antibody conjugated gold nanorods to trigger targeted photothermal lysis on Gram-negative Pseudomonas aeruginosa.135 This study demonstrates that nanoparticles are feasible therapeutic agents for treating antibiotic resistant bacteria.
As an alternative to light-induced cell killing, lipophilic toxins induce cytoxicity via action on the bacterial membrane. Bacteria possess a particularly high transmembrane potential compared to human cells, and the maintenance of this potential is essential for proper cell function. Small lipophilic molecules can distort the cellular membrane and cause ion leakage that results in a change of proton motive force, causing cell death (Fig. 6e).136 Lipophilic toxins, such as daptomycin, have demonstrated the ability to depolarize bacterial membranes and remain the last line of defence for treating drug resistant bacteria.136 However, evidence of bacterial resistance to daptomycin is emerging. One hypothesized mechanism of resistance involves the loss of a docking site for daptomycin on the bacteria membrane.136 The covalent attachment of daptomycin to bacterial cell wall via bioorthogonal chemistry could provide the antibiotic with an anchor to evade current mechanisms of resistance and restore biocidal activity. In addition, targeted delivery of membrane toxins to select bacteria would reduce the risk of nonspecific damage to human cells, potentially revitalizing the utility of lipophilic agents that have been dismissed in clinical trials due to unintended side effects. Therefore, targeting bacterial glycans with conjugated lipophilic agents is worthy of exploration.
In conclusion, the targeted delivery of therapeutics to bacterial glycans has the potential to induce specific cytotoxicity, thus minimizing harm to mammalian cells and beneficial flora. Although there are some direct precedents for targeting bacterial glycans with agents that induce cell death or render bacteria safe, there are significant prospects for growth in this area. Ultimately, the covalent delivery of therapeutics to bacterial glycans could expand new antibiotic strategies.
3.2 Imaging metabolically labelled bacterial glycans
Distinctive glycans that are exclusively present on bacterial cells have the potential to form the basis of molecular imaging strategies to monitor bacterial disease progression in real time. Indeed, the ability to track bacterial glycans in their native physiological setting – ideally in a non-invasive manner – could reveal insights into disease initiation and progression, as well as the effectiveness of particular therapeutics at curing bacterial infections. Further, non-invasive imaging of bacterial glycans in vivo could reveal how bacteria modulate their glycan coat over the course of infection.
Visualizing the bacterial glycan coat in cellular contexts and in animal infection models requires an imaging agent that reports on bacterial glycan structures. Historically, glycan-binding proteins such as lectins and antibodies have been utilized to monitor cellular glycans.137 For example, Mahal and Hsu developed lectin-based microarrays that report on glycan composition of live E. coli.138 Additionally, Dufrêne and colleagues were able to explore the spatial arrangement of wall teichoic acids in living Lactobacillus plantarum cells using fluorescent concanavalin A lectin probes.139 Though lectins have enabled the study of bacterial glycans in cellular settings, they have limited applications in animal infection models due to their poor tissue penetrance.
Alternatively, fluorescent derivatives of cell wall-binding antibiotics have been used to visualize cell wall biosynthesis and disruption. For example, Pereira et al. visualized new peptidoglycan biosynthesis at division septa in S. aureus using a fluorescent analogue of vancomycin.140 Similarly, Carlson and co-workers developed a fluorescent cephalosporin analogue that binds selectively to a subset of penicillin-binding proteins and reveals in vivo labelling and visualization of cell wall biosynthesis in B. subtilis and S. pneumonia.141 While fluorescent antibiotics display far better tissue penetrance than lectins and thus have potential for visualizing bacterial glycans in living organisms, these fluorescent antibiotics perturb bacterial populations under study.
MOE-based approaches have emerged as a complementary means to visualize bacterial glycans. MOE overcomes many of the challenges associated with lectins, antibodies, and fluorescent antibiotics. Advantages include the high tissue permeability endowed by small molecules and the low probability of disrupting bacterial populations under study. In a pioneering example of utilizing MOE as the basis of glycan imaging in live bacteria, Sadamoto et al. visualized peptidoglycan synthesis in both Gram-positive and Gram-negative bacteria by tagging ketone-labelled peptidoglycan with fluorescein-hydrazide probes.56 Since that initial report, several labs have employed MOE to image bacterial glycans. For example, Dumont et al. visualized LPS biosynthesis in live bacteria using azide-Kdo and Cu(I)-catalysed click chemistry with a fluorescent alkyne and a biocompatible copper-chelator; these authors demonstrated that only Gram-negative bacteria were visualized using this approach.60 Additionally, GlcNAz-labelled glycans have been imaged on the surface of H. pylori67 and S. aureus68 cells. Each of these examples demonstrates the utility of MOE as a strategy to image bacterial glycans.
Recent work has enabled imaging of trehalose analogues on mycobacteria. Backus et al. demonstrated that a fluorescein-containing trehalose analogue is incorporated into growing M. tuberculosis, even in the context of a macrophage infection model.142 Thus, this fluorescent glycan visualizes M. tuberculosis infection and has the potential to serve as a possible diagnostic tool to label M. tuberculosis in an infected host. In another report, Swarts et al. found that mycobacterial trehalose biosynthetic pathways are widely tolerant of azide modifications on substrates and can be imaged with fluorescent cyclooctyne-based reporters.62
The use of a bioorthogonal chemical reporter is particularly advantageous over other labelling strategies, as it provides temporal control of probe delivery and offers the possibility to perform pulse-chase experiments. Indeed, VanNieuwenhze and co-workers imaged newly synthesized peptidoglycan in an array of bacterial species in a pulse-chase fashion using a combination of fluorescent and chemical reporter-bearing D-amino acids.58 These authors performed sequential peptidoglycan labelling and executed time-lapse microscopy to visualize real-time peptidoglycan synthesis. Bertozzi and coworkers built upon this work by incorporating alkyne-bearing D-amino acids into Listeria monocytogenes and visualizing peptidoglycan synthesis in real-time during macrophage infection.59 These examples highlight the use of metabolic labelling as a facile, modular approach to probing bacterial glycan dynamics in a spatial and temporal manner.
The next big steps in this area will likely involve moving into animal infection models. Though bacteria have been imaged in mice previously (e.g. magnetic resonance imaging of iron-oxide nanoparticle-coated bacteria143; fluorescence imaging of labelled bacteria144), they have not been visualized in animals in a glycan-specific way. There is, however, significant potential for translating the tracking of bacterial glycans into animal infection models. Pulse-chase, time-lapse imaging of glycans using MOE with azide-cyclooctyne chemistry has been performed in live zebrafish95. Moreover, fluorescence and radionuclide imaging of labelled cellular glycans on cancer cells based on Staudinger ligation and SPAAC have been performed successfully in mice.145, 146 Thus, literature precedents suggest that visualizing bacterial glycans and monitoring bacterial infection in animal models is just around the corner.
Non-invasive imaging of bacteria in animal models will be greatly aided by the development of “smart” probes that are activated upon reaction with bioorthogonal partners. Such “smart” probes enhance the signal to noise ratio while eliminating time required for probe removal. Fortunately, fluorogenic dyes that are activated by Staudinger ligation, Cu(I)-catalysed click chemistry, or SPAAC with azides have been introduced (Fig. 7). For example, Bertozzi and coworkers developed phosphine probes that become fluorescent or bioluminescent after reacting with azides (Fig. 7a-c).83, 147, 148 Moreover, Jewett and Bertozzi developed a fluorogenic cyclooctyne that is activated upon reaction with azides (Fig. 7d).149 Analogously, Wong, Bertozzi, and others introduced fluorogenic dyes that are activated by Cu(I)-catalysed click chemistry (Fig. 7e-h).70, 150, 151 Future steps will likely focus on developing “smart” probes that extend beyond azide-chemical space and visualize some of the newer chemical reporters. Indeed, fluorogenic probes based on photo-click chemistry between tetrazoles and alkenes (Fig. 7i)115 and between tetrazine-cyclopropene cycloaddition (Fig. 7j)152 have been published and are important steps toward this goal. The development of probes activated by a variety of chemical reporters opens the door to tracking multiple bacterial glycans in vivo in parallel, thus enabling a thorough analysis of the bacterial glycan coat.
Fig. 7.

Probes that are activated by bioorthogonal chemistries in cell-based settings. (a, b) Staudinger ligation-based fluorogenic reporters are activated by reaction with azides to yield fluorescent products. The probe in (a) is initially quenched by the phosphine lone pair electrons147, whereas the probe in (b) utilizes a FRET quencher that is released upon Staudinger ligation83. (c) Staudinger ligation-based bioluminescent reporter that yields luciferin upon reaction with azides.148 Once luciferin is released, luciferase-expressing cells catalyse the conversion of luciferin to ocyluciferin in a bioluminescent reaction that yields visible light. (d) Cu-free click chemistry-based reporter becomes fluorescent upon reaction with azides.149 (e-h) Several Cu(I)-catalysed azide alkyne cycloaddition-based reporters become fluorescent when the azide or alkyne-based probe reacts with alkynes or azides, respectively.70, 150, 151 (i) Photoinduced tetrazole-based reporter fluoresces blue upon reaction with alkenes.115 (j) Tetrazine quenches BODIPY fluorescence; the conjugate becomes fluorescent when the tetrazine reacts with cyclopropene to yield a cycloadduct.152
In sum, a number of exciting uses of MOE to image glycans on live bacterial cells have emerged. These examples set the stage for imaging bacterial glycans in normal and pathological settings, both in live bacteria and in animal models. Ultimately, this approach has the potential to enter the clinic for diagnosing disease and monitoring disease progression.
4. Conclusions
There are substantial challenges associated with existing antibiotics, including widespread antibiotic resistance and the unintended consequences of broad-spectrum antibiotics on our beneficial flora. To overcome these challenges, novel antibiotics and diagnostics aimed at specific bacterial populations are needed. Bacterial glycans are attractive targets of therapeutics and diagnostics because they are linked to pathogenesis, have distinctive structures, and are present on only a small number of pathogenic bacteria. MOE offers an innovative approach to modify bacterial glycans with chemical reporters in either a narrow-spectrum or broad-spectrum manner, depending on the choice of metabolic substrate. This highly modular approach can be tailored to applications of interest by modifying the unnatural substrate, the chemical reporter, the bioorthogonal chemistry, and the choice of therapeutic or imaging agent. Therefore, recent advances in MOE and bioorthogonal chemistry have laid the foundation for labelling distinctive bacterial glycan structures with chemical reporters, developing covalent therapeutics that target labelled glycans, and performing multiplex bacterial glycan imaging to monitor disease progression in real time.
Before MOE could be applied in a clinical context, either for therapeutics or diagnostics, there are several paramount challenges that would need to be addressed. In particular, the time required for metabolic labelling of bacterial glycans must be rapid. Further, the delivery and pharmacokinetic properties of the covalent partner need to be optimized. Additionally, any extra steps that have to be performed, such as exposure to light, should be tolerated by patients in a clinical setting. Finally, since some bacteria change their glycan coat to evade host immune detection, the approach could include a panel of sugars to cirvcumvent potential resistance mechanisms. Addressing these constraints would set the stage for looking beyond the scope of bacterial glycans. Indeed, this chemical reporter-based approach has widespread promise for labelling, targeting, and imaging diverse biomolecules in a wide variety of normal and pathological settings.
Acknowledgements
We gratefully acknowledge funding for this work from the Camille & Henry Dreyfus Foundation and by grants from the National Center for Research Resources (5P20RR016463-12) and the National Institute of General Medical Sciences (8 P20 GM103423-12) from the National Institutes of Health. We thank members of the Dube lab for thoughtful comments and discussions.
Biography

Van Tra joined the Dube lab as an undergraduate researcher in Fall 2012, when she initiated an Honors project focused on creating a photo-activatable antibiotic that targets Helicobacter pylori’s surface glycans. She graduated from Bowdoin College with Honors in Biochemistry in May 2013. She is now conducting post-baccalaureate research under the guidance of Professor Dube as an NIH-funded Maine-INBRE Biomedical Researcher. Her research interests are in the areas of chemical biology and mass spectrometry, and she intends to pursue her PhD.

Danielle Dube is an Assistant Professor of Chemistry and Biochemistry at Bowdoin College. She received her PhD in Chemistry from the University of California, Berkeley under the guidance of Professor Carolyn Bertozzi in 2005. She then pursued post-doctoral training with Professor Jennifer Kohler at Stanford University. In 2007 she joined the faculty at Bowdoin College. Her research interests are in the areas of chemical biology and glycobiology, including developing chemical tools to target, alter, and understand glycosylation. Recent work in the lab has focused on harnessing chemical tools to discover and target bacterial glycoproteins.
References
- 1.Arias CA, Murray BE. New England Journal of Medicine. 2009;360:439–443. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 2.Walsh TR, Howe RA. Annual Review of Microbiology. 2002;56:657–675. doi: 10.1146/annurev.micro.56.012302.160806. [DOI] [PubMed] [Google Scholar]
- 3.Clardy J, Fischbach MA, Walsh CT. Nature Biotechnology. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 4.Brogden RN, Carmine A, Heel RC, Morley PA, Speight TM, Avery GS. Drugs. 1981;22:337–362. doi: 10.2165/00003495-198122050-00001. [DOI] [PubMed] [Google Scholar]
- 5.Fischbach MA, Walsh CT. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaser M. Nature. 2011;476:393–394. doi: 10.1038/476393a. [DOI] [PubMed] [Google Scholar]
- 7.Methé BA, Nelson KE, Pop M, Creasy HH, Giglio MG, Huttenhower C, Gevers D, Petrosino JF, Abubucker S, Badger JH. Nature. 2012;486:215–221. [Google Scholar]
- 8.Jakobsson H, Jernber C, Andersson A, Sjolund-Karlsson M, Jansson J, Engstrand L. PLoS ONE. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanevald J, Turnbaugh P, Lozupone C, Ley R, Hamady M, Gordon J, Knight R. Current Opinion in Chemical Biology. 2008;12:109–114. doi: 10.1016/j.cbpa.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riley MA, Robinson SM, Roy CM, Dorit RL. Future Medicinal Chemistry. 2013;5:1231–1242. doi: 10.4155/fmc.13.79. [DOI] [PubMed] [Google Scholar]
- 13.Dube DH, Champasa K, Wang B. Chemical Communications. 2011;47:87–101. doi: 10.1039/c0cc01557a. [DOI] [PubMed] [Google Scholar]
- 14.van Dam V, Olrichs N, Breukink E. ChemBioChem. 2009;10:617–624. doi: 10.1002/cbic.200800678. [DOI] [PubMed] [Google Scholar]
- 15.Park JT, Strominger JL. Science. 1957;125:99–101. doi: 10.1126/science.125.3238.99. [DOI] [PubMed] [Google Scholar]
- 16.Perkins HR. Biochemical Journal. 1969;111:195–205. doi: 10.1042/bj1110195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storm DR, Strominger JL. Journal of Biological Chemistry. 1973;248:3940–3945. [PubMed] [Google Scholar]
- 18.Reid CW, Fulton KM, Twine SM. Future Microbiology. 2010;5:267–288. doi: 10.2217/fmb.09.103. [DOI] [PubMed] [Google Scholar]
- 19.Silhavy TJ, Kahne D, Walker S. Cold Spring Harbor Perspectives in Biology. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown S, Maria JPS, Walker S. In: Annual Review of Microbiology. Gottesman S, editor. Vol. 67. 2013. pp. 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee D. Current Opinion in Chemical Biology. 1997;1:579–588. doi: 10.1016/s1367-5931(97)80055-5. [DOI] [PubMed] [Google Scholar]
- 22.Daffe M, Etienne G. Tubercle and Lung Disease. 1999;79:153–169. doi: 10.1054/tuld.1998.0200. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt MA, Riley LW, Benz I. TRENDS in Microbiology. 2003;11:554–561. doi: 10.1016/j.tim.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Cipolla L, Polissi A, Airoldi C, Gabrielli L, Merlo S, Nicotra F. Current Medicinal Chemistry. 2011;18:830–852. doi: 10.2174/092986711794927676. [DOI] [PubMed] [Google Scholar]
- 25.Hartley MD, Morrison MJ, Aas FE, Borud B, Koomey M, Imperiali B. Biochemistry. 2011;50:4936–4948. doi: 10.1021/bi2003372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horzempa J, Held TK, Cross AS, Furst D, Qutyan M, Neely AN, Castric P. Clinical and Vaccine Immunology. 2008;15:590–597. doi: 10.1128/CVI.00476-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumann H, Tzianabos AO, Brisson JR, Kasper DL, Jennings HJ. Biochemistry. 1992;31:4081–4089. doi: 10.1021/bi00131a026. [DOI] [PubMed] [Google Scholar]
- 28.Schirm M, Soo EC, Aubry AJ, Austin J, Thibault P, Logan SM. Molecular Microbiology. 2003;48:1579–1592. doi: 10.1046/j.1365-2958.2003.03527.x. [DOI] [PubMed] [Google Scholar]
- 29.Hopf PS, Ford RS, Zebian N, Merkx-Jacques A, Vijayakumar S, Ratnayake D, Hayworth J, Creuzenet C. PLoS ONE. 2011;6:e25722–e25722. doi: 10.1371/journal.pone.0025722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goon S, Kelly JF, Logan SM, Ewing CP, Guerry P. Molecular Microbiology. 2003;50:659–671. doi: 10.1046/j.1365-2958.2003.03725.x. [DOI] [PubMed] [Google Scholar]
- 31.Yuriy AK, Evgeny VV, Alexander SS, Boris AD, Nikolay KK, Evgeny SS, Galina MM. European Journal of Biochemistry. 1986;157:129–138. [Google Scholar]
- 32.Chatterjee AN, Young FE. Journal of Bacteriology. 1972;111:220–230. doi: 10.1128/jb.111.1.220-230.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weidenmaier C, Peschel A, Xiong Y-Q, Kristian SA, Dietz K, Yeaman MR, Bayer AS. Journal of Infectious Diseases. 2005;191:1771–1777. doi: 10.1086/429692. [DOI] [PubMed] [Google Scholar]
- 34.Woodruff PJ, Carlson BL, Siridechadilok B, Pratt MR, Senaratne RH, Mougous JD, Riley LW, Williams SJ, Bertozzi CR. Journal of Biological Chemistry. 2004;279:28835–28843. doi: 10.1074/jbc.M313103200. [DOI] [PubMed] [Google Scholar]
- 35.Smedley JG, Jewell E, Roguskie J, Horzempa J, Syboldt A, Stolz DB, Castric P. Infection and Immunity. 2005;73:7922–7931. doi: 10.1128/IAI.73.12.7922-7931.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alemka A, Nothaft H, Zheng J, Szymanski CM. Infection and Immunity. 2013;81:1674–1682. doi: 10.1128/IAI.01370-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howard SL, Jagannathan A, Soo EC, Hui JPM, Aubry AJ, Ahmed I, Karlyshev A, Kelly JF, Jones MA, Stevens MP, Logan SM, Wren BW. Infection and Immunity. 2009;77:2544–2556. doi: 10.1128/IAI.01425-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Girard MP, Preziosi MP, Aguado MT, Kieny MP. Vaccine. 2006;24:4692–4700. doi: 10.1016/j.vaccine.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 39.Black S, Shinefield H, Fireman B, Lewis E, Ray P, Hansen JR, Elvin L, Ensor KM, Hackell J, Siber G, Malinoski F, Madore D, Chang I, Kohberger R, Watson W, Austrian R, Edwards K. Pediatric Infectious Disease Journal. 2000;19:187–195. doi: 10.1097/00006454-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Schuchat A, Robinson K, Wenger JD, Harrison LH, Farley M, Reingold AL, Lefkowitz L, Perkins BA. New England Journal of Medicine. 1997;337:970–976. doi: 10.1056/NEJM199710023371404. [DOI] [PubMed] [Google Scholar]
- 41.Bugg TDH, Braddick D, Dowson CG, Roper DI. Trends in Biotechnology. 2011;29:167–173. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Schoenhofen IC, McNally DJ, Brisson J-R, Logan SM. Glycobiology. 2006;16:8C–14C. doi: 10.1093/glycob/cwl010. [DOI] [PubMed] [Google Scholar]
- 43.Hurdle JG, O’Neill AJ, Chopra I, Lee RE. Nature Reviews Microbiology. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verbrugh HA, Peters R, Rozenberg-Arska M, Peterson PK, Verhoef J. Journal of Infectious Diseases. 1981;144:1–9. doi: 10.1093/infdis/144.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Dube DH, Bertozzi CR. Current Opinion Chemical Biology. 2003;7:616–625. doi: 10.1016/j.cbpa.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Laughlin ST, Bertozzi CR. Nature Protocols. 2007;2:2930–2944. doi: 10.1038/nprot.2007.422. [DOI] [PubMed] [Google Scholar]
- 47.Sletten EM, Bertozzi CR. Accounts of Chemical Research. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prescher JA, Bertozzi CR. Nature Chemical Biology. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 49.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 50.Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 51.Keppler OT, Stehling P, Herrmann M, Kayser H, Grunow D, Reutter W, Pawlita M. Journal of Biological Chemistry. 1995;270:1308–1314. doi: 10.1074/jbc.270.3.1308. [DOI] [PubMed] [Google Scholar]
- 52.Keppler OT, Horstkorte R, Pawlita M, Schmidts C, Reutter W. Glycobiology. 2001;11:11R–18R. doi: 10.1093/glycob/11.2.11r. [DOI] [PubMed] [Google Scholar]
- 53.Goon S, Schilling B, Tullius MV, Gibson BW, Bertozzi CR. Proceedings of the National Academy of Science. 2003;100:3089–3094. doi: 10.1073/pnas.0437851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarkar AK, Fritz TA, Taylor WH, Esko JD. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3323–3327. doi: 10.1073/pnas.92.8.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones MB, Teng H, Rhee JK, Lahar N, Baskaran G, Yarema KJ. Biotechnology and Bioengineering. 2004;85:394–405. doi: 10.1002/bit.10901. [DOI] [PubMed] [Google Scholar]
- 56.Sadamoto R, Niikura K, Sears PS, Liu HT, Wong CH, Suksomcheep A, Tomita F, Monde K, Nishimura SI. Journal of the American Chemical Society. 2002;124:9018–9019. doi: 10.1021/ja026133x. [DOI] [PubMed] [Google Scholar]
- 57.Sadamoto R, Niikura K, Ueda T, Monde K, Fukuhara N, Nishimura S-I. Journal of the American Chemical Society. 2004;126:3755–3761. doi: 10.1021/ja039391i. [DOI] [PubMed] [Google Scholar]
- 58.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, De Pedro MA, Brun YV, Vannieuwenhze MS. Angewandte Chemie International Edition. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siegrist MS, Whiteside S, Jewett JC, Aditham A, Cava F, Bertozzi CR. ACS Chemical Biology. 2013;8:500–505. doi: 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dumont A, Malleron A, Awwad M, Dukan S, Vauzeilles B. Angewandte Chemie International Edition. 2012;51:3143–3146. doi: 10.1002/anie.201108127. [DOI] [PubMed] [Google Scholar]
- 61.Yamagami H, Matsumoto T, Fujiwara N, Arakawa T, Kaneda K, Yano I, Kobayashi K. Infection and Immunity. 2001;69:810–815. doi: 10.1128/IAI.69.2.810-815.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swarts BM, Holsclaw CM, Jewett JC, Alber M, Fox DM, Siegrist MS, Leary JA, Kalscheuer R, Bertozzi CR. Journal of the American Chemical Society. 2012;134:16123–16126. doi: 10.1021/ja3062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F, Aubry AJ, Schoenhofen IC, Logan SM, Tanner ME. Chembiochem. 2009;10:1317–1320. doi: 10.1002/cbic.200900018. [DOI] [PubMed] [Google Scholar]
- 64.Emmadi M, Kulkarni SS. Organic & Biomolecular Chemistry. 2013;11:3098–3102. doi: 10.1039/c3ob40615f. [DOI] [PubMed] [Google Scholar]
- 65.Emmadi M, Kulkarni SS. Nature Protocols. 2013;8:1870–1889. doi: 10.1038/nprot.2013.113. [DOI] [PubMed] [Google Scholar]
- 66.Champasa K, Longwell SA, Eldridge AM, Stemmler EA, Dube DH. Molecular & Cellular Proteomics. 2013;12:2568–2586. doi: 10.1074/mcp.M113.029561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaewsapsak P, Esonu O, Dube DH. ChemBioChem. 2013;14:721–726. doi: 10.1002/cbic.201300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Memmel E, Homann A, Oelschlaeger TA, Seibel J. Chemical Communications. 2013;49:7301–7303. doi: 10.1039/c3cc43424a. [DOI] [PubMed] [Google Scholar]
- 69.Besanceney-Webler C, Jiang H, Wang W, Baughn AD, Wu P. Bioorganic & Medicinal Chemistry Letters. 2011;21:4989–4992. doi: 10.1016/j.bmcl.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sawa M, Hsu TL, Itoh T, Sugiyama M, Hanson SR, Vogt PK, Wong CH. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12371–12376. doi: 10.1073/pnas.0605418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rabuka D, Hubbard SC, Laughlin ST, Argade SP, Bertozzi CR. Journal of American Chemical Society. 2006;128:12078–12079. doi: 10.1021/ja064619y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sletten EM, Bertozzi CR. Angewandte Chemie International Edition. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramil CP, Lin Q. Chemical Communications. 2013;49:11007–11022. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim RKV, Lin Q. Chemical Communications. 2010;46:1589–1600. doi: 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nauman DA, Bertozzi CR. Biochimica et Biophysica Acta-General Subjects. 2001;1568:147–154. doi: 10.1016/s0304-4165(01)00211-2. [DOI] [PubMed] [Google Scholar]
- 76.Dirksen A, Hackeng TM, Dawson PE. Angewandte Chemie International Edition. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 77.Kalia J, Raines RT. Angewandte Chemie. 2008;120:7633–7636. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rideout D. Science. 1986;233:561–563. doi: 10.1126/science.3523757. [DOI] [PubMed] [Google Scholar]
- 79.Rideout D. Cancer Investigation. 1994;12:189–202. doi: 10.3109/07357909409024874. [DOI] [PubMed] [Google Scholar]
- 80.Rideout D, Calogeropoulou T, Jaworski J, McCarthy M. Biopolymers. 1990;29:247–262. doi: 10.1002/bip.360290129. [DOI] [PubMed] [Google Scholar]
- 81.Dube DH, Prescher JA, Quang CN, Bertozzi CR. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4819–4824. doi: 10.1073/pnas.0506855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prescher JA, Dube DH, Bertozzi CR. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 83.Hangauer MJ, Bertozzi CR. Angewandte Chemie. 2008;47:2394–2397. doi: 10.1002/anie.200704847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angewandte Chemie International Edition. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 85.Tornoe CW, Christensen C, Meldal M. Journal of Organic Chemistry. 2002;67 doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 86.Presolski SI, Hong V, Cho S-H, Finn M. Journal of the American Chemical Society. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gaetke LM, Chow CK. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 88.Zödl B, Zeiner M, Marktl W, Steffan I, Ekmekcioglu C. Biological Trace Element Research. 2003;96:143–152. doi: 10.1385/BTER:96:1-3:143. [DOI] [PubMed] [Google Scholar]
- 89.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Agard NJ, Prescher JA, Bertozzi CR. Journal of the American Chemical Society. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 91.Jewett JC, Sletten EM, Bertozzi CR. Journal of the American Chemical Society. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Almeida G. d., Townsend LC, Bertozzi CR. Organic Letters. 2013;15:3038–3041. doi: 10.1021/ol401225n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dehnert KW, Baskin JM, Laughlin ST, Beahm BJ, Naidu NN, Amacher SL, Bertozzi CR. ChemBioChem. 2012;13:353–357. doi: 10.1002/cbic.201100649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dehnert KW, Beahm BJ, Huynh TT, Baskin JM, Laughlin ST, Wang W, Wu P, Amacher SL, Bertozzi CR. ACS Chemical Biology. 2011;6:547–552. doi: 10.1021/cb100284d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Laughlin ST, Bertozzi CR. ACS Chemical Biology. 2009;4:1068–1072. doi: 10.1021/cb900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Proceedings of the National Academy of Sciences. 2010;107:1821–1826. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, Popik VV. Journal of the American Chemical Society. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Geel R, Pruijn GJM, van Delft FL, Boelens WC. Bioconjugate Chemistry. 2012;23:392–398. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- 100.Debets MF, van Berkel SS, Schoffelen S, Rutjes FP, van Hest JC, van Delft FL. Chemical Communications. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 101.Chigrinova M, McKay CS, Bonhomme-Beaulieu L-P, Udachin K, Beauchemin AM, Pezacki JP. Organic Biomolecular Chemistry. 2013;11:3436–3441. doi: 10.1039/c3ob40683k. [DOI] [PubMed] [Google Scholar]
- 102.Kennedy DC, McKay CS, Legault MCB, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z, Pezacki JP. Journal of the American Chemical Society. 2011;133:17993–18001. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- 103.Hong V, Steinmetz NF, Manchester M, Finn M. Bioconjugate Chemistry. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Organic Letters. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 105.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angewandte Chemie International Edition. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Michaels HA, Zhu L. Chemistry – An Asian Journal. 2011;6:2825–2834. doi: 10.1002/asia.201100426. [DOI] [PubMed] [Google Scholar]
- 107.Wang W, Hong S, Tran A, Jiang H, Triano R, Liu Y, Chen X, Wu P. Chemistry – An Asian Journal. 2011;6:2796–2802. doi: 10.1002/asia.201100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brotherton WS, Michaels HA, Simmons JT, Clark RJ, Dalal NS, Zhu L. Organic Letters. 2009;11:4954–4957. doi: 10.1021/ol9021113. [DOI] [PubMed] [Google Scholar]
- 109.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY. Angewandte Chemie. 2012;124:5954–5958. doi: 10.1002/anie.201108181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li J, Lin S, Wang J, Jia S, Yang M, Hao Z, Zhang X, Chen PR. Journal of the American Chemical Society. 2013;135:7330–7338. doi: 10.1021/ja402424j. [DOI] [PubMed] [Google Scholar]
- 111.Melber C, Keller D, Mangelsdorf I. Palladium, Environmental Health Criteria. 2002;226 doi: 10.1078/1438-4639-00180. [DOI] [PubMed] [Google Scholar]
- 112.Agency for toxic substances and disease registry. 2004.
- 113.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. Journal of the American Chemical Society. 2012;134:18638–18643. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 114.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih H-W, Houk KN, Prescher JA. Journal of the American Chemical Society. 2013;135:13680–13683. doi: 10.1021/ja407737d. [DOI] [PubMed] [Google Scholar]
- 115.Song W, Wang Y, Yu Z, Vera CIR, Qu J, Lin Q. ACS Chemical Biology. 2010;5:875–885. doi: 10.1021/cb100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Niederwieser A, Späte A-K, Nguyen LD, Jüngst C, Reutter W, Wittmann V. Angewandte Chemie International Edition. 2013;52:4265–4268. doi: 10.1002/anie.201208991. [DOI] [PubMed] [Google Scholar]
- 117.Stairs S, Neves AA, Stöckmann H, Wainman YA, Ireland-Zecchini H, Brindle KM, Leeper FJ. ChemBioChem. 2013;14:1063–1067. doi: 10.1002/cbic.201300130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stockmann H, Neves AA, Stairs S, Brindle KM, Leeper FJ. Organic & Biomolecular Chemistry. 2011;9:7303–7305. doi: 10.1039/c1ob06424j. [DOI] [PubMed] [Google Scholar]
- 119.Li Q, Dong T, Liu X, Lei X. Journal of the American Chemical Society. 2013;135:4996–4999. doi: 10.1021/ja401989p. [DOI] [PubMed] [Google Scholar]
- 120.Karjalaninen K, Mäkelä O. European Journal of Immunology. 1976;6:88–93. doi: 10.1002/eji.1830060204. [DOI] [PubMed] [Google Scholar]
- 121.Dubrovska A, Kim C, Elliott J, Shen W, Kuo T-H, Koo D-I, Li C, Tuntland T, Chang J, Groessl T. ACS Chemical Biology. 2011;6:1223–1231. doi: 10.1021/cb200222s. [DOI] [PubMed] [Google Scholar]
- 122.McEnaney PJ, Parker CG, Zhang AX, Spiegel DA. ACS Chemical Biology. 2012;7:1139–1151. doi: 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garzoni C, Kelley WL. Trends in Microbiology. 2009;17:59–65. doi: 10.1016/j.tim.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 124.Hagberg L, Hull R, Hull S, Falkow S, Freter R, Edén CS. Infection and Immunity. 1983;40:265–272. doi: 10.1128/iai.40.1.265-272.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown SB, Brown EA, Walker I. The Lancet Oncology. 2004;5:497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]
- 126.Davis ME. Nature Reviews Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 127.Pastan I, Chaudhary V, FitzGerald DJ. Annual Review of Biochemistry. 1992;61:331–354. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- 128.O’Connor AE, Gallagher WM, Byrne AT. Photochemistry and Photobiology. 2009;85:1053–1074. doi: 10.1111/j.1751-1097.2009.00585.x. [DOI] [PubMed] [Google Scholar]
- 129.Moan J, BERG K. Photochemistry and photobiology. 1991;53:549–553. doi: 10.1111/j.1751-1097.1991.tb03669.x. [DOI] [PubMed] [Google Scholar]
- 130.Kharkwal GB, Sharma SK, Huang YY, Dai TH, Hamblin MR. Lasers in Surgery and Medicine. 2011;43:755–767. doi: 10.1002/lsm.21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sperandio F, Huang Y, Hamblin M. Recent Patents on Anti-infective Drug Discovery. 2013;8:108–120. doi: 10.2174/1574891x113089990012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wardlaw J, Sullivan T, Lux C, Austin F. The Veterinary Journal. 2012;192:374–377. doi: 10.1016/j.tvjl.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 133.Doane TL, Burda C. Chemical Society Reviews. 2012;41:2885–2911. doi: 10.1039/c2cs15260f. [DOI] [PubMed] [Google Scholar]
- 134.Rani D, Somasundaram VH, Nair S, Koyakutty M. Journal of the Indian Institute of Science. 2012;92:187–218. [Google Scholar]
- 135.Norman RS, Stone JW, Gole A, Murphy CJ, Sabo-Attwood TL. Nano Letters. 2008;8:302–306. doi: 10.1021/nl0727056. [DOI] [PubMed] [Google Scholar]
- 136.Humphries RM, Pollett S, Sakoulas G. Clinical Microbiology Reviews. 2013;26:759–780. doi: 10.1128/CMR.00030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Laughlin ST, Bertozzi CR. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12–17. doi: 10.1073/pnas.0811481106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hsu KL, Pilobello KT, Mahal LK. Nature Chemical Biology. 2006;2:153–157. doi: 10.1038/nchembio767. [DOI] [PubMed] [Google Scholar]
- 139.Andre G, Kulakauskas S, Chapot-Chartier MP, Navet B, Deghorain M, Bernard E, Hols P, Dufrene YF. Nature Communications. 2010;1:27. doi: 10.1038/ncomms1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pereira PM, Filipe SR, Tomasz A, Pinho MG. Antimicrobial Agents and Chemotherapy. 2007;51:3627–3633. doi: 10.1128/AAC.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kocaoglu O, Calvo RA, Sham LT, Cozy LM, Lanning BR, Francis S, Winkler ME, Kearns DB, Carlson EE. ACS Chemical Biology. 2012;7:1746–1753. doi: 10.1021/cb300329r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Backus KM, Boshoff HI, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE, Davis BG. Nature Chemical Biology. 2011;7:228–235. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hoerr V, Tuchscherr L, Hove J, Nippe N, Loser K, Glyvuk N, Tsytsyura Y, Holtkamp M, Sunderkotter C, Karst U, Klingauf J, Peters G, Loffler B, Faber C. BMC Biology. 2013;11:63. doi: 10.1186/1741-7007-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.White AG, Gray BD, Pak KY, Smith BD. Bioorganic & Medicinal Chemistry Letters. 2012;22:2833–2836. doi: 10.1016/j.bmcl.2012.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, Kwon IC, Jeong SY, Kim K. Angewandte Chemie-International Edition. 2012;51:11836–11840. doi: 10.1002/anie.201206703. [DOI] [PubMed] [Google Scholar]
- 146.Neves AA, Stockmann H, Harmston RR, Pryor HJ, Alam IS, Ireland-Zecchini H, Lewis DY, Lyons SK, Leeper FJ, Brindle KM. FASEB Journal. 2011;25:2528–2537. doi: 10.1096/fj.10-178590. [DOI] [PubMed] [Google Scholar]
- 147.Lemieux GA, de Graffenried CL, Bertozzi CR. Journal of the American Chemical Society. 2003;125:4708–4709. doi: 10.1021/ja029013y. [DOI] [PubMed] [Google Scholar]
- 148.Cohen AS, Dubikovskaya EA, Rush JS, Bertozzi CR. Journal of the American Chemical Society. 2010;132:8563–8565. doi: 10.1021/ja101766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jewett JC, Bertozzi CR. Organic Letters. 2011;13:5937–5939. doi: 10.1021/ol2025026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sivakumar K, Xie F, Cash BM, Long S, Barnhill HN, Wang Q. Organic Letters. 2004;6:4603–4606. doi: 10.1021/ol047955x. [DOI] [PubMed] [Google Scholar]
- 151.Shieh P, Hangauer MJ, Bertozzi CR. Journal of the American Chemical Society. 2012;134:17428–17431. doi: 10.1021/ja308203h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang J, Šečkutẹ J, Cole CM, Devaraj NK. Angewandte Chemie. 2012;124:7594–7597. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]