

Abstract
Age-related macular degeneration (AMD) is the leading cause of vision loss in developed countries. While linked to genetic polymorphisms in the complement pathway, there are many individuals with high risk alleles that do not develop AMD, suggesting that other ‘modifiers’ may be involved. Mitochondrial (mt) haplogroups, defined by accumulations of specific mtDNA single nucleotide polymorphisms (SNPs) which represent population origins, may be one such modifier. J haplogroup has been associated with high risk for AMD while the H haplogroup is protective. It has been difficult to assign biological consequences for haplogroups so we created human ARPE-19 cybrids (cytoplasmic hybrids), which have identical nuclei but mitochondria of either J or H haplogroups, to investigate their effects upon bioenergetics and molecular pathways. J cybrids have altered bioenergetic profiles compared with H cybrids. Q-PCR analyses show significantly lower expression levels for seven respiratory complex genes encoded by mtDNA. J and H cybrids have significantly altered expression of eight nuclear genes of the alternative complement, inflammation and apoptosis pathways. Sequencing of the entire mtDNA was carried out for all the cybrids to identify haplogroup and non-haplogroup defining SNPs. mtDNA can mediate cellular bioenergetics and expression levels of nuclear genes related to complement, inflammation and apoptosis. Sequencing data suggest that observed effects are not due to rare mtDNA variants but rather the combination of SNPs representing the J versus H haplogroups. These findings represent a paradigm shift in our concepts of mt–nuclear interactions.
INTRODUCTION
Mitochondria (mt) are unique organelles that have their own DNA. Human mtDNA is maternally inherited and forms a circle of double-stranded DNA with 16 569 nucleotide pairs. The non-coding mt-Dloop (also called the control region) contains 1121 nucleotides important for replication and transcription. The coding region of mtDNA encodes for 37 genes, including 13 protein subunits essential for oxidative phosphorylation (OXPHOS), 2 ribosomal RNAs and 22 transfer RNAs (1–3). The mtDNA can be categorized into haplogroups that are defined by a group of single nucleotide polymorphism (SNP) variants that have accumulated over tens of thousands of years and correspond to various geographic populations. While some studies have shown that certain mtDNA haplogroups can be either protective or increase risk for human diseases (4–10), others report no haplogroup association with diseases (11–13). However, many of these studies identified the major branch of the haplogroups (e.g. H, J, K, T, and U) but did not further refine the subsets within each haplogroup (e.g. J haplogroup is further divided into numerous J1 and J2 subclades). This may be a reason why disease risk association studies may have not proved useful. One of the proposed mechanisms for mtDNA involvement with diseases is through as yet unknown nuclear and mt interactions, which are being investigated using various models (14–22).
Studies of European mtDNA variants have revealed that J, T and U haplogroups are associated with age-related macular degeneration (AMD) (23–27), while the H haplogroup is protective (27). The J mtDNA haplogroup has attracted interest because these individuals have significantly lower VO2max (maximum oxygen uptake) than do non-J individuals (28,29) and the J haplogroup correlates positively with centenarians (30–32). However, it is still not clear how the J haplogroup mtDNA might affect various cellular functions. Using the osteosarcoma cybrid (cytoplasmic hybrids) model, it has been shown that J haplogroup cybrids have lower electron transport chain (ETC) efficiency and reactive oxygen species (ROS) levels, along with lower mtDNA and mtRNA levels than do other haplogroups (20,33).
While cybrids have been used in other systems, the model has not been used to study the contribution of mtDNA variants to mt–nuclear interactions in human retinal cells. Therefore, we created cybrids using human ARPE-19 Rho0 cells, which lack mitochondria, and are fused with platelets containing mitochondria from either H or J haplogroup individuals. This process yields cybrids that have identical nuclear DNA but have mitochondria representing each haplogroup. Each cybrid has mitochondria from only one individual donor. Using this model, we reported that J haplogroup cybrids as a group have decreased expression levels of four genes related to retinal degenerations [complement factor H (CFH), C3, EFEMP1 and MYO7A], diminished ROS production and increased rates of growth compared with H haplogroup cybrids (34). The CFH and C3 belong to the complement pathway (Fig. 1) and are associated with AMD (35,36). The present study examines nine additional genes associated with the complement pathways to determine if the J mtDNA mediates other aspects of complement activation. Moreover, genes associated with inflammation and apoptosis, both important factors for AMD, were investigated. In addition, H and J cybrids reportedly have differences in ATP and lactate production (34) so this study analyzed their bioenergetic profiles and expression levels for mtDNA-encoded genes of the respiratory complexes.
Figure 1.

Schematic of the three branches of the complement pathway showing sites where CFH, C3, CD55, C1s, C1r and C4b are located. The CFH is an inhibitor of the alternative complement pathway. CD55 is a critical inhibitor of the C3 convertase step. C1s and C1r are major subunits of C1 component and C4b is a component of C4. The C1s, C1r and C4b are components of the classical complement pathway.
Our findings with this cybrid model support the hypothesis that J haplogroup mtDNA variants within cells can mediate both energy pathways and non-energy pathways (complement, inflammatory and apoptotic) via unknown mechanisms. Most importantly, these data support a paradigm shift in thinking about the role that mitochondria, with their unique mtDNA variants representing different populations, influence numerous cellular functions and disease processes.
RESULTS
Expression of mtDNA-encoded genes
A previous study showed that H cybrids have higher ATP production but lower lactate levels than do J cybrids (34). Using the cybrid model, we measured the expression of the mtDNA-encoded genes in respiratory complexes I, III, IV and V of the OXPHOS pathway (Table 1). The H cybrids were normalized to a value of 1 for each of the genes. The J cybrids had significantly lower expression levels in seven genes: Complex I: MT-ND1 (P = 0.018), MT-ND2 (P = 0.003), MT-ND3 (P = 0.013), MT-ND4/ND4L (P = 0.027); Complex IV: MT-CO2 (P = 0.003), MT-CO3 (P = 0.046); Complex V: MT-ATP6 (P = 0.038) compared with the H cybrids. These findings suggest that the J cybrids express lower mtRNA levels of the OXPHOS genes, which is consistent with lower OXPHOS utilization.
Table 1.
Q-PCR expression levels for mtDNA-encoded genes from Complexes I, III, IV and V found in the H versus J cybrids
| Symbol | Gene name | H versus J P-value |
H versus J ΔΔCT/fold |
Complex |
|---|---|---|---|---|
| MT-ND1 | NADH dehydrogenase subunit 1 | 0.018 | −0.68 ± 0.26/0.62 | I |
| MT-ND2 | NADH dehydrogenase subunit 2 | 0.003 | −1.21 ± 0.34/0.43 | I |
| MT-ND3 | NADH dehydrogenase subunit 3 | 0.013 | −0.73 ± 0.26/0.60 | I |
| MT-ND4/ND4L | NADH dehydrogenase subunit 4/4L | 0.027 | −0.81 ± 0.33/0.57 | I |
| MT-ND5 | NADH dehydrogenase subunit 5 | 0.118 | −0.65 ± 0.40/0.63 | I |
| MT-ND6 | NADH dehydrogenase subunit 6 | 0.177 | −0.44 ± 0.31/0.74 | I |
| MT-CYB | Cytochrome b | 0.7524 | −0.10 ± 0.31/0.93 | III |
| MT-CO1 | Cytochrome c oxidase subunit I | 0.093 | −0.39 ± 0.22/0.76 | IV |
| MT-CO2 | Cytochrome c oxidase subunit II | 0.003 | −0.87 ± 0.24/0.55 | IV |
| MT-CO3 | Cytochrome c oxidase subunit III | 0.046 | −0.56 ± 0.26/0.68 | IV |
| MT-ATP6 | ATP synthase F0 subunit 6 | 0.038 | −0.75 ± 0.33/0.59 | V |
| MT-ATP8 | ATP synthase F0 subunit 8 | 0.077 | −0.51 ± 0.27/0.70 | V |
N = 3 with six values for each sample. Bold values represent those which are significantly different between H versus J cybrids.
Fold values >1 indicate upregulation of the gene compared with H cybrids. H cybrids are assigned a value of 1.
Fold values <1 indicate downregulation of the gene compared with H cybrids. H cybrids are assigned a value of 1.
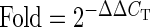 .
.
Measurements of the bioenergetic profiles for cybrids
The Seahorse XF24 extracellular flux analyzer was used to measure basal OCR, representing OXPHOS, and basal ECAR, representing glycolysis, in H versus J cybrids (Fig. 2) (see Extracellular Flux Analysis section in Material and Methods for explanation of abbreviations and description of sequential treatments). The profile shapes were similar between the H and J cybrids, but the values differed quantitatively. The H cybrids had a significantly higher OCR:ECAR ratio than did the J cybrids (24 ± 2.5 versus 14 ± 2.7, P < 0.05). These values represent combined values from three H and three J cybrids, each run in triplicate, and the experiment was repeated twice.
Figure 2.

The OCR and ECAR values were measured after sequential treatments with Oligomycin (1 µm), FCCP (1 µm) and Antimycin A plus Rotenone (1 µm) for the H cybrids (n = 3) and J cybrids (n = 3). The values represent the means ± SEM which were obtained by analyzing four wells per cybrid sample and repeating the experiment twice. Note that the H cybrids versus J cybrids showed similar overall patterns in their responses to the inhibitors and uncoupling agents. However, the OCR:ECAR ratios were significantly different from each other (24 ± 2.5 versus 14 ± 2.7, P < 0.05). OCR, oxygen consumption rate; ECAR, extracellular acidification rate; FCCP, carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone.
We also examined the real-time bioenergetic profiles with the Seahorse flux analyzer by exposing the cybrids to sequential treatments with Oligomycin, FCCP and Rotenone/Antimycin A (Fig. 3A). The XF24 flux analyzer values were used to calculate ATP turnover and spare respiratory capacity of the cybrids (Fig. 3B and C). ATP turnover values for the H cybrids (49.0 ± 1.3) were similar to the J cybrids (46.39 ± 0.5, P = 0.07; Fig. 3B). The spare respiratory capacity of the H cybrids (166.5 ± 4.07) was similar to the J cybrids (156.9 ± 3.3, P = 0.1; Fig. 3C). This indicates that the H and J cybrids respond similarly to inhibitors and uncoupling agents of the respiration pathway, suggesting that the respiration profiles were similar qualitatively and that the main differences between H and J cybrids were in the levels of OCR (aerobic respiration) and ECAR (glycolysis).
Figure 3.

(A) A representative profile measured by the Seahorse XF24 flux analyzer showing OCR and ECAR patterns after sequential treatment with oligomycin (1 µm), FCCP (1 µm) and Antimycin A plus Rotenone (1 µm), showing the regions that define the basal aerobic respiration, ATP turnover and spare respiratory capacity. (B) The H and J cybrids showed similar responses in ATP turnover after Oligomycin treatment. (C) The H and J cybrids showed similar responses in the spare respiratory capacity measured after FCCP treatment. These data are presented as a % response from the basal readings, with non-mt respiration subtracted out and the basal respiratory rate is normalized to 100%. Cybd, cybrid.
Expression levels of genes related to complement activation, inflammation and apoptosis
Previously, we showed by GeneChip analyses that H and J cybrids have different expression levels for genes related to cancer, cell death, morphology, movement and signaling interactions. In this study, we further analyze the GeneChip array data using Q-PCR methods so we could identify additional genes that might differentially expressed.
Both genetic and molecular studies have shown that the complement system (Fig. 1) and inflammation are major pathways associated with AMD. We showed previously that J cybrids have significantly lower expression levels of CFH (0.5-fold, P = 0.0001) and C3 (0.29-fold, P = 0.0003) compared with H cybrids (34). Therefore, we analyzed the data from the Affymetrix GeneChip to identify other genes belonging to the complement pathways and identified seven that were key components (Table 2). With regards to inflammation and innate immunity, the TGFA and IL-33 genes showed a >2-fold difference on the GeneChip array between the H and J cybrids so they were also analyzed further by Q-PCR. As a comparison, because the TGFB2 and IL-6 genes are known to be pro-inflammatory, they were analyzed by Q-PCR even though the GeneChip values did not show a great difference between the H and J cybrids. The RARA gene showed a significant difference between the H and J cybrids on the GeneChip array so it was analyzed by Q-PCR. Finally, we performed Q-PCR on the BBC-3 and BLC2L13 genes since they were known to be associated with mt dysfunction and are localized to mitochondria, respectively (Table 3).
Table 2.
Fold differences between H versus J cybrids for nuclear encoded genes of the different pathways as determined by GeneChip array
| Symbol | Gene name | Representative public ID | Fold value H versus J |
|
|---|---|---|---|---|
| CD55/DAF | CD55 molecule, decay accelerating factor for complement | CA448665 | −1.33 | Complement |
| CD59 | CD59 molecule, complement regulatory protein | NM_000611 | −1.58 | Complement |
| C1QC | Complement component 1, q subcomponent, C chain | AI184968 | 1.01 | Complement |
| C1S | Complement component 1, s subcomponent | M18767 | −1.61 | Complement |
| C4B | Complement component 4A/4B | NM_000592 | −4.2 | Complement |
| C4BPB | Complement component 4-binding protein, beta | NM_000716 | −2.74 | Complement |
| CFHR4 | Complement factor H-related 4 | NM_006684 | −17.5 | Complement |
| TGFA | Transforming growth factor alpha | NM_003236 | −2.03 | Proliferation, Differentiation, Development |
| IL-33 | Interleukin 33 | AB024518 | −3.59 | Innate immunity |
| TGFB2 | Transforming growth factor beta 2 | M19154 | 1.38 | Pro-inflammatory |
| AU145950 | −1.14 | |||
| IL-6 | Interleukin 6 | NM_000600 | 1.03 | Pro-inflammatory |
| RARA | Retinoic acid receptor, alpha | BE383139 | 16.98 | Apoptosis |
| BBC-3 | BCL2-binding component 3 | AF332558 | 1.99 | Apoptosis |
| BCL2L13 | BCL2-like 13 | NM_015367 | −1.08 | Apoptosis |
Positive fold values indicate higher expression levels than H cybrids.
Negative fold values indicate lower expression levels than H cybrids.
Table 3.
Description of primers for genes analyzed by Q-PCR
| Symbol | Gene name | GeneBank accession no. | Function |
|---|---|---|---|
| CFPa | Complement factor properdin | NM_001145252 | Plasma glycoprotein that binds many microbial surfaces and apoptotic cells and stabilizes C3 and C5 convertases to lead to the formation of the membrane attack complex and lysis of target cells in the alternative complement pathway |
| NM_002621 | |||
| CD55/DAFa | Decay accelerating factor for complement | NM_000574 | Involved in the regulation of complement cascade by accelerating the decay of complement proteins and disrupting the cascade. Inhibits C3 convertase and prevents assembly of C3bBb |
| NM_001114543 | |||
| NM_001114544 | |||
| NM_001114752 | |||
| CD59a,b | CD59 molecule, Complement regulatory protein | NM_000611 | Cell surface glycoprotein that regulates complement-mediated cell lysis, inhibits complement membrane attack complex and involved in lymphocyte signal transduction |
| NM_203329 | |||
| NM_203331 | |||
| NM_001127223 | |||
| NM_001127225 | |||
| NM_001127226 | |||
| NM_001127227 | |||
| C1QCb | Complement component 1, q subcomponent, C chain | NM_001114101 | Major constituent of complement subcomponent 1 and associates with C1r and C1s to yield the first component of the classical complement system |
| NM_172369 | |||
| C1Sb | Complement component 1, s subcomponent | NM_001734 | Serine protease, which is the major constituent of complement subcomponent 1 along with C1r and C1q to yield the first component of the classical complement system |
| NM_201442 | |||
| C1Rb | Complement component 1, r subcomponent | NM_001733 | Modular protease in the classical component pathway |
| XM_001124624 | |||
| C4Bb | Complement component 4B (Chido blood group) | NM_001002029 | Classical activation pathway. Provides surface for interaction between antigen–antibody complex and complement components |
| NM_000592 | |||
| C4BPBb | Complement component 4-binding protein, beta | NM_000716 | A multimeric protein that controls activation of the complement cascade through the classical pathway |
| NM_001017364 | |||
| NM_001017365 | |||
| NM_001017366 | |||
| NM_001017367 | |||
| CFHR4a | Complement factor H-related 4 | NM_006684 | CFH-related protein that enhances the cofactor activity of CFH and is involved in complement regulation. It can associate with lipoproteins |
| NM_001201550 | |||
| NM_001201551 | |||
| TGFA | Transforming growth factor, alpha | NM_003236 | Ligand for EGFR which activates signaling pathway for cell proliferation, differentiation and development. Closely related to EGF |
| NM_001099691 | |||
| TGFB2 | Transforming growth factor, beta 2 | NM_003238 | Secreted cytokine that regulates proliferation, differentiation, adhesion, migration and other functions |
| NM_001135599 | |||
| IL-33 | Interleukin 33 | NM_033439 | Member of IL1 family. Critical pro-inflammatory cytokine involved in production of T helper-2-associated cytokines |
| NM_001199640 | |||
| NM_001127180 | |||
| IL-6 | Interleukin 6 | NM_000600 | Cytokine with pro- and anti-inflammatory effects. Involved with maturation of B cells |
| RARA | Retinoic acid receptor, alpha | NM_000964 | Nuclear retinoic acid receptor. Regulates transcription. Involved with apoptosis, differentiation |
| NM_001033603 | |||
| NM_001145301 | |||
| BBC-3 | BCL2-binding component 3 | NM_014417 | Member of BLC-2 family, BH3-only prop-apoptotic subclass to induce mt outer membrane |
| NM_001127240 | |||
| NM_00112741 | permeabilization, apoptosis, mt dysfunction and caspase activation | ||
| BCL2L13 | BCL2-like 13 | NM_015367 | mt localized protein whose overexpression results in apoptosis |
aBelongs to the alternative complement pathway.
bBelongs to the classical complement pathway.
With Q-PCR analyses, we demonstrated the J cybrids had significantly decreased expression levels for CD55/DAF, a major inhibitor of the alternative complement pathway (0.4-fold, P = 0.0003) and three genes related to the classical complement pathway (C1S, 0.14-fold, P = 0.001; C1R, 0.18-fold, P = 0.036; C4B, 0.5-fold, P = 0.01) compared with H cybrids (Table 4). The TGFA and IL-33 genes were expressed at lower levels by the J cybrids (0.43-fold, P = 0.03 and 0.46-fold, P = 0.004) than by the H cybrids. Surprisingly, the H and J cybrids had similar expression levels of both IL-6 and TGFB2 genes. Three apoptosis-related genes (RARA, BBC-3 and BCL2L13) were also evaluated. In the J cybrids, the expression levels were decreased for RARA (0.57-fold, P = 0.007) and BCL2L13 (0.56-fold, P = 0.005) compared with that of the H cybrids.
Table 4.
Differential gene expressions found in the H versus J cybrids
| Symbol | H versus J P-value |
H versus. J ΔΔCT/fold |
Pathways |
|---|---|---|---|
| CFP | 0.07 | 0.56 ± 0.29/1.48 | Complement |
| CD55/DAF | 0.03 | −0.74 ± 0.31/0.60 | |
| CD59 | 0.18 | −0.27 ± 0.19/0.83 | |
| C1QC | 0.59 | 0.3 ± 0.54/1.23 | |
| C1S | 0.001 | −2.86 ± 0.52/0.14 | |
| C1R | 0.036 | −2.44 ± 1.06/0.18 | |
| C4B | 0.01 | −1.01 ± 0.36/0.5 | |
| C4BPB | 0.76 | −0.21 ± 0.65/0.86 | |
| CFHR4 | 0.19 | −0.59 ± 0.44/0.66 | |
| TGFA | 0.03 | −1.21 ± 0.51/0.43 | Inflammation |
| TGFB2 | 0.41 | −0.21 ± 0.25/0.86 | |
| IL-33 | 0.004 | −1.12 ± 0.33/0.46 | |
| IL-6 | 0.09 | −1.39 ± 0.78/0.38 | |
| RARA | 0.007 | −0.82 ± 0.43/0.57 | Apoptosis |
| BBC-3 | 0.11 | −0.52 ± 0.3/0.70 | |
| BCL2L13 | 0.005 | −0.84 ± 0.26/0.56 |
N = 3 with six values for each sample.
Fold values >1 indicate upregulation of the gene compared with H cybrids. H cybrids are assigned a value of 1.
Fold values <1 indicate downregulation of the gene compared with H cybrids. H cybrids are assigned a value of 1.
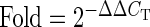 .
.
Sequence variations of the H and J cybrid mtDNA
The entire mtDNA from each of the cybrids were sequenced and compared with the Cambridge Reference Sequence. SNPs we define as unique are not listed in www.MitoMap.org. Private SNPs are those that do not define the haplogroup. The J cybrids were classified into J1d1a, J1c1 and J1c7. There were a total of nine private SNPs in the J cybrids: J1d1a cybrid with m.2305T>C (unique, MT-RNR2) and m.10654C>T (A-V, MT-ND4L); J1c1 cybrid with m.7226G>A (syn, MT-CO1), m.7747C>T (syn, MT-CO2), m.13143T>C (syn, MT-ND5) and m.16209T>C (non-coding); and J1c7 cybrid with m.6734G>A (syn, MT-CO1), m.12072T>C (F-S, MT-ND4) and m.16209T>C (non-coding). Two SNPs were in the MT-CO1 loci and another two were in the non-coding region. All of the rest of the private SNPs were all in different mt loci (MT-RNR2, MT-CO2, MT-ND4L, MT-ND4 and MT-CYB). There were seven J-defining haplogroup SNPs that were non-synonymous SNPs: m.3394T>C (Y-H, MT-ND1), m.4216T>C (Y-H, MT-ND1*), m.6261G>A (A-T, MT-CO1), m.10398A>G (T-A, MT-ND3*), m.13708G>A (A-I, MT-ND5*), m.14798T>C (F-L, MT-CYB) and m.15452C>A (L-I, MT-CYB*). The four SNPs with asterisks are J-defining SNPs found in all three J cybrids. The non-coding region of the J cybrids possessed 15 SNPs, of which 12 defined the J haplogroup (Table 5).
Table 5.
Sequencing results for the entire mtDNA in H and J cybrids
| Loci: MT | AA change | J CY 10-01 | J1d1a | J CY 10-05 | J1c1 | J CY 11-32 | J1c7 |
|---|---|---|---|---|---|---|---|
| RNR1 | 750A>G | a | 750A>G | a | 750A>G | a | |
| RNR1 | 1007G>A | J1d1a | |||||
| RNR1 | 1438A>G | a | 1438A>G | a | 1438A>G | a | |
| RNR2 | Unique | 2305T>C | b | ||||
| RNR2 | 3010G>A | J1 | 3010G>A | J1 | 3010G>A | J1 | |
| ND1 | Y-H | 3394T>C | J1c1 | ||||
| ND1 | Y-H | 4216T>C | JT | 4216T>C | JT | 4216T>C | JT |
| ND2 | SYN | 4769A>G | a | 4769A>G | a | 4769A>G | a |
| CO1 | A-T | 6261G>A | J1c3b1a | ||||
| CO1 | SYN | 6554C>T | J1c7 | ||||
| CO1 | SYN | 6734G>A | b | ||||
| CO1 | SYN | 7226G>A | b | ||||
| CO2 | SYN | 7789G>A | J1d | ||||
| CO2 | SYN | 7747C>T | b | ||||
| CO2 | SYN | 7963A>G | J1d | ||||
| ATP6 | T-A | 8860A>G | a | 8860A>G | a | 8860A>G | a |
| ND3 | T-A | 10398A>G | J | 10398A>G | J | 10398A>G | J |
| ND4L | A-V | 10654C>T | b | ||||
| ND4 | SYN | 11251A>G | JT | 11251A>G | JT | 11251A>G | JT |
| ND4 | F-S | 12072T>C | b | ||||
| ND5 | SYN | 12612A>G | J | 12612A>G | J | ||
| ND5 | SYN | 13143T>C | b | ||||
| ND5 | A-I | 13708G>A | J | 13708G>A | J | 13708G>A | J |
| CYB | F-L | 14798T>C | J1c | 14798T>C | J1c | ||
| CYB | T-A | 15326A>G | a | 15326A>G | a | 15326A>G | a |
| CYB | L-I | 15452C>A | JT | 15452C>A | JT | 15452C>A | JT |
| DLoop | Non-coding | 16069C>T | J | 16069C>T | J | 16069C>T | J |
| DLoop | Non-coding | 16092T>C | b | ||||
| DLoop | Non-coding | 16126T>C | JT | 16126T>C | JT | 16126T>C | JT |
| DLoop | Non-coding | 16193C>T | J1d | ||||
| DLoop | Non-coding | 16209T>C | b | ||||
| DLoop | Non-coding | 16261C>T | J1c6'7 | ||||
| DLoop | Non-coding | 16309A>G | J1d1a | ||||
| DLoop | Non-coding | 152T>C | J1d | ||||
| DLoop | Non-coding | 185G>A | J1c | 185G>A | J1c | ||
| DLoop | Non-coding | 228G>A | J1c | 228G>A | J1c | ||
| DLoop | Non-coding | 263A>G | a | 263A>G | a | 263A>G | a |
| DLoop | Non-coding | 295C>T | J | 295C>T | J | 295C>T | J |
| DLoop | Non-coding | 462C>T | J1 | 462C>T | J1 | ||
| DLoop | Non-coding | 482T> | J1c1 | ||||
| DLoop | Non-coding | 489T>C | J | 489T>C | J | ||
| Loci | AA change | H CY 10-03 | H | H CY 10-04 | H5a | H CY 10-07 | H66a |
| RNR1 | 750A>G | a | 750A>G | a | 750A>G | a | |
| RNR1 | 1198A>G | b | |||||
| RNR1 | 1438A>G | a | 1438A>G | a | 1438A>G | a | |
| RNR1 | Unique | 1477T>C | b | ||||
| RNR2 | 2159T>C | H63 | |||||
| RNR2 | 2706A>G | * | 2706A>G | * | 2706A>G | * | |
| TQ | tRNA | 4336T>C | H5a | ||||
| ND2 | A-D | 4483C>G | b | ||||
| ND2 | SYN | 5051A>G | b | ||||
| ND2 | SYN | 4769A>G | H2a1dc | 4769A>G | H2a1dc | 4769A>G | H2a1dc |
| CO1 | SYN | 7028T>C | H | 7028T>C | H | 7028T>C | H |
| CO1 | SYN | 7337G>A | H66 | ||||
| TK | tRNA | 8343A>G | H5e1 | ||||
| ATP6 | T-A | 8860A>G | a | 8860A>G | a | 8860A>G | a |
| CO3 | SYN | 9305G>A | b | ||||
| CO3 | Unique | 9771T>C | b | ||||
| ND4 | SYN | 11719G>A | H | 11719G>A | H | 11719G>A | H |
| CYB | T-I | 14766T>C | H | 14766T>C | H | 14766T>C | H |
| CYB | T-A | 15326A>G | a | 15326A>G | a | 15326A>G | a |
| CYB | SYN | 15833C>T | H5a1 | ||||
| DLoop | Non-coding | 16093T>C | H1f, H3v1, H4b1, | ||||
| DLoop | Non-coding | 16153G>A | H8c2 | ||||
| DLoop | Non-coding | 16172T>C | H66a | ||||
| DLoop | Non-coding | 16304T>C | H5 | ||||
| DLoop | Non-coding | 73A>G | 73G>A | H | 73G>A | H | |
| DLoop | Non-coding | 263A>G | a | 263A>G | a | 263A>G | a |
a Private: non-haplogroup defining SNP found in both H and J cybrids.
b Private: non-haplogroup defining SNP found in single H or J cybrid.
cBack mutation to ancestral state.
Unique: SNP not listed on www.mitomap.org.
Based upon the sequences, the H cybrids were classified into H, H5a and H66a. The H-defining SNPs were m.73G>A, m.2706G>A, m.7028T>C, m.11719A>G and m.14766T>C. There were six private SNPs in total for the H cybrids. The H cybrid had the following SNPs: m.1198A>G (MT-RNR1), m.1477T>C (unique, MT-RNR1), m.4483C>G (A-D, MT-ND2), m.9305G>A (syn, MT-CO3) and m.9771T>C (unique, MT-CO3). The H5a cybrid had one private SNP, m.5051A>G (syn, MT-ND2). The H66a cybrid had no private SNPs.
None of the SNPs found in either the H or J cybrids were associated with diseases when compared with the www.MitoMap.org: reported Mitochondria DNA Base Substitution in Diseases section or the Coding and Control Region Point Mutations section. The sequencing data support the theory that the observed effects are not due to private polymorphism (SNPs) or rare mtDNA variants but rather due to the combination of SNPs that represent the J haplogroups compared with the H haplogroups.
DISCUSSION
Studies show the complement pathways play a role in AMD and as such, therapies have been developed to target the complement pathway proteins and inhibit activation of these pathways. However, to date, this approach has been unsuccessful and one contributing reason may be that individuals with different mtDNA haplogroups could have differing levels of complement activation so responses to medications would vary.
Recent studies have shown that specific mtDNA haplogroups are associated with age-related diseases, including AMD (23–27), Alzheimer's disease (37), Parkinson's disease (38–40) and osteoarthritis (41). The European J haplogroup is high risk for AMD (23,25,27) while the European H haplogroup is protective (27). These findings suggest that mtDNA population variants may play a role in human disease but it has been difficult to identify the contribution of mtDNA to cellular functions. Therefore, we established human cybrid RPE cell lines that have identical nuclei but vary in their mtDNA profiles, so the influence of H versus J haplogroups can be characterized.
Respiration rates and mtDNA-encoded genes for Complexes I, III, IV and V
The XF24 Flux analyzer allows for simultaneous, real-time measurements of bioenergetic profiles for multiple cybrid samples. In our study, measurements were taken as three different compounds were added in succession to metabolically alter the bioenergetic profiles of the cybrids. Oligomycin acts as an ATP uncoupler which inhibits ATP synthase (Complex V). The FCCP is an ETC accelerator which acts as an uncoupling agent as hydrogen ions are transported across the mt membrane; this disrupts ATP synthesis associated with Complex V activity. The third compound, a combination of Rotenone, a Complex I inhibitor, and Antimycin A, a Complex III inhibitor, shuts down mt respiration and enables both the mt and non-mt fractions that contribute to respiration to be calculated. The ATP turnover and spare respiratory capacity were similar in H and J cybrids, indicating that they responded similarly to inhibitors and uncoupling agents of the respiration pathway, suggesting that the oxidative respiration profiles of the H and J cybrids were qualitatively similar to each other. Our findings suggest that the main difference between the haplogroups was a quantitative higher OCR (aerobic respiration) to ECAR (glycolysis) ratio in the H cybrids. The higher OCR (OXPHOS) in H cybrids is also consistent with the higher ATP production levels reported in ARPE-19 cybrids and osteosarcoma cybrids with the H haplogroup (34,42). It has also been shown that in patients that have both Huntington's disease and the H haplogroup, the CD4+ cells have higher ATP levels than do non-H individuals (43). Interestingly, osteosarcoma cybrids with the UK haplogroup show even higher ATP levels than do the H cybrids, but this is dependent on the substrate (44).
Our results support the idea that the mtDNA ‘directs’ the mode of energy production within a cell. Not only do the J cybrids have lower ATP levels and higher lactate levels (34), they also have lower mtRNA expression levels for seven of the mtDNA-encoded respiratory complex genes. Previously, we demonstrated that the mtDNA copy numbers in the H cybrid and J cybrid cultures were similar at Days 1 and 7 incubation periods (34). The concept that mtDNA haplogroups can mediate the energy production levels is supported by another study which compares H (most common European) versus L (African maternal origin) haplogroups within ARPE-19 cybrids (45). It is important to note that nuclei and culture conditions for the H and L cybrids in that study and J cybrids in this study are all identical. In both studies, the H cybrids were considered the baseline for the L cybrids or J cybrids. In the first study, the L cybrids had lower mtDNA copy numbers but showed nine mtRNA genes with significantly increased expression (1.57- to 2.11-fold) compared with H cybrids and one mtRNA gene with lower expression (0.16-fold). In addition, the L cybrids had significantly decreased ATP turnover and spare respiratory capacity but similar OCR:ECAR ratios (45). These patterns, which were also normalized to H haplogroup cybrids, are very different from what we found in J cybrids, with L cybrids having the highest respiratory activities, the H cybrids intermediate and the J cybrids showing the lowest activities. This indicates that each of the cybrid haplogroups (L versus H versus J) had bioenergetics unique to that haplogroup. This strongly supports the hypothesis that cellular bioenergetics are greatly affected by the mtDNA haplogroups within a cell.
Endogenous production of ROS occurs typically as electrons leak from Complexes I and III of the ETC within the mitochondria (46). Our Q-PCR studies showed that J cybrids had lower expression levels of four mtDNA-encoded genes of Complex I but similar mtRNA expressions for MT-CYB of Complex III compared with H cybrids. This suggests that the diminished ROS production in the J cybrids compared with H cybrids (34) may be due to differences in Complex I rather than Complex III. Our ARPE-19 cybrid findings are similar to those from osteosarcoma cybrids studies (20). One mechanism to explain the unique paradoxical behavior of J haplogroups (46,47) compared with other European haplogroups may be that, although the J haplogroup has had mutation rates similar to other haplogroups, there has been a preferential accumulation of non-synonymous SNPs in the J haplogroups (48). This means that the J haplogroup, with its overaccumulation of non-synonymous SNPs, could have decoupling of OXPHOS, increased heat production and lower ROS formation (46). These non-synonymous SNPs may mediate lower RNA expression of respiratory complex genes, which may lead to diminished oxidative energy production and ROS but to increased activation of inflammatory pathways.
Complement and inflammation genes
Patients with advanced AMD have elevated plasma levels of fragmented, activated complement components (Bb and C5a) and there is an inverse association with CFH, an inhibitor of the alternative complement pathway (49). Investigations have demonstrated that genes within the complement pathways (CFH, C3, CFB, C2 and CFI) are linked strongly to AMD (50–57). However, many subjects with the high risk complement genetic alleles do not ever develop AMD so the nuclear genetic patterns cannot account entirely for this disease. We propose that other factors or modifiers, such as mtDNA haplogroups, likely play a role. For example, specific mtDNA haplogroup backgrounds can increase severity of disease when coupled with nuclear mutations (7) or with a typically mild mtDNA mutation (58). Cybrids created from cells of patients with Huntington's disease and Leber hereditary optic neuropathy have shown important correlations between mtDNA mutations and cellular functions, including apoptosis and ATP production (21,43,59,60). It is possible that similar ‘modifier effects’ may occur in AMD subjects.
High levels of glycolysis, as seen in the J cybrids, are associated often with oxidative stress, so we examined the cybrid lines for expression of genes related to the inflammation and complement pathways. In a previous study, we reported that the J cybrids had significantly lower expression levels (0.5-fold) of CFH inhibitor (34) and we now report that a second inhibitor, CD55/DAF, is 0.6-fold lower in J cybrids (P = 0.03) than H cybrids. The CD55/DAF protein blocks the C3 convertase to accelerate the decay of complement proteins, thereby disrupting cascade activity (Fig. 1). CD55/DAF can modulate autoimmune disease and inflammation. Although CD55/DAF has been immuno-localized to retinal ganglion cells, photoreceptors and choroidal capillaries (61,62), there were no differences in staining intensities between normal retinas and early AMD retinas (62). It is possible that lower levels of CFH and CD55/DAF inhibitors in the RPE cells contribute to higher complement activities and to elevated MAC deposition, similar to that seen in AMD retinas. If we extrapolate our cybrid findings to in vivo RPE cells, then J haplogroup individuals, with lower expression of the CFH and CD55/DAF inhibitors, would theoretically have increased activation of the complement components at the local level, making them more susceptible to the development of AMD. The more protected individuals would be the ones with H haplogroups, with higher levels of CFH and CD55/DAF inhibitors, and thus a lower risk for AMD (27). The cybrid model also showed differences in expression for the more classical pathway genes (C1S, C1R and C4B), again reinforcing that, although the nuclei are identical, the different mtDNA profiles can influence the complement system. This suggests that at some level, activation of the classical pathway may also be mediated by mtDNA variants. It should be noted that in the GeneChip array analyses the transcripts for CFHR4 and RARA showed were not consistent with the Q-PCR values, which is an anomaly common for this type of analyses. The Q-PCR primer for the CFHR4 gene included three GeneBank accession numbers (single accession number primers were not available), whereas the GeneChip ID was for only a single number, NM_006684. For the RARA gene, the GeneChip ID was BE3833139 while the Q-PCR primer represented three different accession numbers. This might accounting for the discrepancy between the GeneChip values and the Q-PCR values and demonstrates the importance of using both techniques.
Cybrids created in osteosarcoma cells show altered expression levels of stress response genes depending upon their mtDNA haplogroup (63,64). In our model, the J cybrids have lower expression of TGF-alpha, a growth factor with 40% homology to epidermal growth factor (EGF) that binds to the EGF receptor. TGF-alpha is present in retinal extracts (65), plays a role in retinal progenitor cell proliferation and differentiation (66), has mitogenic properties for retinal cells in vitro (67) and, when injected into the vitreous of rabbits, can induce c-fos production by retinal Muller cells (68). While the biological functions of TGF-alpha within the human retina and its role in disease processes remain unclear, the fact that the H cybrids have significantly higher levels of TGF-alpha compared with J cybrids implies that it has a functional impact.
The expression levels of IL-33 were significantly lower in J cybrids compared with H cybrids. This newly discovered cytokine is important in immune-inflammatory diseases (69,70) and angiogenesis (71). It is a chemo-attractant for Th2 cells (72) and can activate dendrite cells to induce naïve T cells to become cytokine-producing Th2 cells (73,74). In a mouse model, IL-33 promotes type 2 innate responses (73). If individuals with J versus H haplogroups have different IL-33 expression levels, their innate immunity responses may be different because IL-33 can alter the levels of IL-4, IL-5, IL-13, IgA, IgE and IgG and promote the phenotypic switch from Th-1 cells to Th-2 cells (75–80), which may contribute to the development of more advanced AMD (81). A recent study shows that RPE cells respond to amyloid-beta by increasing IL-33 production, which in turn modulates levels of various pro-inflammatory cytokines (82). So while the role of IL-33 in AMD is not understood, if it is a critical regulator of inflammation, then IL-33 should be investigated as a potential trigger and therapeutic target site to diminish the inflammatory component of AMD. Targeting IL-33 to reduce its level or activity would lower inflammation in RPE-19 cells. However, H haplogroup, which is protected against AMD as compared with J haplogroup, has lower IL-33 already. Lowering IL-33 levels even further would perhaps alleviate inflammation in patients possessing the H haplogroup and could be essential in AMD patients possessing the J haplogroup. While the IL-33 levels are lower in the J cybrids, after oxidative stress, the IL-33 levels are significantly higher than the H cybrids (data not shown). It is interesting that both TGFB2 and IL-6, recognized pro-inflammatory cytokines, were expressed at the same levels for H and J cybrids. It may be that mtDNA influences are through very different pathways than the more well-known inflammatory pathways. Additional experiments are needed to understand if there are ‘mt-driven’ versus ‘nuclear-driven’ inflammatory pathways and the role that IL-33 might play in this cross-talk. We believe that the cybrid models will be a very useful tool in these studies.
Apoptosis genes
The present study shows that J cybrids have reduced expression of RARA compared with H cybrids (P = 0.007). As a protein, RARA can bind to the A2E ligand and induce vascular endothelial growth factor (VEGF) expression (83). Recent studies have shown that treatment with a RARA antagonist blocks VEGF expression and formation of laser-induced choroidal neovascularization (84). These studies further suggest that RARA may be a potential target for anti-angiogenesis therapy. Targeting RARA to reduce its levels may be necessary to reduce neovascularization in patients of the H haplogroup, and it may be essential to reduce even further neovascularization in those who have the J haplogroup. The mechanism(s) by which SNP variants within mtDNA could mediate expression of RARA are unknown. However, it is known that human RPE cells can be dramatically affected by exposure to A2E, a component of lipofuscin, which is also a retinoic acid receptor ligand. As such, elevated levels of A2E can disrupt many functions of the RPE cells (85–88). Our finding that different mtDNA variants can mediate the expression of RARA is very important, since this could potentially contribute to A2E accumulation, cell death and VEGF expression. RARA is an important gene to further characterize and possibly target for therapy.
The J cybrids also have altered levels of BLC2L13 (also known as BCL-rambo), which is a protein that localizes to the mt membrane, is highly conserved in vertebrates and inhibits mt disruption from apoptosis stimuli. In patients with childhood acute lymphoblastic leukemia, elevated BCL2L12 levels can be associated with poor prognosis and variable treatment outcomes of patients (89,90), which makes it a target for therapy. BCL2L13 protein has pro-apoptotic activity that can be blocked with caspase inhibitors. It has an unusual mode of action compared with other BCL2 family members, in that its activity is induced by a membrane-anchored C-terminus domain and is not dependent on the classical mt-induced apoptotic pathway (91). Further work is needed to clarify whether BCL2L13 expression plays an important role in RPE cell death.
Various models are being used to study the mechanisms by which mtDNA variants might influence cell behavior and nuclear gene expression. These conclusions are bolstered by studies in yeast, worms, flies, mice and human cells that establish cause and effect and are not simply correlative (92,93). The Drosophila model has shown that mt genotypes can influence mt metabolism (14) and that combinations of mtDNA haplotypes and different nuclear DNA allelic backgrounds can affect the longevity of Drosophila (15). Moreover, incompatibility between nuclear DNA and mtDNA genotypes affects Aatm, an mt amino-tRNA synthetase for tryrosine, and may contribute to disease penetrance variability (16). Finally, in Drosophila, the effects of temperature upon mt function are mediated by mt–nuclear interactions (17). The Caenorhabditis elegans model has been used to investigate the mt–nuclear interactions associated with longevity (18) and to identify mechanisms for communication between stressed mitochondria and the nucleus (94). With respect to human cells, the cybrids are an extremely useful model (19–22) because the cells have identical nuclei, but vary in their mtDNA haplogroups, so functional consequences of human mtDNA variants can be identified.
Based upon our data and that in the literature, we are proposing that the maternally inherited mtDNA haplogroup (e.g. H versus J), which an individual is born with, acts as a ‘modifier’ for the nuclear genome (Fig. 4). There is constant interaction between the mtDNA and nDNA, and the haplogroup pattern sets a ‘baseline’ of bioenergetics (energy production, ATP turnover, etc.) and somehow affects the nDNA expression levels for specific cellular pathways (e.g. complement, innate immunity, inflammation and apoptosis). The individual then is exposed to a variety of environmental factors (smoking, obesity, light exposures, etc.), which creates oxidative stress within the retinal cells. The H haplogroup cells would be able to handle the stressors differently than the J haplogroups cells. This in turn can influence which cells undergo mitochondria dysfunction and cell death and which cells are protected and have longevity. We believe that the cybrid model can be useful for examining the mt–nuclear interactions and also testing drugs that can increase mt efficiency since numerous studies show mt dysfunction and damage in many age-related diseases, including AMD.
Figure 4.

Schematic of new paradigm for mt–nuclear interactions. We propose that individuals have maternally inherited mtDNA haplogroups (e.g. H versus J), which acts as a ‘modifier’ for the nuclear genome via constant interactions. The haplogroup pattern mediates a ‘baseline’ of bioenergetics (e.g. energy production, ATP turnover) and nDNA expression levels for specific cellular pathways (e.g. complement, inflammation and apoptosis). As the cells are exposed to different environmental oxidative-stress factors (e.g. smoking, obesity, light exposures), the H haplogroup cells might respond differently than the J haplogroups cells because of the baseline expression levels of major pathways (e.g. complement, inflammation, apoptosis). This in turn may influence which cells undergo mitochondria dysfunction and cell death and which cells are protected and have longevity.
MATERIALS AND METHODS
Cybrid cultures and culture conditions
Institutional review board approval was obtained from the University of California, Irvine (#2003–3131). For DNA analyses, 10 ml of peripheral blood was collected via venipuncture in tubes containing 10 mm EDTA. DNA was isolated with a DNA extraction kit (PUREGENE, Qiagen, Valencia, CA, USA) and quantified using the Nanodrop 1000 (Thermo Scientific, Wilmington, DE, USA). Platelets were isolated by a series of centrifugation steps and final pellets were suspended in Tris buffer saline. ARPE-19 cells are a cell line derived from human retinal epithelia (ATCC, Manassa, VA, USA). The original ARPE-19 cells are a human diploid cell line that shows structural and functional properties similar to RPE cells in vivo (95) and are commonly used in retinal research. The ARPE-19 cells were made Rho0 (deficient in mtDNA) by serial passage in low-dose ethidium bromide (96). Cybrids were produced by polyethylene glycol fusion of platelets with Rho0 ARPE-19 cells according to a modified procedure of Chomyn (34,97). The generation of the cybrids has been described in detail in a previous study (34). Briefly, cybrids were cultured until confluent in DMEM-F12 containing 10% dialyzed fetal bovine serum, 100 unit/ml penicillin and 100 µg/ml streptomycin, 2.5 µg/ml fungizone, 50 µg/ml gentamycin and 17.5 mm glucose.
Sequence analyses of cybrid haplogroups
Cybrid DNA was extracted from cell pellets using a Spin Column Kit (DNeasy Blood and Tissue Kit, Qiagen). Allelic discrimination was performed to confirm the haplogroups. The primers for allelic discrimination were synthesized by ABI Assay-by-Design (Applied Biosystems, Grand Island, NY, USA). The samples were run at GenoSeq, the UCLA Genotyping and Sequencing Core, on an ABI 7900HT. Data were analyzed with Sequence Detection Systems software from ABI. All experiments used passage 5 cybrid cells for the assays described below.
Whole mtDNA genome sequencing
The sequencing method was a modified version used by Zaragoza et al. (98). The PCR for whole mtDNA genome sequencing was performed in two parts with a high fidelity PCR system (FailSafe™ PCR System, EpiCentre Biotechnologies, Madison, WI, USA). Part A is with primers hmt L569 [AACCAAACCCCAAAGACAC] and hmtH12111 [AAACCCGGTAATGATGTCGG] while Part B is with primers hmtL11727 [GCCCACGGGCTTACATC] and hmtH1405 [ATCCACCTTCGACCCTTAAG] (Integrated DNA Technologies, Inc., Coralville, IA, USA). The PCR products were run on a 1% agarose gel and then a single step enzymatic elimination of the unincorporated primers and dNTPs was performed (ExoSAP-IT, Affymetrix, Santa Clara, CA, USA). The samples were then sent to ELIM BioPharm for sequencing with internal primers (ELIM Biopharm, Hayward, CA, USA). The sequencing results are analyzed with DNA variant analysis software (Mutation Surveyor, SoftGenetics, State College, PA, USA).
Extracellular flux analysis
The oxygen consumption rate (OCR), extracellular acidification rate (ECAR) and bioenergetic profiles (responses to inhibitors and uncoupling agents) for the cybrids were measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA). Cybrids with haplogroups H (n = cybrids from three different individuals) and J (n = cybrids from three different individuals) were plated overnight at 30 000 cells/well into XF24 plates (Seahorse Bioscience). Plates were washed and placed for 1 h in a 37°C incubator under air in 500 µl of unbuffered DMEM (Dulbecco's modified Eagle's medium, pH 7.4), supplemented with 17.5 mm glucose (Sigma, St. Louis, MO, USA), 200 mm l-glutamine (Invitrogen-Molecular Probes, Carlsbad, CA, USA) and 10 mm sodium pyruvate (Invitrogen-Molecular Probes). The OCR, which represents the basal aerobic respiration of the cells, is determined by measuring the drop in O2 partial pressure over time followed by linear regression to find the slope. The ECAR, which represents glycolysis, is determined by measuring the change in pH levels over time followed by linear regression to find the slope of the line which represents ECAR. The OCR and ECAR values were measured for three H and three J cybrids (each from different individuals) with six separate wells per cybrid. The experiments were repeated twice. These calculations were determined by the Seahorse XF24 program software.
In order to measure the bioenergetic profile of the cells, Oligomycin (1 µm final concentration, which blocks ATP synthase to assess respiration required for ATP turnover), FCCP (carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone, 1 µm final concentration, a proton ionophore which induces chemical uncoupling and maximal respiration) and Rotenone plus Antimycin A (1 µm final concentration of each, which completely inhibits electron transport to measure non-mt respiration) were injected sequentially through ports in the Seahorse Flux Pak cartridges. Data were normalized by total protein from each well.
All data from XF24 assays were collected using the XF Reader software from Seahorse Bioscience. The percentage ATP turnover rate is calculated by the following formula: 100 − (ATP coupler response/basal respiration × 100). The percentage spare respiratory capacity represents a bioenergetic value for cells needing high amounts of ATP in response to demands placed upon them. This is calculated by the formula: ETC accelerator response/basal respiration × 100. Data from these experiments were exported to GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) where they were analyzed, normalized and graphed. Statistical significance was determined by performing two-tailed Student's t-tests and P ≤ 0.05 was considered significant in all experiments.
Isolation of RNA and amplification of cDNA
Cells from cybrid cultures (H cybrids, n = cybrids from three different individuals and J cybrids, n = cybrids from three different individuals) were pelleted, and RNA isolated using the RNeasy Mini-Extraction kit (Qiagen) as described in our previous manuscript (34). One hundred nanograms of individual RNA samples were reverse transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) and used for Q-PCR analyses.
Gene expression arrays and statistical analyses
For gene expression array analyses, RNAs from the three different H haplogroup cybrid cultures or three different J cybrids were combined (250 ng/μl per sample) into two different samples for analyses by the UCLA Clinical MicroArray Core Lab (Affymetrix Human U133 Plus 2.0 Array). The INGENUITY Systems pathway software (Redwood City, CA, USA) was used to analyze the gene expression results. These arrays were originally described in our previous manuscript (34) but these data generated earlier are now being analyzed in detail for specific pathways and genes.
Q-PCR analyses
Q-PCR was performed using 16 different primers (QuantiTect Primer Assay, Qiagen) for genes associated with the complement pathway (CFHR4, CFP, CD55/DAF, CD59, C1QC, C1S, C1R, C4B, C4BPB), inflammatory genes (TGFA, TGFB2, IL33, IL6) and genes related to apoptosis (RARA, BBC-3, BLC2L13; Table 1). Total RNA was isolated from individual pellets of cultured cells of haplogroup H cybrids (n = 3 different individuals) and J cybrids (n = 3 different individuals), as described above. These cDNA samples were not pooled, but, rather, were analyzed individually. The Q-PCR was performed on individual samples using a QuantiFast SYBR Green PCR Kit (Qiagen) on a Bio-Rad iCycler iQ5 detection system. The C1QC, C1S and C4BPB primers were standardized with the housekeeping gene HMBS. The C4B primers were run with the housekeeping gene HPRT1. All other genes were standardized with TBP. The analyses were performed in triplicate. This study compares relative differences in gene expression levels between the H and J cybrids. For the various target genes, housekeeping genes that have comparable amplification efficiencies to the genes of interest were chosen in order to maximize the accuracy of our ΔΔCT values. Statistical analyses of gene expression levels were performed to measure difference between haplogroups using Prism, Version 5.0 (GraphPad Software Inc.).
The MT-DNA primers used for Q-PCR were the following: MT-ND1 F: cccatggccaacctcctactcctc R: agcccgtaggggcctacaacg; MT-ND2 F: aaccctcgttccacagaagct R: ggattatggatgcggttgct; MT-ND3 F: acgagtgcggcttcgaccct R: tcactcataggccagacttagggct; MT-ND4/ND4L F: cccactccctcttagccaatatt R: taggcccaccgctgctt; MT-ND5 F: tgctccgggtccatcatc R: tgagtagtcctcctatttttcgaatatct; MT-ND6 F:gcccccgcaccaataggatcctccc R:cctgaggcatgggggtcaggggt; MT-CO1 F: gcccccgatatggcgtttccccgca R: ggggtctcctcctccggcggggtcg; MT-CO2 F: accaggcgacctgcgactcct R: acccccggtcgtgtagcggt; MT-CO3 F: cccccaacaggcatcaccccgc R:atgccagtatcaggcggcggc; MT-CYB F: cccaccctcacacgattcttta R: ttgctagggctgcaataatgaa; MT-ATP6 F: ttatgagcgggcacagtgatt R: gaagtgggctagggcattttt; MT-ATP8 F: cccaccataattacccccatact R: ggtaggtggtagtttgtgtttaatatttttag. These mtDNA genes were standardized to GAPDH. Our previous study using these cybrids showed that the H cybrids and J cybrids had a similar number of mtDNA copies (34).
Statistical analyses
Data were subjected to statistical analysis by ANOVA, GraphPad Prism, Version 5.0. Newman–Keuls multiple-comparison test was done to compare the data within each experiment. P < 0.05 was considered statistically significant. Error bars in the graphs represent SEM (standard error mean).
FUNDING
This work was supported by the Discovery Eye Foundation, Guenther Foundation, Beckman Macular Research Initiative, Polly and Michael Smith Foundation, Max Factor Family Foundation, Skirball Foundation, Lincy Foundation, Iris and the B. Gerald Cantor Foundation, Challenge Grant from Research to Prevent Blindness and the National Institute on Aging (AG006168 to S.M.J.).
ACKNOWLEDGEMENTS
We thank the subjects who participated in this study.
Conflict of Interest statement.None declared.
REFERENCES
- 1.Wallace D.C. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- 2.Wallace D.C. Mitochondrial DNA mutations in diseases of energy metabolism. J. Bioenerg. Biomembr. 1994;26:241–250. doi: 10.1007/BF00763096. [DOI] [PubMed] [Google Scholar]
- 3.McFarland R., Turnbull D.M. Batteries not included: diagnosis and management of mitochondrial disease. J. Intern. Med. 2009;265:210–228. doi: 10.1111/j.1365-2796.2008.02066.x. [DOI] [PubMed] [Google Scholar]
- 4.Wallace D.C., Lott M.T., Procaccio V. Mitochondrial Genes in Degenerative Diseases, Cancer and Aging. Philadelphis, PA: Churchill Livingstone Elsevier; 2007. [Google Scholar]
- 5.Czarnecka A.M., Bartnik E. The role of the mitochondrial genome in ageing and carcinogenesis. J. Aging Res. 2011;2011:136435. doi: 10.4061/2011/136435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridge P.G., Maxwell T.J., Corcoran C.D., Norton M.C., Tschanz J.T., O'Brien E., Kerber R.A., Cawthon R.M., Munger R.G., Kauwe J.S. Mitochondrial genomic analysis of late onset Alzheimer's disease reveals protective haplogroups H6A1A/H6A1B: the Cache County Study on Memory in Aging. PLoS ONE. 2012;7:e45134. doi: 10.1371/journal.pone.0045134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strauss K.A., DuBiner L., Simon M., Zaragoza M., Sengupta P.P., Li P., Narula N., Dreike S., Platt J., Procaccio V., et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. USA. 2013;110:3453–3458. doi: 10.1073/pnas.1300690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Luca A., Nasi M., Di Giambenedetto S., Cozzi-Lepri A., Pinti M., Marzocchetti A., Mussini C., Fabbiani M., Bracciale L., Cauda R., et al. Mitochondrial DNA haplogroups and incidence of lipodystrophy in HIV-infected patients on long-term antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2012;59:113–120. doi: 10.1097/QAI.0b013e31823daff3.. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Caggiano M., Barallobre-Barreiro J., Rego-Perez I., Crespo-Leiro M.G., Paniagua M.J., Grille Z., Blanco F.J., Domenech N. Mitochondrial DNA haplogroup H as a risk factor for idiopathic dilated cardiomyopathy in Spanish population. Mitochondrion. 2013;13:263–268. doi: 10.1016/j.mito.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Canter J.A., Kallianpur A.R., Fowke J.H. Re: North American white mitochondrial haplogroups in prostate and renal cancer. J. Urol. 2006;176:2308–2309. doi: 10.1016/j.juro.2006.07.067. author reply 2309. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez-Cubero M.J., Saiz Guinaldo M., Martinez-Gonzalez L.J., Alvarez Merino J.C., Cozar Olmo J.M., Acosta J.A. Mitochondrial haplogroups and polymorphisms reveal no association with sporadic prostate cancer in a southern European population. PLoS ONE. 2012;7:e41201. doi: 10.1371/journal.pone.0041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller E.E., Eder W., Mayr J.A., Paulweber B., Sperl W., Horninger W., Klocker H., Kofler B. Mitochondrial haplogroups and control region polymorphisms are not associated with prostate cancer in Middle European Caucasians. PLoS ONE. 2009;4:e6370. doi: 10.1371/journal.pone.0006370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim W., Yoo T.K., Shin D.J., Rho H.W., Jin H.J., Kim E.T., Bae Y.S. Mitochondrial DNA haplogroup analysis reveals no association between the common genetic lineages and prostate cancer in the Korean population. PLoS ONE. 2008;3:e2211. doi: 10.1371/journal.pone.0002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pichaud N., Ballard J.W., Tanguay R.M., Blier P.U. Naturally occurring mitochondrial DNA haplotypes exhibit metabolic differences: insight into functional properties of mitochondria. Evolution. 2012;66:3189–3197. doi: 10.1111/j.1558-5646.2012.01683.x. [DOI] [PubMed] [Google Scholar]
- 15.Tranah G.J. Mitochondrial-nuclear epistasis: implications for human aging and longevity. Ageing Res. Rev. 2011;10:238–252. doi: 10.1016/j.arr.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meiklejohn C.D., Holmbeck M.A., Siddiq M.A., Abt D.N., Rand D.M., Montooth K.L. An Incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 2013;9:e1003238. doi: 10.1371/journal.pgen.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pichaud N., Ballard J.W., Tanguay R.M., Blier P.U. Mitochondrial haplotype divergences affect specific temperature sensitivity of mitochondrial respiration. J. Bioenerg. Biomembr. 2013;45:25–35. doi: 10.1007/s10863-012-9473-9. [DOI] [PubMed] [Google Scholar]
- 18.Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R.W., Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller E.E., Brunner S.M., Mayr J.A., Stanger O., Sperl W., Kofler B. Functional differences between mitochondrial haplogroup T and haplogroup H in HEK293 cybrid cells. PLoS ONE. 2012;7:e52367. doi: 10.1371/journal.pone.0052367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellizzi D., D'Aquila P., Giordano M., Montesanto A., Passarino G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics. 2012;4:17–27. doi: 10.2217/epi.11.109. [DOI] [PubMed] [Google Scholar]
- 21.Brown M.D., Trounce I.A., Jun A.S., Allen J.C., Wallace D.C. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J. Biol. Chem. 2000;275:39831–39836. doi: 10.1074/jbc.M006476200. [DOI] [PubMed] [Google Scholar]
- 22.Pacheu-Grau D., Gomez-Duran A., Iglesias E., Lopez-Gallardo E., Montoya J., Ruiz-Pesini E. Mitochondrial antibiograms in personalized medicine. Hum. Mol. Genet. 2013;22:1132–1139. doi: 10.1093/hmg/dds517. [DOI] [PubMed] [Google Scholar]
- 23.Jones M.M., Manwaring N., Wang J.J., Rochtchina E., Mitchell P., Sue C.M. Mitochondrial DNA haplogroups and age-related maculopathy. Arch. Ophthalmol. 2007;125:1235–1240. doi: 10.1001/archopht.125.9.1235. [DOI] [PubMed] [Google Scholar]
- 24.Canter J.A., Olson L.M., Spencer K., Schnetz-Boutaud N., Anderson B., Hauser M.A., Schmidt S., Postel E.A., Agarwal A., Pericak-Vance M.A., et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE. 2008;3:e2091. doi: 10.1371/journal.pone.0002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Udar N., Atilano S.R., Memarzadeh M., Boyer D., Chwa M., Lu S., Maguen B., Langberg J., Coskun P., Wallace D.C., et al. Mitochondrial DNA haplogroups associated with Age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2009;50:2966–2974. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- 26.SanGiovanni J.P., Arking D.E., Iyengar S.K., Elashoff M., Clemons T.E., Reed G.F., Henning A.K., Sivakumaran T.A., Xu X., DeWan A., et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE. 2009;4:e5508. doi: 10.1371/journal.pone.0005508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller E.E., Schaier E., Brunner S.M., Eder W., Mayr J.A., Egger S.F., Nischler C., Oberkofler H., Reitsamer H.A., Patsch W., et al. Mitochondrial haplogroups and control region polymorphisms in age-related macular degeneration: a case-control study. PLoS ONE. 2012;7:e30874. doi: 10.1371/journal.pone.0030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Redondo D., Marcuello A., Casajus J.A., Ara I., Dahmani Y., Montoya J., Ruiz-Pesini E., Lopez-Perez M.J., Diez-Sanchez C. Human mitochondrial haplogroup H: the highest VO2max consumer--is it a paradox? Mitochondrion. 2010;10:102–107. doi: 10.1016/j.mito.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Marcuello A., Martinez-Redondo D., Dahmani Y., Casajus J.A., Ruiz-Pesini E., Montoya J., Lopez-Perez M.J., Diez-Sanchez C. Human mitochondrial variants influence on oxygen consumption. Mitochondrion. 2009;9:27–30. doi: 10.1016/j.mito.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 30.De Benedictis G., Rose G., Carrieri G., De Luca M., Falcone E., Passarino G., Bonafe M., Monti D., Baggio G., Bertolini S., et al. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB J. 1999;13:1532–1536. doi: 10.1096/fasebj.13.12.1532. [DOI] [PubMed] [Google Scholar]
- 31.Niemi A.K., Hervonen A., Hurme M., Karhunen P.J., Jylha M., Majamaa K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Hum. Genet. 2003;112:29–33. doi: 10.1007/s00439-002-0843-y. [DOI] [PubMed] [Google Scholar]
- 32.Ross O.A., McCormack R., Curran M.D., Duguid R.A., Barnett Y.A., Rea I.M., Middleton D. Mitochondrial DNA polymorphism: its role in longevity of the Irish population. Exp. Gerontol. 2001;36:1161–1178. doi: 10.1016/s0531-5565(01)00094-8. [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Duran A., Pacheu-Grau D., Martinez-Romero I., Lopez-Gallardo E., Lopez-Perez M.J., Montoya J., Ruiz-Pesini E. Oxidative phosphorylation differences between mitochondrial DNA haplogroups modify the risk of Leber's hereditary optic neuropathy. Biochim. Biophys. Acta. 2012;1822:1216–1222. doi: 10.1016/j.bbadis.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Kenney M.C., Chwa M., Atilano S.R., Pavlis J.M., Falatoonzadeh P., Ramirez C., Malik D., Hsu T., Woo G., Soe K., et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3, and EFEMP1 genes: Implications for age-related macular degeneration. PLoS ONE. 2013;8:e54339–e54348. doi: 10.1371/journal.pone.0054339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratnapriya R., Chew E.Y. Age-related macular degeneration-clinical review and genetics update. Clin. Genet. 2013;84:160–166. doi: 10.1111/cge.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynolds R., Rosner B., Seddon J.M. Dietary omega-3 fatty acids, other fat intake, genetic susceptibility, and progression to incident geographic atrophy. Ophthalmology. 2013;120:1020–1028. doi: 10.1016/j.ophtha.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Walt J.M., Dementieva Y.A., Martin E.R., Scott W.K., Nicodemus K.K., Kroner C.C., Welsh-Bohmer K.A., Saunders A.M., Roses A.D., Small G.W., et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 38.van der Walt J.M., Nicodemus K.K., Martin E.R., Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Haines J.L., Koller W.C., Lyons K., et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huerta C., Castro M.G., Coto E., Blazquez M., Ribacoba R., Guisasola L.M., Salvador C., Martinez C., Lahoz C.H., Alvarez V. Mitochondrial DNA polymorphisms and risk of Parkinson's disease in Spanish population. J. Neurol. Sci. 2005;236:49–54. doi: 10.1016/j.jns.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 40.Coskun P., Wyrembak J., Schriner S., Chen H.W., Marciniack C., Laferla F., Wallace D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Moreno M., Soto-Hermida A., Oreiro N., Pertega S., Fenandez-Lopez C., Rego-Perez I., Blanco F.J. Mitochondrial haplogroups define two phenotypes of osteoarthritis. Front. Physiol. 2012;3:129. doi: 10.3389/fphys.2012.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D'Aquila P., Rose G., Panno M.L., Passarino G., Bellizzi D. SIRT3 gene expression: a link between inherited mitochondrial DNA variants and oxidative stress. Gene. 2012;497:323–329. doi: 10.1016/j.gene.2012.01.042. [DOI] [PubMed] [Google Scholar]
- 43.Arning L., Haghikia A., Taherzadeh-Fard E., Saft C., Andrich J., Pula B., Hoxtermann S., Wieczorek S., Akkad D.A., Perrech M., et al. Mitochondrial haplogroup H correlates with ATP levels and age at onset in Huntington disease. J. Mol. Med. (Berl.) 2010;88:431–436. doi: 10.1007/s00109-010-0589-2. [DOI] [PubMed] [Google Scholar]
- 44.Gomez-Duran A., Pacheu-Grau D., Lopez-Gallardo E., Diez-Sanchez C., Montoya J., Lopez-Perez M.J., Ruiz-Pesini E. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 2010;19:3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- 45.Kenney M.C., Chwa M., Atilano S.R., Falatoonzadeh P., Ramirez C., Malik D., Tarek M., Del Carpio J.C., Nesburn A.B., Boyer D.S., et al. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim. Biophys. Acta. 2014;1842:208–219. doi: 10.1016/j.bbadis.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiz-Pesini E., Mishmar D., Brandon M., Procaccio V., Wallace D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- 47.Pierron D., Rocher C., Amati-Bonneau P., Reynier P., Martin-Negrier M.L., Allouche S., Batandier C., Mousson de Camaret B., Godinot C., Rotig A., et al. New evidence of a mitochondrial genetic background paradox: impact of the J haplogroup on the A3243G mutation. BMC. Med. Genet. 2008;9:41. doi: 10.1186/1471-2350-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pierron D., Chang I., Arachiche A., Heiske M., Thomas O., Borlin M., Pennarun E., Murail P., Thoraval D., Rocher C., et al. Mutation rate switch inside Eurasian mitochondrial haplogroups: impact of selection and consequences for dating settlement in Europe. PLoS ONE. 2011;6:e21543. doi: 10.1371/journal.pone.0021543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reynolds R., Hartnett M.E., Atkinson J.P., Giclas P.C., Rosner B., Seddon J.M. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest. Ophthalmol. Vis. Sci. 2009;50:5818–5827. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haines J.L., Hauser M.A., Schmidt S., Scott W.K., Olson L.M., Gallins P., Spencer K.L., Kwan S.Y., Noureddine M., Gilbert J.R., et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 51.Klein R.J., Zeiss C., Chew E.Y., Tsai J.Y., Sackler R.S., Haynes C., Henning A.K., Sangiovanni J.P., Mane S.M., Mayne S.T., et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hageman G.S., Anderson D.H., Johnson L.V., Hancox L.S., Taiber A.J., Hardisty L.I., Hageman J.L., Stockman H.A., Borchardt J.D., Gehrs K.M., et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gold B., Merriam J.E., Zernant J., Hancox L.S., Taiber A.J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G.R., et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edwards A.O., Ritter R., 3rd, Abel K.J., Manning A., Panhuysen C., Farrer L.A. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 55.Maller J., George S., Purcell S., Fagerness J., Altshuler D., Daly M.J., Seddon J.M. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat. Genet. 2006;38:1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 56.Maller J.B., Fagerness J.A., Reynolds R.C., Neale B.M., Daly M.J., Seddon J.M. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat. Genet. 2007;39:1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 57.Fagerness J.A., Maller J.B., Neale B.M., Reynolds R.C., Daly M.J., Seddon J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009;17:100–104. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudson G., Carelli V., Spruijt L., Gerards M., Mowbray C., Achilli A., Pyle A., Elson J., Howell N., La Morgia C., et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am. J. Hum. Genet. 2007;81:228–233. doi: 10.1086/519394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferreira I.L., Nascimento M.V., Ribeiro M., Almeida S., Cardoso S.M., Grazina M., Pratas J., Santos M.J., Januario C., Oliveira C.R., et al. Mitochondrial-dependent apoptosis in Huntington's disease human cybrids. Exp. Neurol. 2010;222:243–255. doi: 10.1016/j.expneurol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Jun A.S., Trounce I.A., Brown M.D., Shoffner J.M., Wallace D.C. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol. Cell. Biol. 1996;16:771–777. doi: 10.1128/mcb.16.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson D.H., Radeke M.J., Gallo N.B., Chapin E.A., Johnson P.T., Curletti C.R., Hancox L.S., Hu J., Ebright J.N., Malek G., et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog. Retin. Eye Res. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fett A.L., Hermann M.M., Muether P.S., Kirchhof B., Fauser S. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol. Histopathol. 2012;27:357–364. doi: 10.14670/HH-27.357. [DOI] [PubMed] [Google Scholar]
- 63.Bellizzi D., Taverna D., D'Aquila P., De Blasi S., De Benedictis G. Mitochondrial DNA variability modulates mRNA and intra-mitochondrial protein levels of HSP60 and HSP75: experimental evidence from cybrid lines. Cell Stress Chaperones. 2009;14:265–271. doi: 10.1007/s12192-008-0081-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bellizzi D., Cavalcante P., Taverna D., Rose G., Passarino G., Salvioli S., Franceschi C., De Benedictis G. Gene expression of cytokines and cytokine receptors is modulated by the common variability of the mitochondrial DNA in cybrid cell lines. Genes Cells. 2006;11:883–891. doi: 10.1111/j.1365-2443.2006.00986.x. [DOI] [PubMed] [Google Scholar]
- 65.Fassio J.B., Brockman E.B., Jumblatt M., Greaton C., Henry J.L., Geoghegan T.E., Barr C., Schultz G.S. Transforming growth factor alpha and its receptor in neural retina. Invest. Ophthalmol. Vis. Sci. 1989;30:1916–1922. [PubMed] [Google Scholar]
- 66.Lillien L., Cepko C. Control of proliferation in the retina: temporal changes in responsiveness to FGF and TGF alpha. Development. 1992;115:253–266. doi: 10.1242/dev.115.1.253. [DOI] [PubMed] [Google Scholar]
- 67.Anchan R.M., Reh T.A., Angello J., Balliet A., Walker M. EGF and TGF-alpha stimulate retinal neuroepithelial cell proliferation in vitro. Neuron. 1991;6:923–936. doi: 10.1016/0896-6273(91)90233-p. [DOI] [PubMed] [Google Scholar]
- 68.Sagar S.M., Edwards R.H., Sharp F.R. Epidermal growth factor and transforming growth factor alpha induce c-fos gene expression in retinal Muller cells in vivo. J. Neurosci. Res. 1991;29:549–559. doi: 10.1002/jnr.490290416. [DOI] [PubMed] [Google Scholar]
- 69.Murphy G.E., Xu D., Liew F.Y., McInnes I.B. Role of interleukin 33 in human immunopathology. Ann. Rheum. Dis. 2010;69(Suppl. 1):i43–i47. doi: 10.1136/ard.2009.120113. [DOI] [PubMed] [Google Scholar]
- 70.Liew F.Y. IL-33: a Janus cytokine. Ann. Rheum. Dis. 2012;71(Suppl. 2):i101–i104. doi: 10.1136/annrheumdis-2011-200589. [DOI] [PubMed] [Google Scholar]
- 71.Kuchler A.M., Pollheimer J., Balogh J., Sponheim J., Manley L., Sorensen D.R., De Angelis P.M., Scott H., Haraldsen G. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am. J. Pathol. 2008;173:1229–1242. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Komai-Koma M., Xu D., Li Y., McKenzie A.N., McInnes I.B., Liew F.Y. IL-33 is a chemoattractant for human Th2 cells. Eur. J. Immunol. 2007;37:2779–2786. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 73.Schmitz J., Owyang A., Oldham E., Song Y., Murphy E., McClanahan T.K., Zurawski G., Moshrefi M., Qin J., Li X., et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 74.Smithgall M.D., Comeau M.R., Yoon B.R., Kaufman D., Armitage R., Smith D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 75.Miller A.M., Xu D., Asquith D.L., Denby L., Li Y., Sattar N., Baker A.H., McInnes I.B., Liew F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008;205:339–346. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rank M.A., Kobayashi T., Kozaki H., Bartemes K.R., Squillace D.L., Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009;123:1047–1054. doi: 10.1016/j.jaci.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carriere V., Roussel L., Ortega N., Lacorre D.A., Americh L., Aguilar L., Bouche G., Girard J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leung B.P., Xu D., Culshaw S., McInnes I.B., Liew F.Y. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J. Immunol. 2004;173:145–150. doi: 10.4049/jimmunol.173.1.145. [DOI] [PubMed] [Google Scholar]
- 79.Seidelin J.B., Bjerrum J.T., Coskun M., Widjaya B., Vainer B., Nielsen O.H. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol. Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Kurowska-Stolarska M., Stolarski B., Kewin P., Murphy G., Corrigan C.J., Ying S., Pitman N., Mirchandani A., Rana B., van Rooijen N., et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 81.Tuo J., Grob S., Zhang K., Chan C.C. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocular Immunol. Inflamm. 2012;20:27–36. doi: 10.3109/09273948.2011.628432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu X.C., Liu X.F., Jian C.X., Li C.J., He S.Z. IL-33 is induced by amyloid-beta stimulation and regulates inflammatory cytokine production in retinal pigment epithelium cells. Inflammation. 2012;35:776–784. doi: 10.1007/s10753-011-9379-4. [DOI] [PubMed] [Google Scholar]
- 83.Iriyama A., Fujiki R., Inoue Y., Takahashi H., Tamaki Y., Takezawa S., Takeyama K., Jang W.D., Kato S., Yanagi Y. A2E, a pigment of the lipofuscin of retinal pigment epithelial cells, is an endogenous ligand for retinoic acid receptor. J. Biol. Chem. 2008;283:11947–11953. doi: 10.1074/jbc.M708989200. [DOI] [PubMed] [Google Scholar]
- 84.Iriyama A., Inoue Y., Takahashi H., Tamaki Y., Jang W.D., Yanagi Y. A2E, a component of lipofuscin, is pro-angiogenic in vivo. J. Cell. Physiol. 2009;220:469–475. doi: 10.1002/jcp.21792. [DOI] [PubMed] [Google Scholar]
- 85.Finnemann S.C., Leung L.W., Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA. 2002;99:3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suter M., Reme C., Grimm C., Wenzel A., Jaattela M., Esser P., Kociok N., Leist M., Richter C. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J. Biol. Chem. 2000;275:39625–39630. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]
- 87.Bergmann M., Schutt F., Holz F.G., Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:562–564. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]
- 88.Sparrow J.R., Nakanishi K., Parish C.A. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest. Ophthalmol. Vis. Sci. 2000;41:1981–1989. [PubMed] [Google Scholar]
- 89.Holleman A., den Boer M.L., de Menezes R.X., Cheok M.H., Cheng C., Kazemier K.M., Janka-Schaub G.E., Gobel U., Graubner U.B., Evans W.E., et al. The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood. 2006;107:769–776. doi: 10.1182/blood-2005-07-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang Y.L., Lin S.R., Chen J.S., Lin S.W., Yu S.L., Chen H.Y., Yen C.T., Lin C.Y., Lin J.F., Lin K.H., et al. Expression and prognostic significance of the apoptotic genes BCL2L13, Livin, and CASP8AP2 in childhood acute lymphoblastic leukemia. Leuk. Res. 2010;34:18–23. doi: 10.1016/j.leukres.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 91.Kataoka T., Holler N., Micheau O., Martinon F., Tinel A., Hofmann K., Tschopp J. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 2001;276:19548–19554. doi: 10.1074/jbc.M010520200. [DOI] [PubMed] [Google Scholar]
- 92.Jazwinski S.M., Kriete A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physiol. 2012;3:139. doi: 10.3389/fphys.2012.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ballinger S.W. Beyond retrograde and anterograde signalling: mitochondrial-nuclear interactions as a means for evolutionary adaptation and contemporary disease susceptibility. Biochem. Soc. Trans. 2013;41:111–117. doi: 10.1042/BST20120227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nargund A.M., Pellegrino M.W., Fiorese C.J., Baker B.M., Haynes C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dunn K.C., Marmorstein A.D., Bonilha V.L., Rodriguez-Boulan E., Giordano F., Hjelmeland L.M. Use of the ARPE-19 cell line as a model of RPE polarity: basolateral secretion of FGF5. Invest. Ophthalmol. Vis. Sci. 1998;39:2744–2749. [PubMed] [Google Scholar]
- 96.Miceli M.V., Jazwinski S.M. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2005;46:1765–1773. doi: 10.1167/iovs.04-1327. [DOI] [PubMed] [Google Scholar]
- 97.Chomyn A. Platelet-mediated Transformation of Human Mitochondrial DNA-Less Cells. Salt Lake City, UT: Academic Press, Inc.; 1996. [DOI] [PubMed] [Google Scholar]
- 98.Zaragoza M.V., Brandon M.C., Diegoli M., Arbustini E., Wallace D.C. Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur. J. Hum. Genet. 2011;19:200–207. doi: 10.1038/ejhg.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]