

Abstract
The availability of enantiomerically enriched carbonyl-containing compounds is essential to the synthesis of biologically active molecules. Since catalytic enantioselective conjugate addition (ECA) reactions directly generate the latter valuable class of molecules, the design and development of such protocols represents a compelling objective in modern chemistry. Herein, we disclose the first solution to the problem of ECA of alkenyl groups to acyclic trisubstituted enones, an advance achieved by adopting an easily modifiable and fully catalytic approach. The requisite alkenylaluminum reagents are synthesized with exceptional site- and/or stereoselectivity by a Ni-catalyzed hydroalumination process, and the necessary enones are prepared through a site- and stereoselective zirconocene-catalyzed carboalumination/acylation reaction. The all-catalytic procedure is complete within four hours, furnishing the desired products in up to 77% overall yield and 99:1 enantiomeric ratio.
Keywords: carboalumination, conjugate additions, copper, N-heterocyclic carbenes, quaternary carbon center
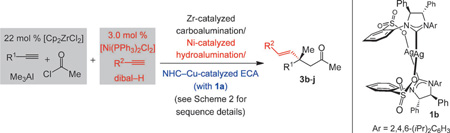
The significance of enantiomerically enriched carbonyl-containing compounds to the availability of biologically active molecules places considerable value on transformations such as catalytic enantioselective conjugate addition (ECA).[1, 2] While progress has been made in the development of efficient ECA protocols, notable shortcomings remain unaddressed. Among the underdeveloped approaches are those that involve the less reactive acyclic α,β-unsaturated carbonyl compounds and furnish products with all-carbon-substituted quaternary stereogenic centers.[3, 4] Of the limited number of reported instances,[5] only a few pertain to linear substrates, and most involve highly activated electrophiles (e.g., nitroalkenes or Meldrum’s acid derivatives).[6,7] As far as we are aware, none of the existing approaches deal with the addition of alkenyl units, readily functionalizable moieties that allow access to a variety of useful enantiomerically enriched derivatives.[8] Here, we disclose the first examples of catalytic ECA of alkenyl metal reagents to acyclic trisubstituted enones, generating products that contain all-carbon-substituted quaternary stereogenic centers. The desired goal has been achieved by means of a strategy that, in the majority of cases, involves catalytic site- and/or stereoselective preparation of the starting materials.
The central role of an effective chiral promoter notwithstanding, the efficiency and selectivity with which the substrates and reagents are accessed is critical in a catalytic enantioselective process. We envisioned a multicomponent ECA protocol entailing catalytic synthesis of the unsaturated carbonyl compounds as well as the nucleophilic alkenyl partners prior to their union, which is in turn promoted by a third catalyst (Scheme 1). The all-catalytic method would ideally exhibit maximum versatility by providing access to an array of enones and nucleophilic entities, generated readily and selectively with inexpensive, easily available, and robust reagents and catalysts. It would be preferable if efforts regarding the isolation and purification of the catalytically generated starting materials were kept to a minimum.
Scheme 1.

The general and fully catalytic strategy for the generation and enantioselective coupling of two fragments by an enantioselective conjugate addition (ECA) reaction.
The multicomponent transformation in Scheme 2 is illustrative of the power of the approach. The alkenylaluminum reagent was obtained in two hours by site- and stereoselective Ni-catalyzed addition of aluminum hydride to phenylacetylene (93% β, >98% E).[9] The same aryl alkyne served as the starting point for the site- and stereoselective Zr-catalyzed aluminum methyl addition/acylation sequence,[10] furnishing the enone substrate after 45 min (2; >98% α, >98% E, 80% yield).[11] The union of the two catalytically generated components was accomplished by subjection of the alkenylaluminum species and enone 2 to 2.5 mol% of the AgI-based N-heterocyclic carbene (NHC) complex 1a[12] and 5.0 mol% of CuCl2·2H2O in tetrahydrofuran (thf) at 22 °C; there was complete conversion to the desired product after 45 min (<2% addition of α-alkenylaluminum species). β-Alkenylketone 3a was accordingly obtained in 64% overall yield and 98:2 e.r. by a process that requires a total of less than four hours. The solution of the alkenylaluminum species was used directly, whereas the enone was purified by silica gel chromatography; when the procedure was carried out without purification of 2, ketone 3a was isolated in 41% overall yield (vs. 64%) and with identical enantioselectivity (98:2 e.r.).
Scheme 2.

Proof-of-principle: Effective combination of three catalytic processes, culminating in the formation of a product containing an all-carbon-substituted quaternary stereogenic center in high enantiomeric purity in less than a total of four hours.
Aryl-, heteroaryl- and alkyl-substituted alkynes can be utilized. The facile modularity[13] of the multicomponent approach permits easy access to a variety of products in high e.r. Examples involving β-alkenylaluminum reagents are presented in Table 1; the desired compounds are isolated in 24–60% overall yield. Products are obtained with e.r. values that typically exceed 95:5. Enones prepared by the carboalumination/acylation of alkynes bearing an electron-donating (e.g., entry 1, Table 1; 40%yield) or an electron-withdrawing aryl unit (e.g., entry 2, 63% yield) are effective substrates. Zirconocene-catalyzed reactions uniformly deliver >98% E selectivity. Heterocyclic alkenylaluminum reagents, represented by 3g, formed in 96.5:3.5 e.r., and alkyl-substituted enones (Table 1, entries 7 and 8) were converted to products in 90.5:9.5–97:3 e.r. and 31–44% overall yield. In some instances, maximum selectivity calls for 1b as the catalyst precursor.
Table 1.
Multicomponent catalytic reactions with β-alkenylaluminum reagents.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | ECA conv. [%][b] |
Prod. | Carbomet./acyl. yield [%];[c] ECA yield [%][d] |
e.r.[e] | |
| 1 | >98 | 3b | 40; 75 | 98:2 | |
| 2 | >98 | 3c | 63; 60 | 96:4 | |
| 3 | >98 | 3d | 80; 60 | 96.5:3.5 | |
| 4 | >98 | 3e | 80; 72 | 98:2 | |
| 5 | >98 | 3 f | 80; 60 | 98:2 | |
| 6 | >98 | 3g | 80; 54 | 96.5:3.5 | |
| 7 | >98 | 3h | 61; 51 | 97:3 | |
| 8 | >98 | 3i | 80; 55 | 90.5:9.5 | |
| 9 | >98 | 3j | 80; 60 | 94.5:5.5 | |
Reactions were performed under N2 atmosphere; enones generated with >98% E selectivity in all cases.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Yield of isolated and purified products (±5%).
Determined by HPLC analysis; see the Supporting Information for details.
NHC-Ag complex 1b was used as catalyst precursor.
Unlike the transformations with β-substituted cyclic enones,[8c] the use of a silyl-substituted alkenylaluminum species is not necessary for obtaining high e.r. values with linear enones. The need for the initial preparation of a silyl-substituted alkyne and subsequent proto-desilylation is obviated. Attempts to effect NHC–Cu-catalyzed ECA with acyclic trisubstituted enones and silyl-containing Al-based reagents led to <2% conversion.[8c] The latter finding underscores the challenge of effecting the formation of products containing all-carbon-substituted quaternary centers from acyclic substrates.
Issues vis-á-vis the diminished reactivity of sizeable nucleophilic entities raise the question of whether acyclic trisubstituted enones would participate in efficient ECA processes involving α-substituted alkenylaluminum reagents (derived from hydroaluminations catalyzed by [Ni-(dppp)Cl2]).[9] Addition of aryl- or heteroaryl-substituted α-alkenylaluminum compounds and aryl- or alkyl-substituted substrates furnish β-alkenyl ketones in better yields and enantioselectivities than the analogous reactions of the less encumbered β-alkenylmetal variants (Table 2; 29–77% overall yield and 92:8–99:1 e.r.).
Table 2.
Multicomponent catalytic reactions with α-alkenylaluminum reagents.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | ECA conv. [%][b] |
Prod. | Carbomet./acyl. yield [%];[c] ECA yield [%][d] |
e.r.[e] | |
| 1 | >98 | 4a | 80; 82 | 99:1 | |
| 2 | >98 | 4b | 40; 95 | 97.5:2.5 | |
| 3 | >98 | 4c | 88; 88 | 99:1 | |
| 4 | 88 | 4d | 60; 48 | 99:1 | |
| 5 | >98 | 4e | 80; 95 | 98:2 | |
| 6 | >98 | 4 f | 80; 80 | 97:3 | |
| 7 | >98 | 4g | 80; 97 | 98:2 | |
| 8 | >98 | 4h | 80; 71 | 99:1 | |
| 9 | >98 | 4i | 40; 81 | 98:2 | |
| 10 | >98 | 4j | 63; 95 | 99:1 | |
| 11 | >98 | 4k | 62; 87 | 95:5 | |
| 12 | >98 | 4l | 80; 85 | 97.5:2.5 | |
| 13 | >98 | 4m | 61; 83 | 92:8 | |
Reactions were performed under N2 atmosphere; enones generated with >98% E selectivity in all cases.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Yield of isolated and purified products (±5%).
Determined by HPLC analysis; see the Supporting Information for details.
NHC-Ag complex 1a was used as catalyst precursor.
TBS = tert-butyl-(dimethyl)silyl.
Enantioselective synthesis of 5 in Scheme 3 illustrates that the catalytic process can be performed with n-alkyl (vs. methyl) ketones; attempts to effect reaction with the bulkier isopropyl ketone resulted in extensive decomposition after 4.0 h. Enynes easily undergo carbometalation/acylation en route to 1,4-dienes such as 6 (78% yield and 96:4 e.r.; Scheme 3). Alkenyl-substituted α-alkenylaluminum species serve equally well, as manifested by the enantioselective formation of 7 (92% yield, 98:2 e.r.). Synthesis of ketones 8 and 9 in high e.r. shows that enones accessed by reaction with commercially available and inexpensive Et3Al are suitable substrates.
Scheme 3.

Products obtained from catalytic multicomponent enantioselective reactions with an enyne, an n-alkyl ketone, and Et3Al. Enones generated with >98% E selectivity in all cases; yields are overall for the entire three-component process. See the Supporting Information for details.
There are instances where the zirconocene-catalyzed carbometalation is inefficient or the terminal alkyne is prohibitively expensive;[14] examples are shown in Scheme 4. Such substrates were prepared by Wittig-type olefinations. Ketones 10–15 were obtained readily in 57–90% yield and 97:3–99:1 e.r. (Scheme 4a). The catalytic ECA with unsaturated keto ester 16 shows that the reaction not only proceeds efficiently (68% yield) and with high enantioselectivity (97:3 e.r.), C–C bond formation is fully site-selective (Scheme 4b); only the product containing the more sterically demanding quaternary carbon stereogenic center β to the ketone unit is formed exclusively (17/18>98:2), underscoring the lower reactivity of enoates.
Scheme 4.

a) Products of NHC–Cu-catalyzed ECA reactions with α,β-unsaturated ketones not efficiently or cost-effectively accessible through Zr-catalyzed carbometalation/acylation. b) ECA reaction proceeds with site-selectivity. Yields refer to catalytic ECA; see the Supporting Information for details.
The sequence in Scheme 5, regarding the enantioselective synthesis of antimicrobial enokipodin B,[15] demonstrates the utility of our method. Conversion of aryl-substituted terminal alkyne 20 and phenylacetylene to 21 proceeds in 57% overall yield and 95:5 e.r.; this is in spite of the carbometalation/acylation of highly electron-rich 20 proceeding in an atypical 67:33 E/Z selectivity.[11] Control experiments indicate that the Z isomer affords the same enantiomer but the ECA is comparatively less efficient.[16] The alkenyl group of unsaturated ketone 21 has been utilized as a convenient precursor to an aldehyde, which was subsequently reacted with the neighboring ketone to generate the desired cyclopentenone.[15] The route in Scheme 5 compares favorably with a recent alternative that generates 21 in 11 steps from commercially available materials[15] (vs. five steps from 19, which can be purchased).
Scheme 5.

Application of the multicomponent catalytic enantioselective process to the synthesis of enokipodin B. Conditions: a) 1.5 equiv ICl, Et2O/CHCl3 (2:1), 22 °C, 3.0 h; 75% yield; b) 1.2 mol% [Pd-(PPh3)2Cl2], 2.0 mol% CuI, 1.2 equiv HC≡CSi(Me)3, 22–85°C 4.0 h; c) KOH, MeOH, thf, 22°C, 3.0 h; 94% overall yield (2 steps). See the Supporting Information for details.
We carried out DFT calculations to gain insight regarding the origins of the high enantioselectivity. The catalytic cycle appears to involve initial conjugate addition of (alkenyl)CuI complex followed by reductive elimination of the (alkenyl) CuIII alkyl intermediate.[17] Transformation through a mechanism whereby the cuprate’s alkenyl unit is directly transferred to the enone substrate in a redox-neutral process was found to be less favored by roughly 7–9 kcal mol−1. Complex A (Scheme 6) represents the lowest energy pathway, consistent with the identity of the major isomers; complex B, leading to the minor enantiomers, is about 1.6 kcal mol−1 higher in energy. In the latter case, simultaneous coordination of the substrate with the Lewis acidic aluminum bridge and Cu center dictates that the enone binds in its energetically more demanding s-trans conformation (vs. s-cis in A), engendering severe A(1,3) strain between the ketone and alkene substituents. In the absence of an aluminum connector, the transition state for addition to the same face as the complex is appreciably higher in energy. The above mechanistic issues underscore the challenges associated with ECA of acyclic compounds where, unlike the more rigid cyclic systems, reaction can proceed via several possible conformers.
Scheme 6.

DFT calculations indicate that the major enantiomer likely arises from the mode of addition A (vs. B).
In closing, we have introduced the first protocol for catalytic ECA reactions that form products with quaternary all-carbon-substituted stereogenic centers and involve alkenyl-based nucleophiles and acyclic enones. In most cases, reagents and substrates are obtained by catalytic processes as well. The above attributes, the variety of functionalizations feasible with alkenes and carbonyls, and the importance of enantiomerically enriched entities with a quaternary carbon stereogenic center underscore the value of the approach presented in this report.
Footnotes
Financial support was provided by the NIH (GM-47480). We thank Dr. F. Haeffner for helpful discussions, Matthew T. Villaume for experimental assistance and Boston College Research Services for providing access to computational facilities.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201309456.
References
- 1.For scholarly reviews on catalytic ECA reactions, see: Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem. Rev. 2008;108:2824–2852. doi: 10.1021/cr068424k. Jerphagnon T, Pizzuti MG, Minnaard AJ, Feringa BL. Chem. Soc. Rev. 2009;38:1039–1075. doi: 10.1039/b816853a. Ji J-X, Chan ASC. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Hoboken: Wiley; 2010. pp. 439–495.
- 2.For applications of catalytic ECA reactions that afford all-carbon-substituted quaternary stereogenic centers to complex molecule natural product synthesis (all involve cyclic enones), see: Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:12904–12906. doi: 10.1021/ja8058414. Peese KM, Gin DY. Chem. Eur. J. 2008;14:1654–1665. doi: 10.1002/chem.200701290. Mendoza A, Ishihara Y, Baran PS. Nat. Chem. 2012;4:21–25. doi: 10.1038/nchem.1196. Tanaka Y, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2010;132:8862–8863. doi: 10.1021/ja1035286.
- 3.For catalytic ECA of alkylmetal reagents to cyclic unsaturated carbonyl compounds that afford all-carbon-substituted quaternary stereogenic centers, see: Hird AW, Hoveyda AH. J. Am. Chem. Soc. 2005;127:14988–14989. doi: 10.1021/ja0553811. d’Augustin M, Palais L, Alexakis A. Angew. Chem. 2005;117:1400–1402. doi: 10.1002/anie.200462137. Angew. Chem. Int. Ed.2005, 44, 1376–1378; Lee K-s, Brown MK, Hird AW, Hoveyda AH. J. Am. Chem. Soc. 2006;128:7182–7184. doi: 10.1021/ja062061o. Martin D, Kehrli S, d’Augustin M, Clavier H, Mauduit M, Alexakis A. J. Am. Chem. Soc. 2006;128:8416–8417. doi: 10.1021/ja0629920. Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem. 2007;119:1115–1118. doi: 10.1002/anie.200604511. Angew. Chem. Int. Ed.2007, 46, 1097–1100; May TL, Brown MK, Hoveyda AH. Angew. Chem. 2008;120:7468–7472. Angew. Chem. Int. Ed.2008, 47, 7358–7362; Matsumoto Y, Yamada K-i, Tomioka K. J. Org. Chem. 2008;73:4578–4581. doi: 10.1021/jo800613h. Ladjel C, Fuchs N, Zhao J, Bernardinelli G, Alexakis A. Eur. J. Org. Chem. 2009. pp. 4949–4955. Kehrli S, Martin D, Rix D, Mauduit M, Alexakis A. Chem. Eur. J. 2010;16:9890–9904. doi: 10.1002/chem.201000471. Sidera M, Roth PMC, Maksymowicz RM, Fletcher SP. Angew. Chem. 2013;125:8153–8157. doi: 10.1002/anie.201303202. Angew. Chem. Int. Ed.2013, 52, 7995–7999. For related studies involving nitroalkanes as reagents, see: Kwiatkowski P, Dudziński K, Łyźwa D. Org. Lett. 2011;13:3624–3627. doi: 10.1021/ol201275h.
- 4.For catalytic ECA of aryl-based nucleophiles to cyclic α,β-unsaturated carbonyls to generate all-carbon-substituted quaternary stereogenic centers, see: with Rh complexes and arylboronic acids, Shintani R, Duan W-L, Hayashi T. J. Am. Chem. Soc. 2006;128:5628–5629. doi: 10.1021/ja061430d. with Cu complexes and arylaluminum reagents, b) Ref. [3f]; Hawner C, Li CK, Cirriez V, Alexakis A. Angew. Chem. 2008;120:8334–8337. doi: 10.1002/anie.200803436. Angew. Chem. Int. Ed.2008, 47, 8211–8214; with Rh complexes and sodium tetraarylborates, Shintani R, Tsutsumi Y, Nagaosa M, Nishimura T, Hayashi T. J. Am. Chem. Soc. 2009;131:13588–13589. doi: 10.1021/ja905432x. with Rh complexes and triarylboroxines, Shintani R, Takeda M, Nishimura T, Hayashi T. Angew. Chem. 2010;122:4061–4063. doi: 10.1002/anie.201000467. Angew. Chem. Int. Ed.2010, 49, 3969–3971; with Rh complexes and arylaluminum reagents, Hawner C, Müller D, Gremaud L, Felouat A, Woodward S, Alexakis A. Angew. Chem. Angew. Chem. Int. Ed. 2010;2010;12249:7935–7938. 7769–7772. doi: 10.1002/anie.201003300. with Pd complexes and arylboronic acids, Kikushima K, Holder JC, Gatti M, Stoltz BM. J. Am. Chem. Soc. 2011;133:6902–6905. doi: 10.1021/ja200664x. Gottumukkala AL, Matcha K, Lutz M, de Vries JC, Minnaard AJ. Chem. Eur. J. 2012;18:6907–6914. doi: 10.1002/chem.201200694.
- 5.For a review on enantioselective synthesis of quaternary carbon stereogenic centers, see: Das JP, Marek I. Chem. Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a.
- 6.For catalytic ECA of C-based nucleophiles to especially activated acyclic substrates affording all-carbon-substituted quaternary stereogenic centers, see: with alkylmetal reagents: Wu J, Mampreian DM, Hoveyda AH. J. Am. Chem. Soc. 2005;127:4584–4585. doi: 10.1021/ja050800f. Fillion E, Wilsily A. J. Am. Chem. Soc. 2006;128:2774–2775. doi: 10.1021/ja056692e. Mauleón P, Carretero JC. Chem. Commun. 2005:4961–4963. doi: 10.1039/b508142d. Wilsily A, Fillion E. Org. Lett. 2008;10:2801–2804. doi: 10.1021/ol800923q. Wilsily A, Fillion E. J. Org. Chem. 2009;74:8583–8594. doi: 10.1021/jo901559d. for studies involving nitroalkanes as nucleophiles, see: Kawai H, Yuan Z, Kitayama T, Tokunaga E, Shibata N. Angew. Chem. 2013;125:5685–5689. doi: 10.1002/anie.201301123. Angew. Chem. Int. Ed.2013, 52, 5575–5579; for catalytic ECA with a CN-based nucleophile, see: g) one example reported in: Mazet C, Jacobsen EN. Angew. Chem. 2008;120:1786–1789. doi: 10.1002/anie.200704461. Angew. Chem. Int. Ed.2008, 47, 1762–1765; h) Ref. [2d].
- 7.For ECA of Me3Al and Et3Al to β,β-substituted acyclic enones, see: Endo K, Hamada D, Shibata YT. Angew. Chem. 2013;125:634–638. Angew. Chem. Int. Ed.2013, 52, 606–610; for reactions with alkyl- and arylaluminum reagents, see: Dabrowski JA, Villaume MT, Hoveyda AH. Angew. Chem. 2013;125:8314–8317. doi: 10.1002/anie.201304035. Angew. Chem. Int. Ed.2013, 52, 8156–8159.
- 8.For Cu-catalyzed ECA with alkenyl-based nucleophiles and β-substituted cyclohexenones, see: Müller D, Hawner C, Tissot M, Palais L, Alexakis A. Synlett. 2010:1694–1698. Müller D, Tissot M, Alexakis A. Org. Lett. 2011;13:3040–3043. doi: 10.1021/ol200898c. For additions to β-substituted cyclopentenones and cyclohexenones, see: May TL, Dabrowski JA, Hoveyda AH. J. Am. Chem. Soc. 2011;133:736–739. doi: 10.1021/ja110054q. for isolated (single) cases of involving β-substituted cyclohexenones, see: Vuagnoux-d’Augustin M, Alexakis A. Chem. Eur. J. 2007;13:9647–9662. doi: 10.1002/chem.200701001. Hawner C, Li K, Cirriez V, Alexakis A. Angew. Chem. 2008;120:8334–8337. doi: 10.1002/anie.200803436. Angew. Chem. Int. Ed.2008, 47, 8211–8214; Palais L, Alexakis A. Chem. Eur. J. 2009;15:10473–10485. doi: 10.1002/chem.200901577.
- 9.Gao F, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10961–10962. doi: 10.1021/ja104896b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Negishi E, Kondakov DY, Chouiery D, Kasai K, Takahashi T. J. Am. Chem. Soc. 1996;118:9577–9588. Wipf P, Lim S. Angew. Chem. 1993;105:1095–1097. Angew. Chem. Int. Ed. Engl.1993, 32, 1068–1071; carbometalation/acylation of acetylenes can be performed at lower catalyst loadings, but at lower yields (e.g., with 10 mol% [Cp2ZrCl2], 2 was obtained in 70% yield after purification). Since the Zr-based salt is inexpensive, we opted to follow the higher yielding procedure with the higher loading.
- 11.Synthesis of trisubstituted enones by established procedures is not straightforward. Horner–Emmons-type transformations are severely inefficient (vs. the derived carboxylic esters); for example, reaction with acetophenone under standard conditions (1.2 equiv NaH, 22°C, 18 h) leads to <2% conversion to the desired enone. Another approach includes addition of a Grignard reagent to acetoacetone in the course of a day, followed by treatment with oxalic acid at reflux for 30 min; the stereoisomeric purity of the resulting enones has not been reported (N. J. A. Martin, B. List, J. Am. Chem. Soc. 2006, 128, 13368–13369). Yet a different procedure calls for an acid chloride as the starting material, entails a three-step process that requires a total of 36 h, and generates the stereoisomerically pure α,β-unsaturated ketone in ca. 26% overall yield after purification (Ref. [2d]).
- 12.See the Supporting Information for data on catalyst screening.
- 13.In principle, any entity or procedure can be modified; however, it is the ease with which alterations can be implemented that is critical. We underscore such distinction by the use of the term “facile modularity”.
- 14.The alkynes required for synthesis of 10–13 are too costly. In the case of p-trifluorophenyl-substituted 13–15, the carbometalation procedure proved to be inefficient (ca. 15% conv. under the conditions shown in Scheme 2).
- 15. Yoshida M, Shoji Y, Shishido K. Org. Lett. 2009;11:1441–1443. doi: 10.1021/ol9001637. for an earlier enantioselective route to enokipodins, see: Kuwahara S, Saito M. Tetrahedron Lett. 2004;45:5047–5049.
- 16.The reason for the low stereoselectivity in this particular Zr-catalyzed carbomethylation/acylation sequence is unclear. Control experiments indicate that the Z enone is first converted to the E isomer, which undergoes ECA more readily. However, the latter sequence leads to the generation of some unidentified impurities and thus a lower overall yield.
- 17.Yoshikai N, Nakamura E. Chem. Rev. 2012;112:2339–2372. doi: 10.1021/cr200241f. [DOI] [PubMed] [Google Scholar]