

Abstract
Rationale
Our previous study has shown that YAP plays a crucial role in the phenotypic modulation of vascular smooth muscle cells (SMCs) in response to arterial injury. However, the role of YAP in vascular SMC development is unknown.
Objective
The goal of this study was to investigate the functional role of YAP in cardiovascular development in mice and determine the mechanisms underlying YAP’s actions.
Methods and Results
YAP was deleted in cardiomyocytes and vascular SMCs by crossing YAP flox mice with SM22α-Cre transgenic mice. Cardiac/SMC-specific deletion of YAP directed by SM22α-Creresulted in perinatal lethality in mice due to profound cardiac defects including hypoplastic myocardium, membranous ventricular septal defect, and double outlet right ventricle. The cardiac/SMC-specific YAP knock-out mice also displayed severe vascular abnormalities including hypoplastic arterial wall, short/absent brachiocephalic artery and retro-esophageal right subclavian artery. Deletion of YAP in mouse vascular SMCs induced expression of a subset of cell cycle arrest genes including Gpr132. Silencing Gpr132 promoted SMC proliferation while over-expression of Gpr132 attenuated SMC growth by arresting cell cycle in G0/G1 phase, suggesting ablation of YAP induced impairment of SMC proliferation was mediated, at least in part, by induction of Gpr132 expression. Mechanistically, YAP recruited the epigenetic repressor, HDAC4 to suppress Gpr132 gene expression via an MCAT element in the Gpr132 gene.
Conclusions
YAP plays a critical role in cardiac/smooth muscle cell proliferation during cardiovascular development by epigenetically regulating expression of a set of cell cycle suppressors.
Keywords: Smooth muscle, defects, myocardium, vasculature, transcription factors, vascular smooth muscle, vascular biology, developmental biology
INTRODUCTION
Vascular wall assembly is critical in the process of the cardiovascular development. This developmental process requires a coordination of multiple cell types including endothelial and smooth muscle cells (SMCs). After establishment of a nascent capillary-like vascular network by endothelial cells, vascular SMC progenitors begin to invest the vessel wall through migration and proliferation. Meanwhile vascular SMCs acquire a unique array of differentiated contractile markers such as SM α-actin, calponin, Hic-51, SM myosin heavy chain, 130-kDa myosin light chain kinase (MLCK) and SM22α2. Defects of molecular pathways that control the migration, proliferation and differentiation of vascular SMCs will produce malformations of blood vessels and aberrant activation of signaling pathways in response to vascular diseases. Therefore, a better understanding of the mechanisms underlying SMC development is essential to not only advance our knowledge with vascular wall biology, but also for identifying genes whose defects cause congenital vascular defects or vascular wall diseases in humans2, 3
The Hippo pathway is crucial for controlling organ size and tumorigenesis4–6. The mammalian core components of Hippo pathway include Mst1/2, Lats1/2, signaling down-stream effector YAP and YAP binding partner TEAD family proteins. Upon stimulation, Mst1/2 phosphorylates down stream Lats1/2, which subsequently phosphorylates YAP and retains YAP in the cytoplasm. Unphosphorylated YAP translocates into the nucleus where it binds TEAD family proteins, to induce genes that promote cell growth and oncogenic transformation7. Recently, several studies showed that the Hippo-YAP pathway plays a critical role in mouse cardiac development and for basal heart homeostasis in adult mice. Cardiac-specific deletion of Hippo kinase Mst1/2, Mst kinase co-activator Salvador and Lats2 in mice resulted in over-grown hearts with elevated cardiomyocyte proliferation8. Consistently, over-expression of YAP in the mouse embryonic heart increased heart size by promoting cardiomyocyte proliferation, whereas inactivation of YAP in the mouse embryonic heart impeded cardiomyocyte proliferation, causing myocardial hypoplasia and embryonic lethality9, 10. Moreover, ablation of YAP in the postnatal mouse heart resulted in dilated cardiomyopathy and premature death11. Conversely, forced expression of YAP in the adult mouse heart stimulated cardiac regeneration and improved contractility after myocardial infarction12. Despite is significant progress in defining the importance of Hippo-YAP signaling in the mouse heart, the functional role of YAP in the vascular development remains elusive.
Our recent study showed that YAP plays a novel integrative role in smooth muscle phenotypic modulation by inhibiting smooth muscle-specific gene expression while promoting smooth muscle proliferation and migration in vitro and in vivo13. Specifically, knock-down of YAP expression in rodent carotid artery injury models attenuated injury-induced smooth muscle dedifferentiation and neointima formation13.
During vasculogenesis SMCs are highly proliferative and migratory, similar to the “synthetic” phenotype that SMCs exhibit in response to arterial injury. Indeed, at the molecular level, non-muscle myosin heavy chain IIB, which is highly expressed in embryonic SMCs during early vasculogenesis and turned off in mature vascular SMCs, is re-expressed in proliferating SMCs after arterial injury and in atherosclerosis14, 15. Due to the similarities between embryonic SMCs and phenotypically modified adult SMC, we sought to explore the function of YAP in vascular development in vivo using YAP conditional mutant mice in which YAP is specifically ablated in cardiomyocytes and vascular SMCs. We found that loss of YAP in murine cardiac muscle and vascular SMCs resulted in perinatal lethality. YAP conditional deletion mice exhibited severe vascular phenotypes including thinning of the vascular wall, retro-esophageal right subclavian artery (RE-RSA) and short/absent brachiocephalic artery. YAP conditional knockout mice also exhibited complex cardiac phenotypes including hypoplastic myocardial wall, membranous ventricular septal defects (VSD) and double outlet right ventricle (DORV) due to the transient Cre activation in embryonic mouse heart. Moreover, we found that YAP deletion had no effects on vascular smooth muscle differentiation but instead significantly attenuated SMC proliferation, at least in part, through up-regulation of the cell cycle arrest gene Gpr132. YAP recruited histone deacetylase HDAC4 to form a transcriptional repressor complex on the Gpr132 gene promoter, to repress Gpr132 expression. Collectively, this study not only suggests a crucial role of YAP in smooth muscle development, but also provides a novel mechanistic insight into YAP-mediated SMC proliferation.
METHODS
Cardiac/smooth muscle-specific YAP knock-out mice were generated by crossing YAP flox mice16 with SM22α-Cre transgenic mice17. Both YAP flox and SM22α-Cre mice were maintained in C57BL/6 strain background. Full materials and methods are detailed in the online Supplemental Material.
RESULTS
Conditional deletion of YAP in cardiac/SMCs in mice results in perinatal lethality
Early lethality observed in global YAP-null embryos precluded assessment of the function of YAP in vascular smooth muscle development18. In order to determine the role of YAP in cardiac/vascular SMC development, YAPF/F mice were crossed with transgenic mice expressing Cre recombinase under the control of the SM22α gene promoter. This resulted in deletion of YAP in embryonic heart and vascular SMCs17 (Online Figure IA). Progenies of SM22α-Cre+/YAPF/W mice intercrossed with YAPF/F mice exhibited the expected 25% Mendelian ratio from E10.5 to E15.5. Starting from E16.5 to postnatal day (P) 0, a significant reduction of SM22α-Cre+/YAPF/F mutants (KO) observed. From P1 to P14, only 1 mutant among 107 offspring survived but died on P21 (Online Table I). These data demonstrated that conditional deletion of YAP in embryonic mouse cardiac/vascular SMCs results in perinatal lethality. The P0 neonatal mutants died within hours after birth with marked cyanosis (Online Figure II), suggesting the perinatal lethality may be attributable to cardiovascular defects, as anticipated.
Specific ablation of YAP in cardiac/vascular SMCs results in cardiac defects that are associated with reduced proliferation
Although SM22α is well documented as a marker of smooth muscle, it is, like most other markers for smooth muscle lineage, transiently expressed in the heart between E8.5 to E12.5 in mice19. Therefore, in addition of vascular SMCs, we expect there will also be deletion of YAP in embryonic heart. Compared to littermate control embryos there was a significant decrease of immuno-blot and/or immunohistochemistry signal in cardiac and vascular SMCs of YAP mutants, although some YAP signal remained due to a possible incomplete deletion or/and mix of cell types in lysates used for western blotting (Online Figure IB and C). Consistent with previous studies demonstrating critical role of YAP in cardiac development in mice9, 10, histology analysis revealed that all of YAP mutant embryos and new born pups examined (100%, n=21/21, E14.5 to P0) displayed membranous ventricular septal defect (VSD) and double-outlet right ventricle (DORV) with hypoplastic myocardial wall (Figure 1). YAP mutants also exhibited significant thinning of right and left ventricular wall from E11.5 to P0 (Online Figure III), at least partially resulting from reduced cardiomyocyte proliferation as shown by decreased Ki67 and phospho-histone (PH) 3 positive cardiomyocytes (Online Figure IV A, B, C and D). Similarly, the VSD phenotype could be a result of hypoplasia of septal cardiomyocytes as there was significantly less Ki67 and PH3 staining in this region of heart (Online Figure IV E and F).
Figure 1. Cardiac pathology in conditional YAP deletion mutants.
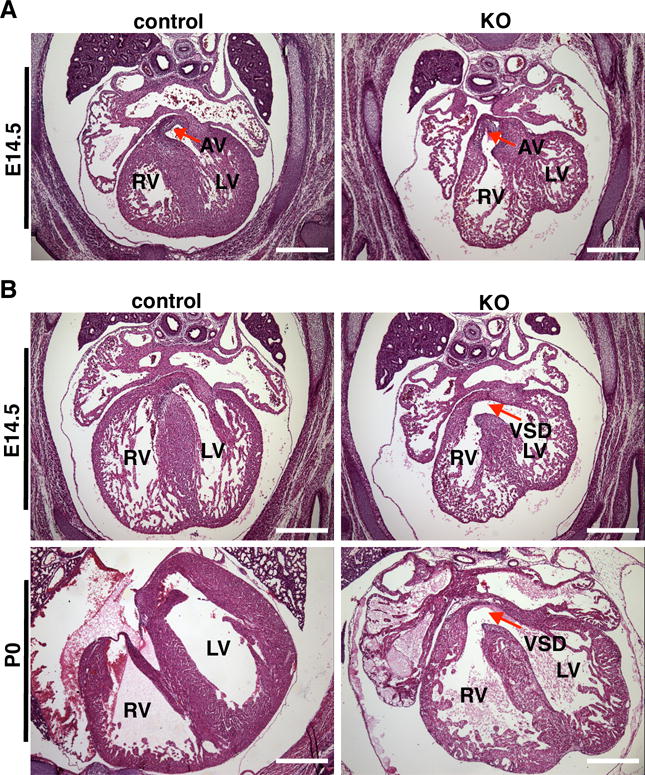
A. Example of an E14.5 YAP knock-out heart that demonstrates double outlet right ventricle with the aorta rising from the right ventricle (RV) whereas the aorta derives from left ventricle (LV) in control embryos. AV: aortic valve (arrow). Scale bar: 500um. B. Representative HE stained images showing membranous ventricular septal defect (VSD, arrow) in the hearts of E14.5 and postnatal day 0 (P0) YAP knock-out mice. Scale bar: 500um.
Specific knock-out of YAP in cardiac/vascular SMCs leads to vascular morphological defects, thinning of vessel walls and aneurysms
Approximately 33% (n=8 of 24) of YAP knock-out mice displayed retro-esophageal right subclavian artery (RE-RSA) whereby the RSA abnormally arose from the aortic arch in E14.5 and E18.5 mutant embryos (Figure 2A). Of note, the abnormal origin and route of the RSA caused the trachea to be off its normal midline location thereby “squeezing” the esophagus in the E18.5 mutant embryos (Figure 2A). 17% (n=4 of 24) of YAP mutants displayed short (Figure 2B) or absent (Figure 2C) brachiocephalic artery in the E18.5 or E13.5 mutant embryos respectively. HE staining of the left carotid artery and thoracic artery revealed a marked thinning of the arterial wall and aneurismal enlargement of vessel lumen in E15.5 YAP mutant embryos (Figure 3). Similar changes were observed in E13.5 to P0 mutant embryos (data not shown).
Figure 2. Conditional deletion of YAP in cardiac and vascular SMCs leads to vascular abnormalities.
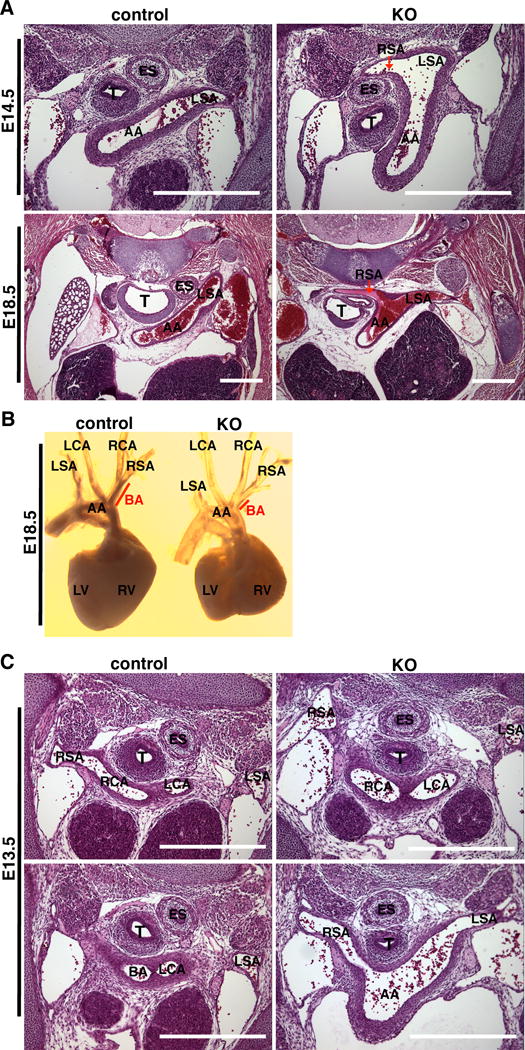
A. Representative HE staining sections to show the retro-esophageal right subclavian artery phenotype in E14.5 and E18.5 YAP mutants in which the right subclavian artery (RSA) abnormally branches from the aortic arch (AA) and travels behind of esophagus (ES) and trachea (T). LSA: left subclavian artery. Scale bar: 500um. B. Posterior view of dissected control and YAP mutant heart and outflow tract showing a short brachiocephalic artery (BA) and abnormally shaped heart in the KO embryo. C. HE staining of serial sections of E13.5 KO and control embryos. In the mutant embryo the branchiocephalic artery (BA) is absent and the right subclavian artery (RSA) directly arises from the aortic arch (AA) whereas in control embryos the right subclavian artery (RSA) and right common carotid artery (RCA) form BA. Scale bar: 500um.
Figure 3. YAP KO mice have thin arterial walls and dilated arteries.
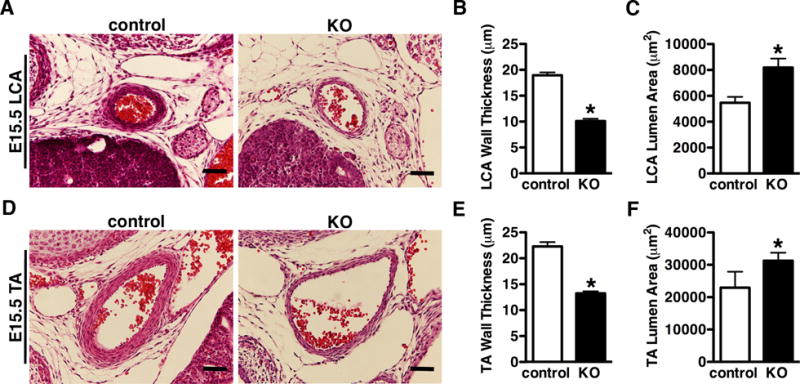
A. HE stained images of the left carotid artery (LCA) or thoracic artery (TA) (D) from E15.5 control and YAP KO embryos. The thickness of medium wall and size of lumen area were quantified and plotted as depicted in panel “B”, “C”, and “E”, “F”, respectively. Data were collected from 3 control and 5 KO embryos. *p<0.05. Scale bar: 50um.
Deletion of YAP in vascular SMCs impairs SMC proliferation in a cell-autonomous manner
As specific deletion of YAP in vascular SMCs resulted in a thinning of the arterial wall, we next sought to determine if this was due to an impairment of SMC proliferation. SM α-actin and Ki67 co-immunostaining revealed, YAP KO mutants exhibited significantly lower SMC numbers and decreased smooth muscle proliferation in the left carotid artery (Figure 4A, B and C; Online Figure V B). To further investigate the effect of YAP deficiency on smooth muscle proliferation, SMCs were isolated from the dorsal aorta of E15.5 mutant and control littermate embryos. First we confirmed >99% SMC purity in the culture by SM MHC staining (Online Figure VI). Next Brdu incorporation assay was carried out to assess SMC proliferation in vitro. Data from this experiment demonstrated there was significantly less BrdU incorporation into SMCs derived from YAP specific-KO embryos compared to control littermates (Figure 4D and E). Furthermore, deletion of YAP in mouse vascular SMCs significantly impaired cell growth during in vitro cell culture (Figure 4F). Taken together, these data demonstrate that YAP is required for vascular SMC proliferation in a cell-autonomous manner.
Figure 4. Deletion of YAP inhibits vascular SMC growth in a cell-autonomous manner.
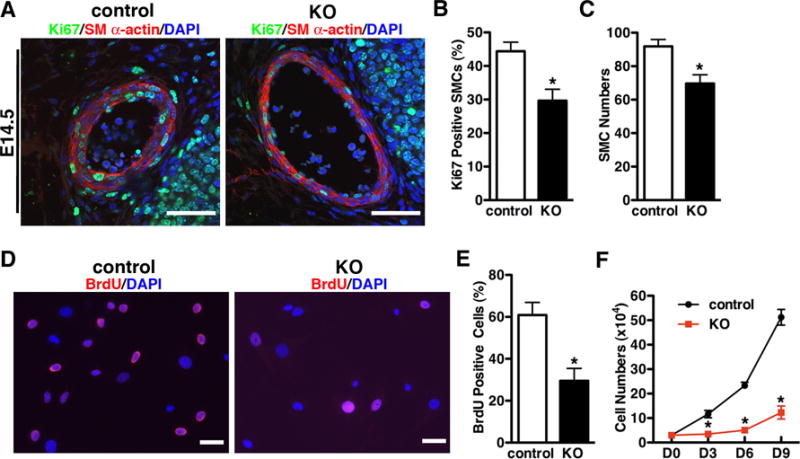
A. Sections were prepared from left carotid artery of E14.5 control or YAP KO embryos and then immuno-stained with antibodies for the smooth muscle differentiation marker SM α-actin (red) and the cell proliferative marker, Ki67 (green) as indicated. Samples were counter-stained with DAPI (blue) to visualize nuclei. The percentage of Ki67 positive SMCs and numbers of SM α-actin positive cells in the arterial wall were quantitated and plotted in panel “B” and “C”, respectively. N=3 embryos per genotype. *p<0.05. Scale bar: 50um. D. Vascular SMCs were prepared from E15.5 control or YAP KO dorsal aorta and treated with BrdU for 16 hours and immune-stained with anti-BrdU antibody for BrdU positive cells (BrdU, red; DAPI, blue). The fraction of BrDU positive cells was quantitated and shown “E”. Scale bar: 50um. F. Vascular SMCs from E15.5 control or YAP KO dorsal aorta were plated at equal density at day (D) 0 and then counted at D3, D6, D9 as indicated. *p<0.05.
Deletion of YAP in vascular SMCs specifically enhances expression of a subset of cell cycle arrest genes without affecting SMC differentiation
Previously we have shown that knock-down of YAP in cultured SMCs and in rat balloon injury model significantly up-regulates smooth muscle marker expression13. However, immunofluorescence staining (Figure 4A, Online Figure V A and C), qRT-PCR (Figure 5A) and western blotting (Figure 5B and C) did not show any significant change in smooth muscle differentiation markers in YAP mutant aortic arteries. These data suggest YAP is dispensable for vascular SMC differentiation whereas is critical for SMC proliferation during development in vivo. No obvious differences in SMC apoptosis were observed between mutant and control embryos as determined by TUNEL staining (data not shown). To explore the mechanism by which deletion of YAP impair SMC proliferation, a PCR-based array was performed to analyze expression of cell cycle related genes in aortic tissues from E15.5 YAP KO and control littermates (Online Table II). qRT-PCR and western blotting were then utilized to confirm the array results (Figure 5). A subset of cell cycle arrest genes including Gadd45α, Slfn1, Gpr132, Trp63 and Sfn were significantly induced in YAP KO aortic tissues, whereas there were no changes detected in the cell cycle genes that promote cell proliferation including Ccnd1, Ccna1 and Foxm1 etc. (Figure 5A, B and C). We chose Gpr132 for further characterization as this gene was the most significantly up-regulated in YAP mutants and its function is unknown in vascular SMCs. To confirm the effects of YAP on Gpr132 expression, retrovirus expressing YAP was transduced into rat primary SMCs and western blot was performed to examine endogenous Gpr132 expression. Data from this experiment revealed that overexpression of YAP significantly suppresses Gpr132 expression (Figure 5D). Taken together, data from these in vivo loss-of-function and in vitro gain-of-function assays demonstrat that YAP is a potent repressor for expression of cell cycle arrest gene Gpr132.
Figure 5. Deletion of YAP induces a subset of cell cycle arrest gene expression without affecting expression of SM-specific genes.
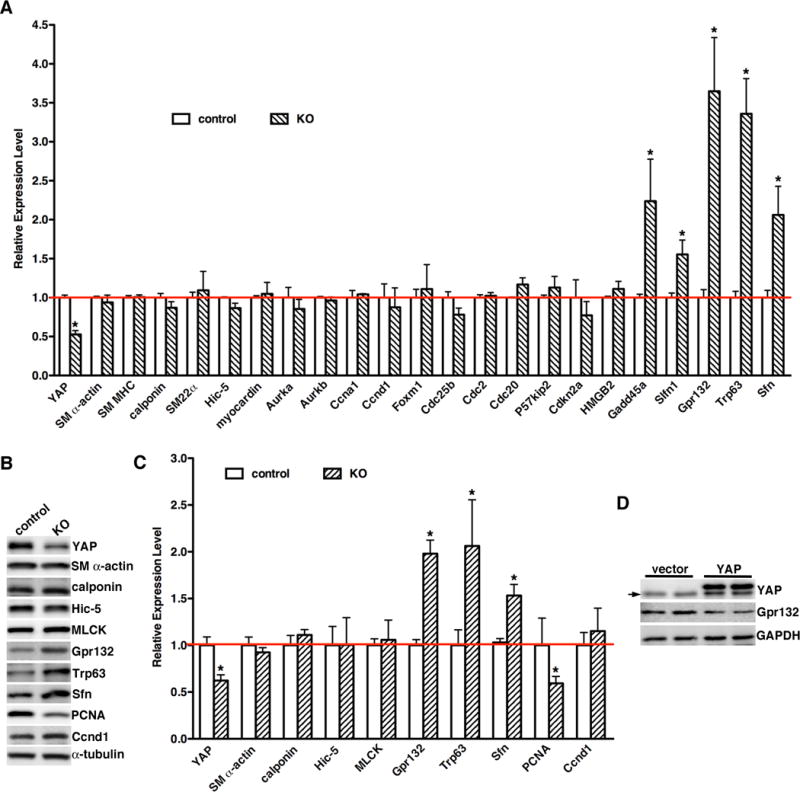
A. Total RNA was harvested from E15.5 control (open bars) YAP KO (hatched bars) dorsal aorta and qRT-PCR was performed to assess gene expression as indicated. The level of gene expression in control vessels was set to 1 (red line). N=5–7 embryos per genotype. *p<0.05. B. A representative western blot using protein lysates from E15.5 control or YAP KO dorsal aorta tissue is shown. C. Immunoblot signals were normalized to α-tubulin loading control then expressed relative to signals from control aortae (set to 1, red line). N=4 embryos per genotype. *p<0.05. D. Empty retrovirus or retrovirus encoding YAP was transduced into rat primary aortic SMCs and then Gpr132 expression was analyzed by western blotting. An arrow points to the endogenous YAP band.
Gpr132 suppresses SMC proliferation
Data described above indicated that ablation of YAP in vascular SMCs induces cell cycle arrest gene Gpr132 expression. We first sought to determine if decreasing Gpr132 would mimic the effects of YAP to promote SMC proliferation13. Knock-down of Gpr132 in rat vascular SMCs resulted in a significant increase of BrdU incorporation and SMC growth (Figure 6 A, B and C). Since the Gpr132 was induced after deletion of YAP in mice, we next sought to examine whether over-expression of Gpr132 can attenuate SMC proliferation. Cell cycle analysis demonstrated that Gpr132 over-expressing SMCs had a higher proportion of cells in G0/G1 phase but a lower proportion in G2/M phase (Figure 6D), indicating a cell cycle arrest in G0/G1 phase. This cell cycle retardation led to a significant decrease cell growth in the SMCs over-expressing Gpr132 (Figure 6E). Furthermore, silencing Gpr132 rescued the decrease in proliferation observed in YAP KO SMCs while promoted the growth of WT SMCs (Figure 6F). Taken together, these data suggest that YAPdeletion induced impairment of SMC proliferation is, at least in part, mediated through induction of the cell cycle arrest gene Gpr132.
Figure 6. Gpr132 suppresses SMC growth.
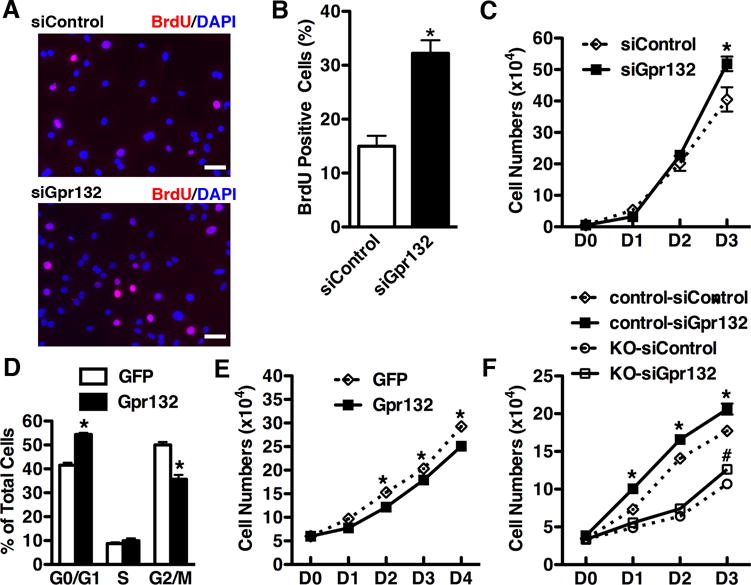
A. After transfected with control or Gpr132 siRNA duplexes, rat primary aortic SMCs were incubated with BrdU for 24 hours and then immuno-staining was performed to BrdU positive SMCs by using an anti-BrdU antibody (red) and co-stained with DAPI (blue) for nuclei. Scale bar: 50um. B. The fraction of BrdU positive SMCs was counted and plotted. C. After silencing Gpr132, rat primary aortic SMCs were seeded at equal density at day (D) 0 and counted at each time point as indicated. N=3. *p<0.05. D. After transduction with Gpr132 or control GFP adenovirus, rat primary aortic SMCs were harvested for propidium iodide staining to analyze cell cycle by a flow cytometry. *p<0.05. E. Following infection with Gpr132 or GFP control adenovirus, rat primary aortic SMCs were seeded at equal density and counted at each time point indicated. *p<0.05. F. E15.5 dorsal aortic SMCs from YAP KO or control embryos were transfected with scrambled control or Gpr132 silencing duplexes. After 24 hours cells then were plated and counted at each time point as indicated. *p<0.05, control-siControl versus control-siGpr132; #p<0.05, KO-siControl versus KO-siGpr132.
YAP inhibits Gpr132 expression via binding to Gpr132 gene promoter
YAP promoted cardiomyocyte proliferation via interaction with TEAD19, thus we next sought to explore whether YAP suppresses Gpr132 expression through TEAD1. In support of this possibility, analysis of the mouse Gpr132 revealed an evolutionally conserved TEAD binding MCAT CATN(T/C)(T/C) element inthe first intron of the Gpr132 gene (Figure 7A). To experimentally validate that YAP regulates Gpr132 through the MCAT element, we generated WT or MCAT mutation Gpr132 luciferase reporter genes and dual luciferase assays were performed. Over-expression of YAP slightly, although significantly, inhibited Gpr132 reporter gene activity by 20–30% but not MCAT mutation reporter (Figure 7B). As YAP has a moderate suppressing effects on Gpr132 promoter activity but knock-out of YAP led to more significant Gpr132 expression in vivo (Figure 5), we reasoned that endogenous YAP may cooperate with repressors to down-regulate Gpr132 expression. Previous study has shown that selective inhibition of class I and II HDACs inhibits SMC proliferation20, 21. As, HDAC4 was not only well known for gene transcriptional inactivation but also highly expressed in vascular SMCs and reported to regulate SMC proliferation21, 22. we next determined whether HDAC4 is involved in YAP-mediated Gpr132 gene promoter repression. Luciferase reporter assays demonstrated that indeed, HDAC4 augmented YAP-mediated repression of Gpr132 promoter activity compared to YAP alone (Figure 7B). In contrast, the MCAT mutant Gpr132 promoter was completely refractory to YAP or HDAC4 mediated repression (Figure 7B). Moreover, data from ChIP assays unveiled an approximate 3–4 fold enrichment of YAP over the MCAT region but not exon 3 of Gpr132 gene, suggesting YAP specifically binds to the MCAT region of the Gpr132 gene (Figure 7C). Furthermore, over-expression of YAP promoted HDAC4 binding to MCAT region of Gpr132 gene (Figure 7D). As these data suggested that YAP recruits HDAC4 to inhibit Gpr132 gene promoter activity, we sought to determine whether YAP can bind to HDAC4 to form a complex in vivo. YAP was transduced into rat primary aortic SMCs and endogenous HDAC4 was immunoprecipitated with anti-HDAC4 antibody. Western blotting of the HDAC4 immunoprecipitates revealed the presence of YAP and TEAD1 together with HDAC4 in the vascular SMCs (Figure 7E). Moreover, data from sequential ChIP assays revealed that YAP and HDAC4 co-occupied over the MCAT region within the Gpr132 gene in the primary vascular SMCs (Figure 7F). Collectively, these data demonstrate that YAP recruits HDAC4 binding to the MCAT element within the Gpr132 gene to repress Gpr132 expression thereby rendering cycle arrest. Conversely, knock-out of YAP liberates the HDAC4 mediated inhibitory effects on Gpr132 gene expression, resulting in a decreased SMC growth due to the cell cycle arrest and ultimately leading to a hypoplastic arterial wall phenotype (Figure 7G).
Figure 7. YAP recruits HDAC4 to an MCAT element to suppress Gpr132 promoter activity.
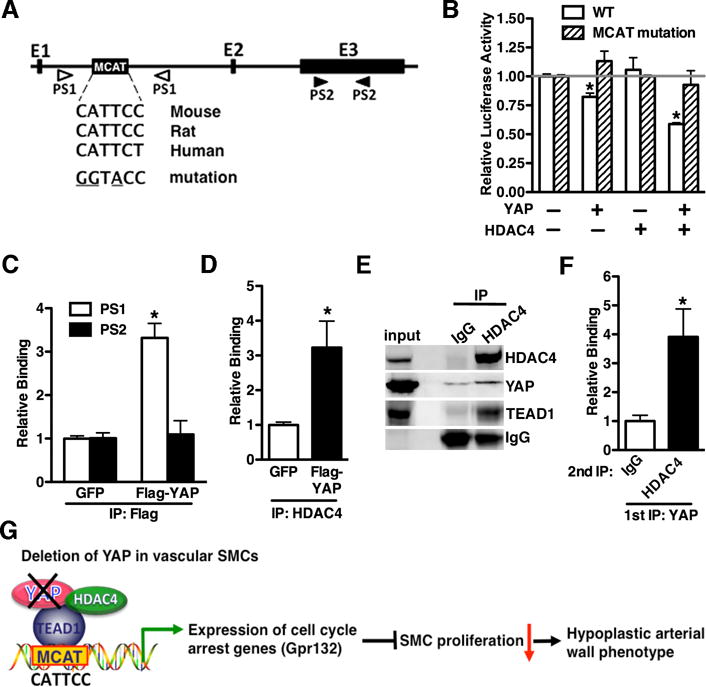
A. Schematic diagram of the Gpr132 gene structure showing a conserved MCAT element within intron 1. A wild-type or MCAT mutant reporter (mutated nucleotide in MCAT element is underscored) containing a 1.5k bp fragment within Gpr132 gene intron 1 was generated into pGL2E luciferase vector. The location of 2 setts of primers (PS), localized either in intron 1 or exon (E) 3 used for ChIP assays in “C”, is shown. B. Wild-type or MCAT mutant Gpr132 promoter reporter genes were transfected into PAC1 SMCs together with YAP in the absence or presence of HDAC4 expression plasmid. Values are presented as relative luciferase activity compared with control vector (set to 1) and are the mean ± S.E. of 6 samples. C. Adenovirus expressing GFP or Flag tagged YAP was transduced into rat primary rat aortic SMCs and then chromatin was harvested for immunoprecipitation with anti-Flag antibody. The precipitated DNA was amplified by a real time PCR using Gpr132 gene-specific primers that span the intronic MCAT region (PS1) or exon 3 (PS2) as depicted in “A”. The YAP binding is expressed relative to GFP control sample (set to 1). *p<0.05. D. ChIP assay was performed as described in “C” except anti-HDAC4 antibody was utilized to immunoprecipitate endogenous HDAC4 and real time PCR was performed using PS1 primers. *p<0.05. E. Adenovirus expressing YAP was transduced into rat aortic SMCs and endogenous HDAC4 was immune-precipitated by using anti-HDAC4 antibody. Western blot was then performed by using the indicated antibodies. A species-matched IgG served as control. F. Chromatin was harvested from SMCs over-expressing. The chromatin was immunoprecipitated first with anti-YAP antibody, eluted and then immunoprecipitated with either IgG or anti-HDAC4 antibody. Immunoprecipitated DNA was then analyzed by quantitative PCR using PS1 primers. *p<0.05. G. Schematic diagram depicting the findings of this study.
DISCUSSION
This study provides the first evidence demonstrating a critical role for YAP during smooth muscle development through regulating SMC proliferation. We found that loss of YAP murine cardiovascular system results in perinatal lethality (Online Figure II and Online Table I). YAP conditional knockout mice exhibit severe vascular abnormalities including thinning vascular walls, RE-RSA and short/absent brachiocephalic arteries (Figure 2). Consistent with attenuated SMC proliferation during development after loss of YAP, we have previously shown that YAP promotes SMC proliferation following vascular injury13. Although in the current study we found YAP deletion had no effects on vascular smooth muscle differentiation during embryonic development (Figure 4, 5 and Online Figure V), we previously found that YAP suppresses expression of smooth muscle differentiation genes in adult smooth muscle tissues and cells13. One possible explanation for this difference could be that there is an incomplete deletion of YAP in vascular SMCs mediated by SM22α promoter driven cre transgene. In support of this notion, data from qRT-PCR (Figure 5A), western blotting (Figure 5B and C) and immunohistochemistry staining (Online Figure IB) showed there is a significant amount of residual YAP in mutant vascular SMCs. Alternatively it is possible that programs governing smooth muscle development and those governing smooth muscle de-differentiation in response to arterial injury in adult mice are distinct. Although there are some common mechanisms shared by both processes, the transient phenotypic switch associated with repair of vascular injury does not exactly recapitulate regulatory programs involved in controlling SMC differentiation during vascular SMC development23. In support of this we found Gpr132 expression was down-regulated after arterial injury in concomitant with up-regulation of YAP. However, knock-down of YAP in the arterial injury model did not reverse the Gpr132 expression (data not shown), suggesting there are distinct regulatory mechanisms for Gpr132 expression in either YAP-dependent or -independent manner.
SM22α-Cre transgenic mice generate highly efficient Cre-mediated recombination as early as E9.5 in cardiomyocytes, mesoderm and neural crest-derived SMCs17, 24, 25. Neural crest cells significantly contribute to craniofacial and cardiovascular development by migrating through the pharyngeal arches. Cardiac neural crest, a specific population of neural crest cells, gives rise to the third, fourth and sixth pharyngeal arches. Among them, the right fourth pharyngeal artery forms the base of the right subclavianartery26. Previous studies have shown that disruption of the migration, proliferation and differentiation of neural crest-derived SMCs in pharyngeal arteries will cause a variety of congenital outflow tract defects26. The observed specific defects such as RE-RSA and short/absent brachiocephalic artery in SM22α-Cre+/YAPF/F mutants suggest that YAP is specifically required for the right fourth pharyngeal artery to form a proper pattern of right subclavian artery. Further studies are needed to specifically delete YAP in neural crest to determine the role of YAP in neural crest mediated pharyngeal artery development. Furthermore, due to the embryonic and perinatal lethality of the YAP mutant mice directed by the SM22α promoter driven cre transgene, further study on the role of YAP in adult SMCs will require use of an inducible KO model.
In this study we identified a novel mechanism whereby YAP promotes smooth muscle proliferation through inhibiting expression of a sub-set of cell cycle arrest genes. Previous studies have shown that YAP stimulates cardiomyocyte proliferation through induction of a number of cell-cycle promoting genes including Aurka, Aurkb, Cdc2 and Cdc208, 9. Furthermore, our recent study showed that YAP can induce cultured SMC proliferation in vitro by induction of Ccnd113. In the current study, however, we showed that none of these cell cycle promoting genes were altered in the vascular SMCs of YAP mice (Figure 5A). Instead, we observed increased expression of a subset of cell cycle arrest genes including Gadd45α, Slfn1, Gpr132, Trp63 and Sfn (Figure 5A and B). Among these genes identified, Gadd45α and Trp63 have been shown to be involved in PDGF-BB and estrogen mediated smooth muscle growth, respectively27, 28. Thus far no studies have been shown that Gpr132 plays a role in SMC proliferation. In this study we found forced expression of YAP specifically suppressed Gpr132 expression (Figure 5 D) but not other cell cycle arrest genes (data not shown). Furthermore we found that silencing Gpr132 promoted vascular SMC growth (Figure 6A, B and C), a result similar to that seen following over-expression of YAP in vascular SMCs13. Consistent with previous report showing ectopic expression of Gpr132 leads to a growth inhibition29, we found over-expression of Gpr132 attenuated vascular SMC growth by arresting the cell cycle at G0/G1 phase (Figure 6D, E and F). These data suggest that of YAP induced vascular hypoplasia, at least in part, is through up-regulation of cell cycle arrest gene Gpr132.
Gpr132, originally identified as a G protein-coupled receptor for which lysophosphatidylcholine (LPC) is a high affinity ligand, belongs to a newly defined lysophospholipid receptor subfamily30. Previous studies demonstrated that Gpr132 differentially couples to multiple G proteins including Gαs, Gαq, and Gα13, and stimulates intracellular Ca2+ transients and extracellular signal-related kinase (ERK) phosphorylation31. Further studies are wanted to determine the role of LPC ligand and dissect the down-stream signaling of Gpr132 in the SMC proliferation during smooth muscle development.
In addition to the Gpr132 identified from the array, we found that several other cell cycle arrest genes such as Gadd45α and Trp63 were up-regulated in the YAP KO arteries. Mechanistically, YAP recruits histone deacetylase HDAC4 to an MCAT element to form a transcriptional repressor complex with TEAD1 on the Gpr132 gene promoter and the MCAT element within Gpr132 gene promoter is indispensible to confer the YAP inhibitory effect (Figure 7). By a bioinformatics search we found several putative consensus MCAT elements within the Gadd45α and Trp63 gene promoters (data not shown). Thus the hypoplastic artery phenotype observed in YAP KO mice is likely due to the combined effects from these cell cycle arrest genes through a similar MCAT-dependent mechanism. Previous studies demonstrated that HDAC2 and HDAC5 play important roles in the smooth muscle differentiation marker SM α-actin expression32, 33, but we found HDAC2 and 5 have no effects on Gpr132 gene promoter activity (data not shown). This would suggest HDAC2 and HDAC5 are critical for SMC differentiation by regulating SM-specific gene SM α-actin expression whereas HDAC4 is important for SMC proliferation by regulating Gpr132 expression. In support of this notion, Usui et al reported recently HDAC4 promoted neointimal hyperplasia through stimulating proliferation of vascular SMCs21. Taken together, these studies suggested HDAC family proteins play distinct roles in smooth muscle differentiation or proliferation.
In summary, this study not only suggests a crucial role of YAP in vascular smooth muscle development in mice, but also identifies a novel mechanism through which YAP mediates vascular SMC proliferation.
Supplementary Material
Novelty and Significance.
What Is Known?
Vascular SMCs are major components of blood vessels and contribute to the maintenance of vascular homeostasis.
YAP, a downstream effector of Hippo signaling, is critical for organ size control and tumorigenesis.
YAP promotes SMC proliferation and migration, thereby mediating neointima formation after arterial injury. However, the role of YAP in vascular SMC development is unknown.
What New Information Does This Article Contribute?
Deletion of YAP in vascular SMCs during mouse embryogenesis leads to perinatal lethality and severe vascular defects associated with impaired proliferation of SMCs.
Deletion of YAP attenuates SMC proliferation through induction of the cell cycle arrest genes by recruiting the epigenetic repressor HDAC4.
YAP is critical for vascular development by promoting SMC proliferation.
YAP is known for controlling organ size and tumor formation by promoting cell growth. Previously, we found that YAP plays a crucial role in the phenotypic switch of vascular SMCs in response to arterial injury. However, the role of YAP in vascular SMC development is unknown. By utilizing a genetic approach to inactivate YAP in vascular SMCs during mouse embryogenesis, we demonstrate that YAP is critical for vascular SMC development. SMC-specific deletion of YAP results in profound vascular abnormalities, including thin arterial wall, due to the impaired SMC proliferation. In contrast to previous studies showing YAP activates cell growth by induction of cell cycle promoting gene expression, we found that YAP promotes SMC growth, by repression of cell cycle arrest gene expression through a novel mechanism by which YAP recruits the epigenetic repressor HDAC4. These results suggest that YAP plays a critical role in cardiovascular development by regulating cell cycle progression. Therefore, these findings could provide the molecular basis for a potential diagnostic strategy for congenital cardiovascular disorders in humans.
Acknowledgments
We thank Dr. Paul Herring for a critical reading of the manuscript and Dr. DJ Pan for sharing the YAP flox mice.
SOURCES OF FUNDING
The project described was supported by a grant (R01HL109605) from the National Heart, Lung, and Blood Institute, NIH to J. Z.
Nonstandard Abbreviations and Acronyms
- YAP
yes associated protein
- SMCs
smooth muscle cells
- SM
smooth muscle
- Gpr132
G-protein coupled receptor 132
- HDAC4
histone deacetylase 4
- MCAT
muscle CAT element
- Hic-5
hydrogen peroxide-inducible clone 5
- MLCK
myosin light chain kinase
- Mst1/2
mammalian sterile 20-like 1 or 2
- Lats1/2
large tumor suppressor 1 or 2
- TEAD1
TEA domain family member 1
- RE-RSA
retro-esophageal right subclavian artery
- E
embryonic day
- P
postnatal day
- KO
Knock-out
- VSD
ventricular septal defect
- DORV
double-outlet right ventricle
- PH3
phosphor-histone 3
- SM MHC
smooth muscle myosin heavy chain
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- GADD45α
growth arrest and DNA-damage-inducible protein alpha
- Slfn1
schlafen 1
- Trp63
transformation related protein 63
- Sfn
Stratifin
- PDGF-BB
platelet-derived growth factor BB
- LPC
lysophosphatidylcholine
- ERK
extracellular signal-related kinase
Footnotes
DISCLOSURES
None
References
- 1.Wang X, Hu G, Betts C, Harmon EY, Keller RS, Van De Water L, Zhou J. Transforming growth factor-beta1-induced transcript 1 protein, a novel marker for smooth muscle contractile phenotype, is regulated by serum response factor/myocardin protein. J Biol Chem. 2011;286:41589–41599. doi: 10.1074/jbc.M111.250878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hungerford JE, Little CD. Developmental biology of the vascular smooth muscle cell: Building a multilayered vessel wall. Journal of vascular research. 1999;36:2–27. doi: 10.1159/000025622. [DOI] [PubMed] [Google Scholar]
- 3.Majesky MW, Dong XR, Regan JN, Hoglund VJ. Vascular smooth muscle progenitor cells: Building and repairing blood vessels. Circ Res. 2011;108:365–377. doi: 10.1161/CIRCRESAHA.110.223800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B, Li L, Lei Q, Guan KL. The hippo-yap pathway in organ size control and tumorigenesis: An updated version. Genes & development. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B, Lei QY, Guan KL. The hippo-yap pathway: New connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20:638–646. doi: 10.1016/j.ceb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT. Yap1, the nuclear target of hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–2399. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by yap governs cardiomyocyte proliferation and embryonic heart size. Science signaling. 2011;4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D, Sadoshima J. Yes-associated protein isoform 1 (yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288:3977–3988. doi: 10.1074/jbc.M112.436311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN. Hippo pathway effector yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–13844. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Hu G, Gao X, Wang Y, Zhang W, Harmon EY, Zhi X, Xu Z, Lennartz MR, Barroso M, Trebak M, Chen C, Zhou J. The induction of yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler Thromb Vasc Biol. 2012;32:2662–2669. doi: 10.1161/ATVBAHA.112.254730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuro-o M, Nagai R, Nakahara K, Katoh H, Tsai RC, Tsuchimochi H, Yazaki Y, Ohkubo A, Takaku F. Cdna cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J Biol Chem. 1991;266:3768–3773. [PubMed] [Google Scholar]
- 15.Majesky MW, Giachelli CM, Reidy MA, Schwartz SM. Rat carotid neointimal smooth muscle cells reexpress a developmentally regulated mrna phenotype during repair of arterial injury. Circ Res. 1992;71:759–768. doi: 10.1161/01.res.71.4.759. [DOI] [PubMed] [Google Scholar]
- 16.Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. The merlin/nf2 tumor suppressor functions through the yap oncoprotein to regulate tissue homeostasis in mammals. Developmental cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-a prevents the acute but not chronic effects of anp on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG, Magnuson TR, O’Neal W, Milgram SL. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of yap65. Mol Cell Biol. 2006;26:77–87. doi: 10.1128/MCB.26.1.77-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Miano JM, Cserjesi P, Olson EN. Sm22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78:188–195. doi: 10.1161/01.res.78.2.188. [DOI] [PubMed] [Google Scholar]
- 20.Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31:851–860. doi: 10.1161/ATVBAHA.110.221952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Usui T, Morita T, Okada M, Yamawaki H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.113.01843. In press. [DOI] [PubMed] [Google Scholar]
- 22.Ginnan R, Sun LY, Schwarz JJ, Singer HA. Mef2 is regulated by camkiidelta2 and a hdac4-hdac5 heterodimer in vascular smooth muscle cells. The Biochemical journal. 2012;444:105–114. doi: 10.1042/BJ20120152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 24.Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial brg1 represses adamts1 to maintain the microenvironment for myocardial morphogenesis. Developmental cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in sm22alpha-cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- 26.Stoller JZ, Epstein JA. Cardiac neural crest. Seminars in cell & developmental biology. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Kettenhofen R, Hoppe J, Eberhard G, Seul C, Ko Y, Sachinidis A. Regulation of gadd45a mrna expression in vascular smooth muscle under growth and stress conditions. Cellular signalling. 2001;13:787–799. doi: 10.1016/s0898-6568(01)00198-x. [DOI] [PubMed] [Google Scholar]
- 28.Zhao J, Imbrie GA, Baur WE, Iyer LK, Aronovitz MJ, Kershaw TB, Haselmann GM, Lu Q, Karas RH. Estrogen receptor-mediated regulation of microrna inhibits proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2013;33:257–265. doi: 10.1161/ATVBAHA.112.300200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weng Z, Fluckiger AC, Nisitani S, Wahl MI, Le LQ, Hunter CA, Fernal AA, Le Beau MM, Witte ON. A DNA damage and stress inducible g protein-coupled receptor blocks cells in g2/m. Proc Natl Acad Sci U S A. 1998;95:12334–12339. doi: 10.1073/pnas.95.21.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin P, Ye RD. The lysophospholipid receptor g2a activates a specific combination of g proteins and promotes apoptosis. J Biol Chem. 2003;278:14379–14386. doi: 10.1074/jbc.M209101200. [DOI] [PubMed] [Google Scholar]
- 31.Kabarowski JH, Zhu K, Le LQ, Witte ON, Xu Y. Lysophosphatidylcholine as a ligand for the immunoregulatory receptor g2a. Science. 2001;293:702–705. doi: 10.1126/science.1061781. [DOI] [PubMed] [Google Scholar]
- 32.Shang Y, Yoshida T, Amendt BA, Martin JF, Owens GK. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. The Journal of cell biology. 2008;181:461–473. doi: 10.1083/jcb.200711145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida T, Gan Q, Shang Y, Owens GK. Platelet-derived growth factor-bb represses smooth muscle cell marker genes via changes in binding of mkl factors and histone deacetylases to their promoters. American journal of physiology. Cell physiology. 2007;292:C886–895. doi: 10.1152/ajpcell.00449.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.