

Abstract
Intronic expansion of a hexanucleotide GGGGCC repeat in the chromosome 9 open reading frame 72 (C9ORF72) gene is the major cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. However, the cellular function of the C9ORF72 protein remains unknown. Here, we demonstrate that C9ORF72 regulates endosomal trafficking. C9ORF72 colocalized with Rab proteins implicated in autophagy and endocytic transport: Rab1, Rab5, Rab7 and Rab11 in neuronal cell lines, primary cortical neurons and human spinal cord motor neurons, consistent with previous predictions that C9ORF72 bears Rab guanine exchange factor activity. Consistent with this notion, C9ORF72 was present in the extracellular space and as cytoplasmic vesicles. Depletion of C9ORF72 using siRNA inhibited transport of Shiga toxin from the plasma membrane to Golgi apparatus, internalization of TrkB receptor and altered the ratio of autophagosome marker light chain 3 (LC3) II:LC3I, indicating that C9ORF72 regulates endocytosis and autophagy. C9ORF72 also colocalized with ubiquilin-2 and LC3-positive vesicles, and co-migrated with lysosome-stained vesicles in neuronal cell lines, providing further evidence that C9ORF72 regulates autophagy. Investigation of proteins interacting with C9ORF72 using mass spectrometry identified other proteins implicated in ALS; ubiquilin-2 and heterogeneous nuclear ribonucleoproteins, hnRNPA2/B1 and hnRNPA1, and actin. Treatment of cells overexpressing C9ORF72 with proteasome inhibitors induced the formation of stress granules positive for hnRNPA1 and hnRNPA2/B1. Immunohistochemistry of C9ORF72 ALS patient motor neurons revealed increased colocalization between C9ORF72 and Rab7 and Rab11 compared with controls, suggesting possible dysregulation of trafficking in patients bearing the C9ORF72 repeat expansion. Hence, this study identifies a role for C9ORF72 in Rab-mediated cellular trafficking.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is characterized by degeneration of upper and lower motor neurons in the brain, brainstem and spinal cord, leading to progressive paralysis. Frontotemporal dementia (FTD) is the second most common cause of presenile dementia (1) and increasing evidence suggests that ALS and FTD overlap, occupying two opposite poles of disease continuum (2–4). Hexanucleotide (GGGGCC) repeat expansions in a non-coding region of chromosome 9 open reading frame 72 (C9ORF72) are the major cause of familial ALS (∼33%) and FTD (∼25%) worldwide and are present in 8% of sporadic ALS cases, highlighting a major role for C9ORF72 in neurodegeneration (5–7). The normal cellular function of C9ORF72 remains unknown but it is highly conserved and expressed in many tissues, including the cerebellum, cortex and spinal cord. Similarly, it remains unclear how C9ORF72 repeat expansions trigger ALS pathology, although haploinsufficiency due to impaired transcription/splicing, leading to reduced C9ORF72 protein expression (up to 50%) (8), RNA dysfunction, and unconventional translation of the repeat to generate insoluble polypeptides, are possible mechanisms (9,10).
Rab GTPases regulate membrane trafficking events and efficient intracellular trafficking is essential for cellular viability (11). Rab GTPases are master regulators of nearly all membrane traffic through their interactions with vesicular coat components, motor proteins and SNARE proteins. In humans, there are >60 members of the Rab family that are localized to distinct intracellular membranes. Rabs alternate between two conformational states: the activated guanosine tri-phosphate (GTP)-bound form and the guanosine di-phosphate (GDP)-bound inactive form. Exchange of GDP with GTP is catalyzed by Rab guanine nucleotide exchange factors (GEFs) that act at specific membranes and facilitate GDP release, thus locally activating their targets. DENN (differentially expressed in normal and neoplastic cells) domain-containing proteins are RabGEFs that activate mostly endocytotic Rabs (12). Two recent bioinformatics studies predicted that C9ORF72 possesses DENN domains (13,14), raising the possibility that it regulates Rab-dependent intracellular trafficking (12). The endosomal system is necessary for regulating, sorting and degrading proteins via autophagy or the ubiquitin–proteasome system (UPS) (15). Multiple Rabs have been implicated in autophagy including Rab1, Rab5, Rab7 and Rab11 (16–18).
Defects in protein degradation are increasingly implicated in ALS pathogenesis (19) and mutations in ubiquilin-2 (20), which regulates autophagy and the UPS by binding/transport of protein cargo (21), also cause ALS/FTD. Inhibition of the proteasome induces the formation of stress granules (SGs) (22), a cellular hallmark of ALS (23). Recently, mutations in heterogeneous nuclear ribonucleoproteins hnRNPA2/B1 and hnRNPA1 were identified in ALS patients (24). hnRNPs granules are major components of SGs that mediate nucleocytoplasmic trafficking of mRNA and RNA metabolism (25). Inhibition of the proteasome triggers alternative splicing of hnRNPs and mRNA-bound hnRNPA1 is recruited to cytoplasmic SGs (26).
Elucidation of the function of C9ORF72 is essential to understand its role in ALS/FTD. Here, we demonstrate a role for C9ORF72 in endosomal trafficking. C9ORF72 colocalized with ubiquilin-2 and Rab proteins implicated in autophagy, and co-migrated with lysosome-positive vesicles. Depletion of C9ORF72 using siRNA dsyregulated autophagy and inhibited endocytosis. Mass spectrometry identified other proteins linked to ALS as interacting partners of C9ORF72: hnRNPA1, hnRNPA2/B1, ubiquilin-2 and actin. Proteasome inhibition and C9ORF72 overexpression led to the formation of nuclear C9ORF72 aggregates and cytoplasmic SGs positive for hnRNPA1 and hnRNPA2/B1. Hence, this study defines novel functions for C9ORF72 in cellular trafficking and protein degradation.
RESULTS
C9ORF72 colocalizes and interact with Rabs in neuronal cell lines
To characterize C9ORF72, we examined expression of endogenous C9ORF72 in neuronal cell lines; murine neuro2a and human SH-SY5Y. Immunofluorescence was present diffuse in the nucleus and as cytoplasmic vesicles (Fig. 1A and B). Similarly, in cells transfected with a construct-encoding GFP-tagged C9ORF72 (Fig. 1C), both nuclear protein and cytoplasmic vesicles were observed (Fig. 1C). The cellular distribution of endogenous C9ORF72 was investigated further using subcellular fractionation of SH-SY5Y cell lysates. Using immunoblotting, we detected that significantly more C9ORF72 was present in the nucleus compared with the cytoplasm (Fig. 1D and E); 64%, P< 0.05). C9ORF72 in the nuclear fraction was present as the 50 kDa isoform; however, in the cytoplasmic fraction, lower molecular weight bands were present in addition to the 50 kDa band, possibly representing the 25 kDa isoform (Fig. 1D). We also detected C9ORF72 in conditioned medium, suggesting active secretion (Fig. 1F), presumably via non-classical means because bioinformatics analysis predicted that a signal leader peptide was not present in C9ORF72 (Secretome P) (27). This was confirmed by immunoblotting of human CSF (Fig. 1G).
Figure 1.

C9ORF72 expression and secretion in neuronal cell lines. (A) Murine Neuro2a cells were fixed and immunostained with anti-C9ORF72 antibodies (green) and DAPI (blue); scale bar: 10 μm. White arrows indicate C9ORF72-positive vesicular-type structures present in the cytoplasm; expression of C9ORF72 is diffuse in the nucleus. (B) Human SH-SY5Y cells were fixed and immunostained with anti-C9ORF72 antibodies (green) and DAPI (blue), expression of C9ORF72 is similar to (A). Scale bar: 10 μm. White arrows indicate C9ORF72-positive vesicular-type structures present in the cytoplasm. (C) Overexpression of C9ORF72-Variant 1 tagged with GFP in SH-SY5Y cells forms cytoplasmic vesicles. (D) Subcellular fractionation of human SH-SY5Y neuroblastoma cells; immunoblotting of nuclear and cytoplasmic fractions. Histone H3 and GAPDH were used as subcellular markers and loading controls for nucleus and cytoplasm, respectively. C9ORF72 is expressed primarily as the 50 kDa isoform in the nucleus, additional bands are present in the cytoplasmic fraction at 25 and 36 kDa. (E) Quantification of endogenous C9ORF72 present in the nuclear and cytoplasmic fraction by densitometry of immunoblots. Data are represented as mean ± SEM; *P < 0.05, n = 3 nuclear versus cytoplasmic by unpaired t-test. (F) C9ORF72 isoforms (50 and 25 kDa) are present in conditioned medium of Neuro2a cells immunoprecipitated using an anti-C9ORF72 antibody; cell lysate fraction shown as a control. (G) C9ORF72 is secreted in human CSF. Immunoblotting with C9ORF72 antibody detects both C9ORF72 isoforms, corresponding to 50 and 25 kDa proteins.
C9ORF72 expression in vesicles suggests a possible role in cellular trafficking, consistent with RabGEF function. This was investigated using immunocytochemistry for Rab proteins. C9ORF72 colocalized strongly with Rabs implicated in autophagy and endosomal transport: Rab1, Rab7, Rab5 and Rab11 (Fig. 2A). Similarly, C9ORF72 co-immunoprecipitated with Rab1, Rab7 and Rab11 (Fig. 2B); control immunoprecipitations using isotype-matched antibodies were negative. This was confirmed by precipitations using GFP-Trap in the reverse direction, with cell lysates transfected with Rab-GFP constructs (Supplementary Material, Fig. S1). Similarly, C9ORF72 was precipitated using GFP-Trap from cells transfected with Rab5-GFP, although we could not determine if Rab5 precipitated with C9ORF72 antibodies due to species cross-reactivity with the available antibodies. However, these findings suggest a physical interaction between C9ORF72 and Rab proteins, consistent with predictions that C9ORF72 is a RabGEF.
Figure 2.

C9ORF72 colocalizes with endosomal Rabs; Rab1, Rab5, Rab7 and Rab11 in neuronal cell lines. (A) Neuro2a cells were fixed and immunostained with anti-C9ORF72 antibodies (red) and either anti-Rab7 or anti-Rab1 antibodies (green), or transfected with constructs encoding either GFP-Rab5 or GFP-Rab11 for 48 h, followed by DAPI staining (blue). The white arrows indicate regions of colocalization between C9ORF72 and the respective Rabs. Scale bar: 10 μm, applied to all fields. Insets demonstrate higher magnification (×100) of the areas highlighted to illustrate colocalization with vesicular structures. (B) Co-immunoprecipitation followed by western blotting revealed that Rab7 and Rab11 are pulled down using anti-C9ORF72 antibodies, and that C9ORF72 is pulled down using anti-Rab1 antibodies. Control immunoprecipitations using an isotype-matched, irrelevant IgG antibody were negative, indicating no cross-reactivity with the antibodies used. 1% input also shown.
We examined the expression of C9ORF72 further in primary cortical neurons obtained from C57Bl/6 mice. C9ORF72 was present both in the nucleus and in vesicles, similar to its expression in neuronal cell lines (Fig. 3A). Immunocytochemistry using Rab5, Rab7 and Rab11 antibodies revealed that C9ORF72-positive vesicles frequently colocalized with Rab5, Rab7 and Rab11 (Fig. 3B). Interestingly, we detected colocalization of C9ORF72 with Rab7 in the axon and the cell body. These data provide further evidence that C9ORF72 has a role in protein trafficking with multiple Rab proteins (Fig. 3A and B). To further validate the interaction between Rab proteins and C9ORF72, C9ORF72-GFP was expressed in neuro2a cells and immunostaining with Rab5 was performed. Rab5 and GFP-tagged fluorescent C9ORF72 vesicles were clearly colocalized in these cells (Supplementary Material, Fig. S2).
Figure 3.

C9ORF72 colocalizes with Rab5, Rab7 and Rab11 in primary neurons. Primary cortical neuronal cells were obtained from the cerebrum of C56/Bl6 mice at E15.5; cortical tissue was dissected from E15.5 mouse embryos and dissociated using 0.0125% trypsin. Tissue was washed in cell plating media, and cells were plated onto poly-l-lysine-coated 12 mm coverslips in 24-well plates at a concentration of 30 000 cells per coverslip. (A) Cells were fixed and immunostained with anti-C9ORF72 antibodies (red), neuronal marker microtubule-associated protein 2 (MAP2) (green), and DAPI (blue). Scale bar: 10 μm. White arrows indicate C9ORF72-positive vesicular structures present in the cytoplasm and axons. (B) Cells were fixed and immunostained with anti-C9ORF72 antibodies (red) and either anti-Rab5 or anti-Rab7 or anti-Rab11 antibodies (green), the white arrows indicate regions of C9ORF72 and the respective Rab immunoreactivity. Scale bar: 10 μm, applied to all fields.
Rab7 and Rab11 colocalize in C9ORF72 human spinal cord motor neurons
These findings were investigated further using immunohistochemistry of human spinal cord sections. Quantitative analysis of 30+ motor neurons in control patients revealed that C9ORF72 colocalized with Rab5 (60%), Rab7 (70%) and Rab11 (60%), consistent with the findings obtained from cell lines (Fig. 4A and B), confirming that C9ORF72 colocalizes with Rab proteins. Quantitation of motor neurons from an ALS patient with the C9ORF72 hexanucleotide repeat expansion revealed an increased proportion of cells with colocalization of C9ORF72 with Rab7 or Rab11 compared with controls (P < 0.05) (Fig. 4B). These data suggest possible dysregulation of endosomal trafficking in ALS patients with the C9ORF72 hexanucleotide repeat expansion.
Figure 4.
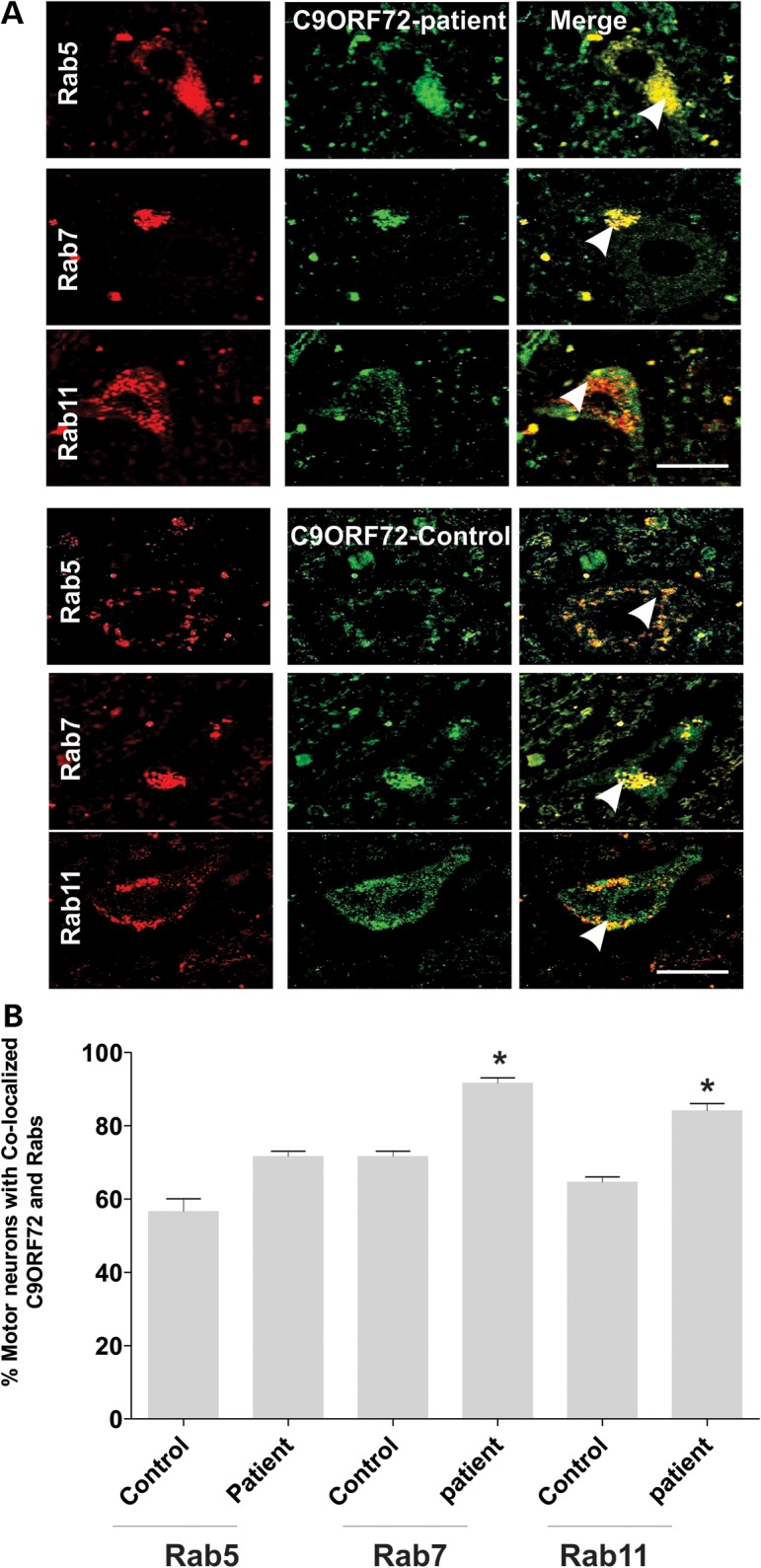
Colocalization of Rab 5, Rab 7 and Rab 11 in human spinal cord motor neurons in an ALS patient with C9ORF72-intronic repeat expansion mutation. (A) Immunohistochemistry of human postmortem spinal cord sections from a control individual without neurological disorders and a human ALS patient bearing C9ORF72-intronic mutation. Human postmortem spinal cord sections (5 µm) were immunostained with anti-Rab11, anti-Rab5 or anti-Rab7 (first column) antibodies and anti-C9ORF72 antibodies (second column). Merge (third column) indicates overlays of the fluorescent confocal images of C9ORF72 and each Rab. White arrow indicates areas of colocalization between C9ORF72 and Rabs in both control and ALS patient tissues. Scale bar: 20 μm, applied to all fields. (B) Quantification of motor neurons containing colocalized C9ORF72 and Rabs reveals an increased proportion of motor neurons in which C9ORF72 colocalized with Rab11 or Rab7 in tissues from an ALS patient bearing the C9ORF72 repeat expansion compared with a control patient. Fifty motor neuron cells were scored for each population. Data are represented as mean ± SEM; *P < 0.05, ALS versus control by unpaired t-test.
C9ORF72 regulates endocytosis in neuronal cell lines
Next, to examine if C9ORF72 regulates endocytosis, human neuronal SH-SY5Y cells were treated with short interfering RNA (siRNA) duplexes to silence human C9ORF72 expression. Using immunoblotting, C9ORF72 levels were depleted by 30%, i.e. to 70% of the original expression level, without obvious off-target effects, as indicated by no change in the levels of β-actin (Fig. 5A). Endocytosis was examined using Shiga toxin subunit B (STxB), an endocytosis marker, conjugated to Cy3, which traffics from the plasma membrane to Golgi network (28). Mander's overlap coefficient (29) was calculated to quantify colocalization between STxB and cis-Golgi marker GM130, detected immunocytochemically. In cells depleted of C9ORF72, transport of StxB-Cy3 to the Golgi apparatus was inhibited by 18% (Fig. 5B and C; P < 0.01) compared with cells treated with control siRNA. Hence, C9ORF72 regulates an endocytotic pathway used by Shiga toxin to reach the cis-Golgi network. To confirm this finding, we also examined endocytosis of TrkB receptor by cell surface biotinylation in SH-SY5Y cells depleted of C9ORF72 by 80% using two successive siRNA transfections (Fig. 5D). C9ORF72 depleted cells expressing FLAG-tagged TrkB were biotinylated for 30 min at 4°C. Endocytosis of biotinylated TrKB receptor was then induced by incubation with BDNF ligand. Internalized TrkB receptor was precipitated with streptavidin beads and quantified by immunoblotting with an anti-FLAG antibody. In cells 80% depleted of C90RF72, endocytosis of TrkB was inhibited by 87% compared with control cells, confirming that C9ORF72 regulates endocytosis (Fig. 5E and F).
Figure 5.

C9ORF72 mediates endocytosis of Shiga toxin-CY3 and TrkB receptor. (A) SH-SY5Y cells were transfected with C9ORF72-targeted siRNA and control siRNA for 72 h. Cell lysates were harvested and immunoblotting followed by densitometry quantification revealed that C9ORF72 expression was reduced by 30% in cells treated with C9ORF72 siRNA compared with control siRNA-treated cells. (B) Purified Shiga toxin conjugated to CY3 (SxTB-CY3) (red) was added to the medium of cells treated with C9ORF72 and control siRNA for 30 min. Endocytosis was examined after 60 min using immunocytochemistry for Golgi marker GM130 (green). (C) The colocalization between C9ORF72 and GM130 was quantified using Mander's coefficient, revealing 18% inhibition of endocytosis of Shiga toxin in C9ORF72-siRNA-treated cells. For each of two replicate experiments, 50 cells were scored for each population. Data are represented as mean ± SEM; **P < 0.001, using unpaired t-test. (D) SH-SY5Y cells were transfected with C9ORF72-targeted siRNA and control siRNA followed by a second transfection 24 h later. Cell lysates were harvested and immunoblotting followed by densitometry quantification revealed that C9ORF72 expression was reduced by 80% compared with control siRNA-treated cells. (E) C90RF72 depleted and control cells expressing FLAG-tagged TrKB receptor were treated with biotin NHS at 4°C to biotinylate cell surface proteins. The cells were then incubated at 37°C and medium containing BDNF was added to induce endocytosis of TrKB receptor. The rate of TrKB receptor endocytosis was examined after 2 h by precipitating biotinylated and internalized TrKB receptor from cell lysates with strepatividin beads and immunoblotting with an anti-FLAG antibody. (F) Densitometric quantification of immunoblots reveals inhibition of endocytosis of TrkB receptor by 87% in cells depleted of C9ORF72 compared with control cells. Biotin precipitation and immunoblotting were performed in triplicate. Data are represented as mean ± SEM; *P < 0.05, using unpaired t-tests.
C9ORF72 regulates autophagy in neuronal cell lines
To examine if C9ORF72 regulates autophagy, cells were cotransfected with constructs-encoding C9ORF72-GFP and DsRed-tagged microtubule-associated protein 1 light chain 3 (LC3), a marker for autophagosomes, in neuro2a cells. The appearance of C9ORF72 as punctate structures that colocalized with DsRed-LC3 in 23% of cells (counted from at least 100 cells), suggested that C9ORF72 associates with autophagosome-like structures under basal conditions (Fig. 6A). Blockage of autophagosome fusion to the lysosome by bafilomycin yielded a significantly increased proportion of cells with LC3-positive structures, suggestive of autophagosomes (73%, P < 0.01), providing further evidence that C9ORF72 associates with autophagosomes (Fig. 6B–D). Immunoblotting for LC3I relative to its phosphatidylethanolamine-modified product, LC3II, was investigated in human SH-SY5Y cells depleted of C9ORF72 by 75% using siRNA (Fig. 6E). The LC3II:LC3I ratio was increased by 45% (P < 0.001) in siRNA-treated cells, indicating dysregulated autophagosome formation. In contrast, there was no change in LC3II:LC3I ratio in cells treated with control siRNA (Fig. 6F) compared with untreated cells. Similarly, in the presence of bafilomycin, LC3II levels increased in SH-SY5Y cells, indicating efficient autophagic flux in this cell line (Fig. 6D), thus confirming the specificity of the findings with C9ORF72 depletion. C9ORF72-positive vesicles also colocalized with Lysotracker, a marker of lysosomes, in every cell examined (Fig. 6G), and co-migrated with Lysotracker-stained vesicles in live cells, indicating that C9ORF72 was present in acidic vesicles of the endolysomal system (Supplementary Material, Fig. S2). Hence, taken together, these data suggest that C9ORF72 regulates autophagy-mediated trafficking.
Figure 6.

C9ORF72 regulates autophagy in neuronal cell lines. (A) Neuro2a cells were cotransfected with DsRed-LC3 for 48 h. Fluorescence microscopy revealed that C9ORF72 appeared as punctate structures that colocalized with DsRed-LC3, indicating autophagosomes, inset demonstrates higher magnification (×100) of the areas highlighted to illustrate autophagosomes, Scale bar: 10 μm. (B) Cells were treated with 100 nm bafilomycin for 4 h to inhibit fusion of autophagosomes to the lysosome. White arrow represents C9ORF72 colocalization with autophagosomes. Scale bar: 10 μm. (C) Quantification of the percentage of cells in which C9ORF72 colocalized with DsRed-LC3 revealed elevation in the numbers of LC3-positive structures, indicating autophagosomes, colocalizing with C9ORF72 in bafilomycin treated cells. Data are represented as mean ± SEM; *P < 0.05 versus untreated cells by unpaired t-test, 50 cells were scored, n = 2. (D) Human SH-SY5Y cells were treated with 100 nm bafilomycin for 4 h to test examine autophagic flux by LC3 immunoblotting. (E) Human SH-SY5Y cells were transfected with human C9ORF72-targeting or control siRNA for 72 h. Immunoblotting revealed depletion of C9ORF72 by 75% compared with control siRNA-treated cells. Immunoblotting for LC3 was performed on lysates harvested from cells treated with control or C9ORF72-targetd siRNA, indicating the formation of autophagosomes. (F) Quantification of the ratio of phosphatidylethanolamine-modified product of LC3II, relative to LC3I by densitometry of immunoblots, demonstrated a reduction in this ratio by 45% in C9ORF72-siRNA-treated cells, whereas there was no change in control siRNA-treated cells compared with untreated cells. Data are represented as mean ± SEM; **P < 0.001 versus untreated cells by unpaired t-test, n = 3. (G) C9ORF72 is associated with lysosomes in Neuro 2A cells. Cells were transfected with C9ORF72-GFP and treated with Lysotracker for 20 min, Scale bar: 10 μm.
Endogenous C9ORF72 interacts with ubiquilin-2, hnRNPA1 and hnRNPA2/B1
To gain further insight into the function of C9ORF72, we used mass spectrometry to identify novel proteins that interact with C9ORF72 in immunoprecipitated cell lysates. These studies identified five additional interacting partners of C9ORF72; ubiquilin-1, ubiquilin-2, hnRNA1, hnRNA2/B1 and actin (cytoplasmic), with Mascot scores 284.78, 579.71, 155.58, 185.47 and 621.91, respectively (Supplementary Material, Table S1). The interaction between C9ORF72 and ubiquilin-2 was investigated further using immunocytochemistry and immunoprecipitation. Immunocytochemistry and subsequent quantification revealed that C9ORF72 and ubiquilin-2 colocalized in 25% of neuro2a cells (Fig. 7A and C) under basal conditions. Also, ubiquilin-2 was coprecipitated from neuro2a cell lysates using C9ORF72 antibodies: control reactions containing buffer only or an isotype-matched control antibody were negative (Fig. 7D). The autophagy and proteasome systems are closely linked; therefore, we treated cells with an inhibitor of the 26S proteasome, lactacystin, to increase autophagic flux (Fig. 7B). This treatment significantly increased the proportion of cells with ubiquilin-2 and C9ORF72 colocalization (Fig. 7B; P < 0.001). Hence these data suggest that C9ORF72 and ubiquilin-2 interact, and hence may function together in autophagy.
Figure 7.

C9ORF72 colocalizes with hnRNPA1, hnRNPA2/B1, ubiquilin-2 and actin. (A) Neuro2a cells were treated with 10 μm lactacystin for 16 h. Both treated and untreated cells were immunostained with anti-C9ORF72 (red) and anti-ubiquillin-2 antibodies (green) and stained with DAPI to locate the nucleus (blue). White arrow in the merge image shows areas of colocalization of ubiquilin-2 and C9ORF72. Scale bar: 10 μm, applied to all fields. Lac, lactacystin. (B) Inhibition of the proteasome by lactacystin promotes the colocalization of C9ORF72 and ubiquillin-2. Manders coefficient was used to calculate the degree of colocalization between C9ORF72 and ubiquillin-2. Data are represented as mean ± SEM; **P < 0.001 versus untreated cells by unpaired t-test. (C) Ubiquilin-2 coprecipitates using anti-C9ORF72 antibodies in Neuro2a cells, revealed by immunoblotting for ubiquillin-2. Control immunoprecipitations using buffer only or irrelevant, isotype-matched control IgG antibody indicates there is no non-specific cross-reactivity. (D) Immunocytochemistry of SH-SY5Y cells using anti-C9ORF72 antibodies (green), anti-hnRNPA1 or anti-hnRNPA2/B1 antibodies (red). White arrow indicates the colocalization between C9ORF72 and hnRNPA2/B1 and hnRNPA1; scale bar: 10 μm. (E) hnRNPS coprecipitate using anti-C9ORF72 antibodies in SH-SY5Y cells, revealed by immunoblotting with anti-hnRNPA1 or anti-hnRNPA2/B1 antibodies. Control immunoprecipitations using buffer only or irrelevant, isotype-matched control IgG antibody indicate there is no non-specific cross-reactivity. (F) Colocalization of C9ORF72 (red) and actin (green) in neuro 2a cells and stained with DAPI to locate the nucleus (blue), white arrows indicate areas of colocalization. Inset demonstrates higher magnification (×100) of the areas highlighted to illustrate colocalization. Scale bar: 10 μm.
Using immunocytochemistry, C9ORF72 also colocalized with both hnRNPA1 and hnRNPA2/B1 in SH-SY5Y cells (Fig. 7E), and C9ORF72 was co-immunoprecipitated using hnRNA1 and hnRNA2/B1 antibodies in cell lysates, confirming the mass spectrometry findings (Fig. 7F). Similarly, immunocytochemistry revealed that C9ORF72 colocalized with actin (Fig. 7G) thus providing further evidence linking C9ORF72 to cellular trafficking pathways.
Inhibition of the proteasome in cells overexpressing C9ORF72 induces the formation of SGs
We next transfected neuro2a cells with a construct-encoding C9ORF72 tagged with GFP. The distribution appeared similar to endogenous C9ORF72, primarily nuclear with cytoplasmic vesicles. However, we also observed the presence of fluorescent aggregate structures containing C9ORF72 in the nucleus in 40% of cells (Fig. 8A), suggesting possible disturbance to protein degradation pathways in cells overexpressing C9ORF72. This was investigated further using lactacystin treatment to inhibit the 26S proteasome; the proportion of cells overexpressing C9ORF72 with nuclear C9ORF72 aggregates significantly increased to 74% (Fig. 8B; P < 0.001) and the number of nuclear aggregates per cell significantly increased in treated cells (Fig. 8C; P < 0.001). Hence, the nuclear aggregates are linked to proteasome function, indicating possible cellular stress. This was investigated by examining the formation of cytoplasmic SGs using immunocytochemistry for hnRNPA1 and hnRNPA2/B as SG markers. SGs were not present in untransfected cells or control GFP cells with or without lactacystin treatment. However, in cells overexpressing C9ORF72, cytoplasmic SGs were present, identified by hnRNPA1 (12% cells) and hnRNPA2/B1 (7% cells) staining, respectively (Fig. 8D), and the proportion of SGs stained by hnRNPA2/B1 was significantly increased with proteasome inhibition (Fig. 8E; P < 0.05). Hence, proteasome inhibition promotes the formation of nuclear C9ORF72-aggregates and cytoplasmic SGs in cells overexpressing C9ORF72.
Figure 8.

Inhibition of the proteasome in cells overexpressing C9ORF72 promotes the formation of nuclear aggregates and cytoplasmic SG's. (A) Neuro2a cells were transfected with C9ORF72-GFP for 48 h, followed by 10 µm lactacystin treatment for 16 h and DAPI staining to locate the nucleus. C9ORF72-positive nuclear aggregate structures are present in the nucleus, with and without lactacystin treatment. Scale bar: 10 μm. (B) Quantification demonstrates that lactacystin increased the percentage of cells bearing nuclear C9ORF72 aggregates from 37% to 70%. Data are represented as mean ± SEM; **P < 0.001, n = 2 treated versus untreated by unpaired t-test. (C) The number of nuclear aggregates per cell increased by 60% in lactacystin treated versus untreated cells. For each of three replicate experiments, 50 cells were scored for each population. Data are represented as mean ± SEM; *P < 0.005 versus untreated by one-way unpaired t-test. (D) SH-SY5Y cells were transfected with C9ORF72-EGFP and immunostained using either hnRNPA1 or hnRNPA2/B1 antibodies (red) and DAPI (blue) to locate the nucleus. Arrows indicate hnRNPs-positive SGs formed in the cytoplasm. Scale bar: 10 μm. (E) The percentage of cells displaying hnRNPA1 or hnRNPA2/B1-positive SGs was quantified, for each of two replicate experiments, 50 cells were scored for each population. Cytoplasmic SGs positive for hnRNPA1 (12%) and hnRNPA2/B1 (7%) were present in C9ORF72-expressing cells. This was significantly increased in the propotion of hnRNPA2/B1-positive SGs treated by proteasome inhibition, lactacystin. Data are represented as mean ± SEM; *P < 0.005 versus untreated by unpaired t-test.
DISCUSSION
Despite the importance of C9ORF72 in neurodegeneration, its normal cellular function remains undefined. Clearly, understanding this function is necessary for elucidating its role in disease. In this study, we demonstrate that C9ORF72 regulates intracellular trafficking processes in the endosomal and autophagy-lysosomal compartments. We observed that depletion of C9ORF72 using siRNA inhibited endocytosis and increased the ratio of LC3II:LC3I, suggesting dysregulation of autophagosome formation. C9ORF72 also colocalized with autophagosomes, lysosomes and ubiquilin-2, implicating C9ORF72 in trafficking essential for autophagy.
Rabs function as molecular switches that alternate between two conformational states: the activated GTP-bound form and the GDP-bound inactive form. Exchange of GDP with GTP is catalyzed by RabGEFs which facilitate GDP release and thus activate Rabs. One would expect that C9ORF72 would colocalize with Rabs in the appropriate compartments and to physically interact with Rab proteins if C9ORF72 possesses RabGEF activity. Consistent with this hypothesis, we observed colocalization and coprecipitation of C9ORF72 with four Rab proteins implicated in endolysosomal trafficking in both neuronal cell lines and primary cortical neurons: Rab1, Rab5, Rab7 and Rab11. Hence, this study is consistent with previous bioinformatics studies that reported the presence of a DENN domain in C9ORF72, part of a hitherto undetected group of DENN proteins that mediate autophagy and vascular–vesicle interactions (13,14). This raises the possibility that C9ORF72 functions as a RabGEF that regulates Rab-dependent intracellular trafficking. This study therefore provides the first experimental evidence to support this notion.
Each of the Rab proteins we investigated has a specific function in endocytosis and/or autophagy. Endocytic cargo is initially present in Rab5-containing early endosomes that undergo maturation to become Rab7-containing late endosomes (18). Rab7 is a critical regulatory component that regulates transport from early-to-late endosomes, biogenesis of lysosomes and maturation of autophagosomes (30–33). Similarly, Rab1 is necessary for the formation of the autophagosome (18), and Rab11 regulates recycling of endocytosed proteins via recycling endosomes and promotes autophagy by providing the autophagosome membrane (17). Hence, association with these four Rab proteins suggests that C9ORF72 may have a broad function in facilitating endocytic trafficking events. Our study is therefore consistent with previous reports that Rab effectors exhibit broad specificity for Rab proteins and interact with multiple Rab isoforms (34). However, whilst our findings support the notion that C9ORF72 is a RabGEF, more specific studies are required to determine this conclusively.
Ubiquilin-2 is part of a family of ubiquitin-like proteins involved in protein degradation that bind and transport ubiquitinated cargo to the proteasome and autophagosome (35). Mass spectrometry, immunoprecipitation and immunocytochemistry identified ubiquilin-2 as an interacting partner of C9ORF72, and this interaction was enhanced by proteasome inhibition, thus providing further evidence that the function of C9ORF72 is linked to protein degradation. These data therefore imply that C9ORF72 mediates trafficking of cargo to the autophagosome and/or proteasome in conjunction with ubiquilin-2. Consistent with this notion, ALS patients with the C9ORF72 repeat expansion also possess p62/ubiquitin-positive pathology (20,36,37).
It remains unclear how the hexanucleotide repeat expansions in C9ORF72 are linked to ALS, although reduced levels of C9ORF72 have been reported in C9ORF72-patient motor neurons (19). Our finding that endocytosis and autophagy are inhibited in cells depleted of C9ORF72 therefore identifies cellular processes affected by reduction in C9ORF72 expression. Membrane trafficking processes are vital for cellular maintenance and viability, hence inhibition of these events would significantly impact on cellular functions. In addition, motor neurons maybe particularly vulnerable to disruption in cellular transport due to their large size and long axons. We also found that in motor neurons in tissue obtained from ALS patients bearing the C9ORF72 GGGGCC mutation, the association of C9ORF72 with Rab7 and Rab11 was increased, implying possible dysregulation of endosomal trafficking in C9ORF72-ALS. Defects in Rab-mediated trafficking are previously described in diseases affecting motor neurons. Mutations in Rab7 cause Charcot-Marie-Tooth disease type 2B (38), and Rab7-positive endosome abnormalities impair trafficking in Wobbler mice (39). Mutations in Alsin, a RabGEF for Rab5, cause juvenile onset ALS and primary lateral sclerosis (40) and enlarged Rab5-positive endosomes are present in spinal ALS motor neurons (41). Dysfunction in dynein, which powers endocytic trafficking, has been described in ALS (42) and depletion of dynein also results in endocytic Rab accumulation and endosomal pathology (43). We also recently demonstrated that mutant SOD1 induces dysfunction in the trafficking between the ER and Golgi compartments, another trafficking process regulated by Rab1 (44).
Similarly, disturbance to protein degradation pathways, including autophagy, are increasingly implicated in ALS (19). Autophagosomes accumulate in the spinal cord of sporadic ALS patients (45) and enhancement of autophagy and decrease in autophagic flux is present in ALS mouse and cell lines expressing mutant SOD1 (46). Multiple ALS causative or associated genes are also linked to autophagy: SQSTM1, DCTN1, VCP, FIG4 and Rab7A (47). We also identified actin as an interacting partner of C9ORF72 by mass spectrometry, another vesicular trafficking protein that provides the structural unit of microfilaments. Moreover, actin plays a role at the early stage of autophagosome formation (48), and autophagosomes are surrounded by an actin filament network that is required for autophagosome/lysosome fusion (49), providing additional evidence for a role for C9ORF72 in autophagy. Furthermore, mutations in profilin, which regulates actin polymerization, were also recently linked to ALS (50). We also provide evidence that C9ORF72 is present extracellularly in neuronal cell lines, human CSF and within the axons of primary neurons. Similarly, C9ORF72 is highly concentrated in synaptic terminals in brains of ALS/FTD patients (51). Hence together, these data imply a possible role for C9ORF72 in synaptic vesicle trafficking.
In this study, we also demonstrate a relationship between C9ORF72 and hnRNPA1 and hnRNPA2B1. Whereas a recent report found that C9ORF72 RNA with the repeat expansion bound to three different hnRNPs, hnRNPA1, hnRNPA2/B1 and hnRNPA3 (10), here we show that wild-type C9ORF72 also interacts with hnRNPs. Hence, this interaction is independent of the presence of the C9ORF72 GGGGCC mutation. hnRNPs shuttle continuously between the nucleus and cytoplasm and during cellular stress they accumulate in cytoplasmic SGs (52). Hence, it is possible that C9ORF72 may facilitate the transport of hnRNPs between the nucleus and cytoplasm, although we did not examine nuclear-cytoplasmic transport in this study. Alternatively, another intriguing possibility is that C9ORF72 may have a functional role with hnRNPs, as an RNA-binding protein with possible roles in RNA metabolism or stability, as proteasome inhibition modifies RNA splicing (26).
The UPS plays an active role in the nucleus at the quality control and transcriptional levels. We observed that expression of C9ORF72 induces cytoplasmic SG's, and this was enhanced by proteasome inhibition, suggesting that wild-type C9ORF72 may induce cellular stress. This may occur via a direct effect on C9ORF72 misfolding or indirectly via other cellular proteins or pathways. Elevated expression of wild-type versions of other proteins linked to ALS, SOD1 and TDP-43, trigger ALS-like pathology (53–55). Targets of the nuclear proteasome form nuclear aggregates reminiscent of aggresomes when the proteasome is inhibited (56). We also identified the presence of nuclear aggregate structures in cells expressing C9ORF72, although these aggregates remain to be characterized. However, TDP-43 and FUS form nuclear foci and also interact with hnRNPs (57), TDP-43-positive pathology is present in C9ORF72 patients (58), and sequesteration of nuclear RNA-binding proteins leads to cytoplasmic TDP-43 aggregates in ALS patients with the C9ORF72 mutation (59).
Hence, we show in this study that C9ORF72 is involved in intracellular trafficking pathways linked to protein degradation. We summarize a hypothetical scenario to illustrate convergence of C9ORF72 function in protein degradation (Fig. 9). C9ORF72 normally mediates endocytic trafficking to facilitate both autophagy and endosomal transport, at least partially in conjunction with ubiquilin-2. C9ORF72 also mediates shuttling of hnRNPA2/B1 and hnRNPA1 between the nucleus and cytoplasm, and may also be linked to RNA metabolism. Further experiments in this area are necessary as understanding the normal cellular function of C9ORF72 is an important step in understanding how dysregulation of this function may lead to both ALS and FTD.
Figure 9.
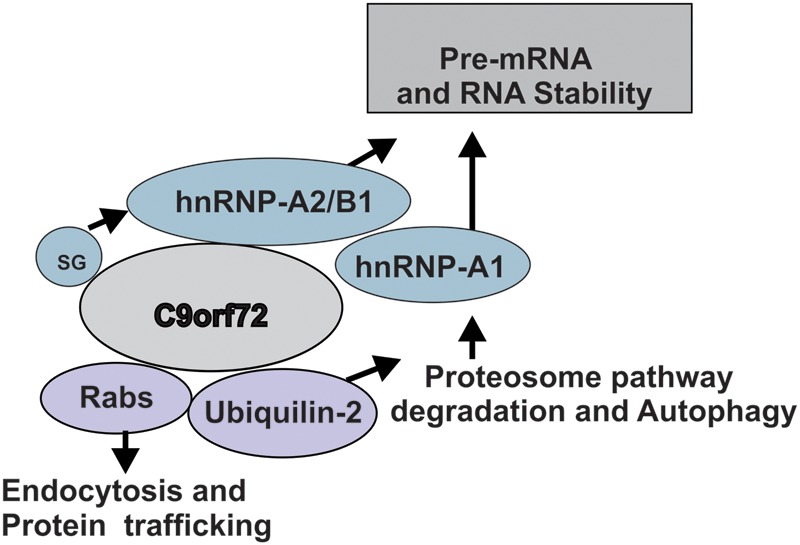
Possible functions of C9ORF72. C9ORF72 mediates endocytic trafficking to facilitate autophagy and proteasome function, at least partially with ubiquilin-2. However, during conditions of cellular stress and SGs, C9ORF72 interacts with hnRNPA2/B1 and hnRNPA1 that regulate splicing and RNA metabolism and may cause RNA instability in ALS.
MATERIALS AND METHODS
Constructs
A construct C9ORF72-Variant 1 tagged with AC-GFP-tagged (NM_018325) was obtained from OriGene, encoding the full length of human C9ORF72 cDNA and constructs-encoding GFP-tagged Rab5, Rab7, Rab11 and DsRed-LC3, were as previously described (60,61). FLAG-tagged TrkB receptor was a kind gift from Drs Junhua Xiao and Simon Murray from the Department of Anatomy and Cell Biology, University of Melbourne.
Cell culture and transfection
Neuro2a or SH-SY5Y neuroblastoma cells were subcultured in 24-well plates at a density of 2 × 104 cells/well and were transfected transiently with plasmids (1 μg DNA per well) using lipofectamine reagent. Cells were examined with an inverted fluorescent microscope (Olympus, NSW, Australia) 72 h after transfection unless otherwise stated. Cells were transfected with C9ORF72-Variant 2 and DsRed-LC3 (61) using lipofectamine 2000 (Invitrogen/Life Technologies) according to the manufacturer's instructions. For proteosomal inhibition, cells were treated with lactacystin (Sigma, L6785) for 16 h before analysis. Fixed cells were immunostained with DAPI to visualize the nucleus, and at least 50 cells were analyzed in each category. To knockdown endogenous human C9ORF72 expression, human SH-SY5Y cells were transfected with C9ORF72 siRNA (Dharma; L-013341–01), following the manufacturers instruction's.
Subcellular fractionation
Subcellular fractionation of SH-SY5Y neuroblastoma cells was performed after lysis of cells using 500 μl of fractionation buffer (250 mm sucrose, 20 mm HEPES (7.4), 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA with protease inhibitors). Cell lysates were passed through a 25 G needle 10 times and left on ice for 20 min. Homogenates were centrifuged at 720 g for 5 min to isolate the nuclear fraction, and washed by adding 500 μl of fractionation buffer. The pellet was passed through a 25 G needle 10 times and centrifuged at 720 g for 10 min. After washing, the pellet was resuspended in standard TN lysis buffer and stored as the nuclear fraction. The supernatant was spun at 100 000 g at 4°C, to obtain the cytosolic fraction. Protein measurements of the obtained fractions were determined and analyzed by immunoblotting, as described.
Primary cells from the central nervous system
Primary neurons were harvested from the cells from the cortex of C57BL/6 mouse embryos at embryonic (E) day 15.5. The procedure for the culture of primary neurons was as described previously (62,63). Briefly, cortical tissue was dissected from E15.5 mouse embryos and dissociated using 0.0125% trypsin (5 min at 37°C). Tissue was washed in cell plating media (consisting of Neurobasal™ medium (Gibco), 2% B27 supplement, 10% fetal calf serum (Gibco), 0.5 mm glutamine, 25 µm glutamate and 1% antibiotic/antimycotic (Gibco), followed by mechanical tituration using a 1 ml pipette. Cell viability and density were assessed using a trypan blue exclusion assay. Cells were plated onto poly-l-lysine-coated 12 mm coverslips (Marienfeld) in 24-well plates at a concentration of 30 000 cell per coverslip. The following day, media were changed to cell maintenance media consisting of plating media without the fetal calf serum and glutamate. Cells were grown in 5% CO2 at 37°C. All animal experiments were performed under the approval of the University of Tasmania animal ethics committee (Ethics # A12780). The purity of primary cortical neurons was confirmed by immunostaining using specific antibodies: neuronal marker microtubule-associated protein 2, MAP2 (Millipore, MAB3418 clone AP20, 1:1000). Immunocytochemistry staining was performed to show the colocalization of C9ORF72 and Rabs by using anti-C9ORF antibody and anti-Rabs antibodies using standard methods as described below.
Immunocytochemistry and endocytosis
Neuro2a or SH-SY5Y neuroblastoma cells were grown on coverslips, washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 min. Cells were permeabilized in 0.1% Triton X-100 in PBS for 2 min, blocked for 30 min with 1% BSA in PBS, and incubated with primary anti-C9ORF72 (1:100, SantaCruz, sc-138763), anti-Rab 1 (1:100, SantaCruz sc-28566), anti-Rab 7, (Abcam, ab50533, 1:50), anti-hnPNP-A1 (Sigma, R4528) or anti hnRNP-A2/B1 (Sigma, R4653), for 16 h at 4°C. Secondary AlexaFluor-594 conjugated anti-mouse or anti-rabbit antibodies (1:200, Molecular Probes) were then incubated for 1 h at room temperature, and cells were counter-stained with DAPI and mounted. Images were acquired using an Olympus inverted confocal laser-scanning microscope or an Olympus inverted fluorescence microscope. To examine endocytosis, purified Shiga toxin conjugated to CY3 (SxTB-CY3) as previously described (28) was added to the medium of cells treated with C9ORF72 and control siRNA for 30 min. Shiga toxin was added for 30 min, and then examined after 60 min by immunocytochemistry for Golgi marker GM130. Endocytosis was confirmed by quantifying the internalization of TrkB receptor by cell surface biotinylation as described previously (64–66) . Briefly, SH-SY5Y cells were transfected with C9ORF72 siRNA, and 24 h later transfected again with siRNA to increase the efficiency of knockdown. The C9ORF72 depleted cells were then transfected with a construct-encoding FLAG-tagged TrKB receptor for 48 h. Cells were then washed twice with ice-cold PBS, and incubated with 0.5 mg/ml biotin NHS (Thermo Scientific) in PBS for 30 min at 4°C to biotinylate all cell surface proteins. Unreacted biotin was quenched and removed with 50 mm NH4Cl for 5 min. Biotinylated cells were then transferred to prewarmed medium containing 50 ng/ml BDNF (Millipore) for 2 h to induce endocytosis of TrkB receptor, and the cells were immediately lysed in TN buffer with 0.1% SDS. Internalized biotinylated TrKB receptor was then precipitated from cell lysates with 10 µl of streptavidin-agarose beads (Cell signaling) and quantified by immunoblotting with anti-FLAG antibody (Sigma).
Live cell imaging
Neuro 2A cells transfected with C9ORF72-GFP were stained for 20 min with 1 µm LysoTracker (L7528, Molecular Probes) and the movement of C9ORF72 vesicles was examined by time-lapse imaging. Images were acquired every 7 s for 15 min using a Zeiss LSM 510 confocal microscope.
Protein extraction
Cells were lysed in Tris–NaCl (TN) buffer (50 mm Tris–HCl, pH 7.5, 150 mm NaCl) with 0.1% (v/v) NP-40, 0.1% (w/v) SDS and 1% (v/v) protease inhibitor mixture (Sigma) for 10 min on ice. Cellular lysates were clarified by centrifugation at 16g for 10 min. Proteins were quantified using the BCA assay kit (Pierce).
Immunoprecipitation and immunoblotting
For immunoprecipitation, cell lysates were incubated with anti-C9ORF72 antibody, and 30 µl of 50% (w/v) protein A-Sepharose CL-4B (Amersham Biosciences) in Tris buffer [50 mm Tris–HCl, pH 7.5, 0.02% (w/v) NaN3] on a rotating wheel overnight at 4°C. Proteins eluted from the beads were separated on 10% tris–glycine polyacrylamide gels. For western blotting, protein measurements of all obtained fractions were determined by BCA assay. Between 10 and 20 μg of protein was loaded and separated in 8–15% SDS–PAGE gels. Proteins were then electrophoretically transferred onto nitrocellulose membranes (Bio-Rad). These membranes were subsequently blocked with 5% (w/v) non-fat milk powder in Tris-buffered saline (TBS) or with 3% bovine serum albumin (BSA) in TBS containing 0.1% Tween 20 (TBS-T) at room temperature for 1 h, followed by incubation with the primary antibodies diluted in 1% BSA/TBS-T overnight at 4°C with shaking. C9ORF72 (1:500, SantaCruz sc-138763), or Rab 5 (1:500, Abcam ab50523), or Rab 11 (1:500, BD Transduction Labs, 610657), or Rab 7 (1:500, ab50533), or LC3 antibody (1:1000, Novus Biologicals; NB100–2220), ubiquilin-2 antibody (1:700, Abnova, H00029978), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:10,000, Ambion AM4300), Histone H3 96C10, Cell Signalling, 36385) and β-actin (1:5000, Sigma). Membranes were washed with TBS-T and then incubated for 1 h at room temperature with secondary antibodies (1:4000, HRP-conjugated goat anti-rabbit or goat anti-mouse antibodies, Chemicon). Protein bands were detected by using enhanced chemiluminescence reagents (Roche), as described by the manufacturer.
Immunohistochemistry
Human postmortem cervical spinal cord sections (5 µm) from a male fALS patients carrying the hexanucleotide GGGGCC repeat expansion was obtained. This patient developed limb onset ALS at 74 years old, with a disease duration of 13 months. He was not diagnosed with FTD, but deterioration of memory was reported. A sex and age matched neurologically normal individual was used as a control. Formalin-fixed, paraffin-embedded spinal cord tissues deparaffinised with xylene, and rehydrated with a descending series of diluted ethanol and water. Antigen retrieval was conducted by boiling the sections for 20 mins in 10 mm citric buffer (pH 6.0). Sections were blocked using 3% goat serum/0.3% Tween 20 for 1 h and then immunostained with different Rabs (1:50) and C9ORF72 (1:100) antibodies and left overnight at 4°C. Sections were washed twice in PBS Tween 20 (0.1%) and secondary antibodies were added; anti-rabbit/mouse Alexa Fluor (1:200, Molecular Probes, Invitrogen, VIC, Australia), for 1 h at RT. Sections were again washed twice in PBS Tween 20 and then a cover slip was applied using Prolong Gold antifade reagent (Invitrogen, VIC, Australia). Images were acquired using an Leica confocal laser-scanning microscope.
Mass spectrometry
Untransfected Neuro 2A cell lysates (500 ug) were immunoprecipitated using anti-C9ORF72 antibody (SantaCruz, sc-138763) and 30 µl of 50% (w/v) protein A-Sepharose CL-4B (Amersham Biosciences), on a rotating wheel overnight at 4°C. Samples were centrifuged at 15 800 g for 1 min, and Sepharose pellets were washed twice in Tris buffer for 10 min each. Protein samples for mass spectrometry were eluted from the beads by heating for 5 min, followed by centrifugation to remove cellular debris, then separated on 10% SDS–PAGE gel, and stained using Coomassie blue. Gel lanes were cut into slices, reduced, alkylated and digested with trypsin: only the five most abundant protein bands were excised. The resulting peptides were injected onto a trapping column (Dionex Acclaim Pepmap100, Nano trap 100 μm × 2 cm, C18, 5 μm, 100 Å) using A-buffer (2% acetonitrile in 0.1% formic acid (aqeous); Sigma-Aldrich). Following a 6 min wash, the sample was transferred onto a resolving column (Dionex Acclaim Pepmap RSLC, 75 μm × 15 cm, C18, 5 μm, 100 Å) and eluted with a gradient of B-buffer (98% acetonitrile in A-buffer) over 70 min. The eluent from the column was directly electrosprayed into the microTof-Q-MS instrument (Bruker-Daltonics, Germany). Mass data were continuously acquired and for each MS spectrum three MS/MS were recorded of the most intense peaks. The score for each protein reflects the combined scores of all observed peptide mass spectra that can be matched to the amino acid sequences within that protein. The peptide score reflects the probability of matching the experimental spectra to the swiss data base. The data were annotated and deconvoluted using the Data Analysis program (Bruker-Daltonics). The acquired MS/MS spectra of peptides were searched protein database from SwissProt (ExPASy, http://www.expasy.org/proteomics) using the Mascot search engine (Matrix Science, London, UK).
Statistical analysis
All analyses were performed using GraphPad Prism 5 software. Statistical significance was calculated using one-way ANOVA followed by Tukey's post-test or two-tailed ‘unpaired t-test’, from two or three independent experiments. P < 0.05 was considered significant. Data are presented as mean ± SEM.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by funding from National Health and Medical Research Council of Australia (project grants 1006141, 1004670 and 1030513), MND Research Institute of Australia and Bethlehem Griffiths Research Council. Funding to pay the Open Access publication charges for this article was provided by LaTrobe University.
Supplementary Material
ACKNOWLEDGEMENTS
The C9ORF72 construct was kindly provided by Dr Justin Yerbury, University of Wollongong, and the human CSF was kindly provided by Professor Malcolm Horne, Florey Institute of Neuroscience and Mental Health. We thanks Prof. Gleeson for providing all the GFP-Rab constructs, and Dr Xiao and Dr Murray for providing Flag-tagged TrkB-FL. We also thank Dr Gert Hoy Talbo, Head of the mass spectrometry facility at La Trobe University.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Josephs K.A., Hodges J.R., Snowden J.S., Mackenzie I.R., Neumann M., Mann D.M., Dickson D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122:137–153. doi: 10.1007/s00401-011-0839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson-Stone C., Hallupp M., Bartley L., Shepherd C.E., Halliday G.M., Schofield P.R., Hodges J.R., Kwok J.B.J. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology. 2012;79:995–1001. doi: 10.1212/WNL.0b013e3182684634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sha S.J., Takada L.T., Rankin K.P., Yokoyama J.S., Rutherford N.J., Fong J.C., Khan B., Karydas A., Baker M.C., Dejesus-Hernandez M., et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology. 2012;79:1002–1011. doi: 10.1212/WNL.0b013e318268452e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Swieten J.C., Grossman M. FTD/ALS families are no longer orphaned: the C9ORF72 story. Neurology. 2012;79:962–964. doi: 10.1212/WNL.0b013e318268471a. [DOI] [PubMed] [Google Scholar]
- 5.Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Majounie E., Renton A.E., Mok K., Dopper E.G., Waite A., Rollinson S., Chio A., Restagno G., Nicolaou N., Simon-Sanchez J., et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fratta P., Mizielinska S., Nicoll A.J., Zloh M., Fisher E.M., Parkinson G., Isaacs A.M. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W., III, Rademakers R., et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer S., Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- 12.Cherfils J., Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 13.Levine T.P., Daniels R.D., Gatta A.T., Wong L.H., Hayes M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29:499–503. doi: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang D., Iyer L.M., He F., Aravind L. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 2012;3:283. doi: 10.3389/fgene.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korolchuk V.I., Menzies F.M., Rubinsztein D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 16.Nishida Y., Arakawa S., Fujitani K., Yamaguchi H., Mizuta T., Kanaseki T., Komatsu M., Otsu K., Tsujimoto Y., Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–U699. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 17.Longatti A., Lamb C.A., Razi M., Yoshimura S., Barr F.A., Tooze S.A. TBC1D14 regulates autophagosome formation via Rab11-and ULK1-positive recycling endosomes. J. Cell Biol. 2012;197:659–675. doi: 10.1083/jcb.201111079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zoppino F.C., Militello R.D., Slavin I., Alvarez C., Colombo M.I. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11:1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 19.Thomas M., Alegre-Abarrategui J., Wade-Martins R. RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain. 2013;136:1345–1360. doi: 10.1093/brain/awt030. [DOI] [PubMed] [Google Scholar]
- 20.Deng H.X., Chen W., Hong S.T., Boycott K.M., Gorrie G.H., Siddique N., Yang Y., Fecto F., Shi Y., Zhai H., et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothenberg C., Monteiro M.J. Ubiquilin at a crossroads in protein degradation pathways. Autophagy. 2010;6:979–980. doi: 10.4161/auto.6.7.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett E.J., Bence N.F., Jayakumar R., Kopito R.R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Li Y.R., King O.D., Shorter J., Gitler A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H.J., Kim N.C., Wang Y.D., Scarborough E.A., Moore J., Diaz Z., Maclea K.S., Freibaum B., Li S., Molliex A., et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bekenstein U., Soreq H. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: from structural insights to post-transcriptional regulatory roles. Mol. Cell. Neurosci. 2013;56:436–446. doi: 10.1016/j.mcn.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Bieler S., Hammer E., Gesell-Salazar M., Volker U., Stangl K., Meiners S. Low dose proteasome inhibition affects alternative splicing. J. Proteome Res. 2012;11:3947–3954. doi: 10.1021/pr300435c. [DOI] [PubMed] [Google Scholar]
- 27.Bendtsen J.D., Jensen L.J., Blom N., Von Heijne G., Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 28.Fuchs E., Haas A.K., Spooner R.A., Yoshimura S., Lord J.M., Barr F.A. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J. Cell Biol. 2007;177:1133–1143. doi: 10.1083/jcb.200612068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manders E.M.M., Verbeek F.J., Aten J.A. Measurement of colocalization of objects in dual-color confocal images. J. Microsc-Oxford. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 30.Harrison R.E., Bucci C., Vieira O.V., Schroer T.A., Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol. Cell. Biol. 2003;23:6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jager S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., Eskelinen E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 32.Gutierrez M.G., Munafo D.B., Beron W., Colombo M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 33.Bucci C., Thomsen P., Nicoziani P., McCarthy J., van Deurs B. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuda M., Kanno E., Ishibashi K., Itoh T. Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol. Cell. Proteomics. 2008;7:1031–1042. doi: 10.1074/mcp.M700569-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Rothenberg C., Srinivasan D., Mah L., Kaushik S., Peterhoff C.M., Ugolino J., Fang S., Cuervo A.M., Nixon R.A., Monteiro M.J. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 2010;19:3219–3232. doi: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brettschneider J., Van Deerlin V.M., Robinson J.L., Kwong L., Lee E.B., Ali Y.O., Safren N., Monteiro M.J., Toledo J.B., Elman L., et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012;123:825–839. doi: 10.1007/s00401-012-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Troakes C., Maekawa S., Wijesekera L., Rogelj B., Siklos L., Bell C., Smith B., Newhouse S., Vance C., Johnson L., et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2012;32:505–514. doi: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- 38.Meggouh F., Bienfait H.M.E., Weterman M.A.J., de Visser M., Baas F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology. 2006;67:1476–1478. doi: 10.1212/01.wnl.0000240068.21499.f5. [DOI] [PubMed] [Google Scholar]
- 39.Palmisano R., Golfi P., Heimann P., Shaw C., Troakes C., Schmitt-John T., Bartsch J.W. Endosomal accumulation of APP in wobbler motor neurons reflects impaired vesicle trafficking: implications for human motor neuron disease. BMC Neurosci. 2011;12:24. doi: 10.1186/1471-2202-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Topp J.D., Gray N.W., Gerard R.D., Horazdovsky B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004;279:24612–24623. doi: 10.1074/jbc.M313504200. [DOI] [PubMed] [Google Scholar]
- 41.Hadano S., Benn S.C., Kakuta S., Otomo A., Sudo K., Kunita R., Suzuki-Utsunomiya K., Mizumura H., Shefner J.M., Cox G.A., et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006;15:233–250. doi: 10.1093/hmg/ddi440. [DOI] [PubMed] [Google Scholar]
- 42.LaMonte B.H., Wallace K.E., Holloway B.A., Shelly S.S., Ascano J., Tokito M., Van Winkle T., Howland D.S., Holzbaur E.L.F. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 43.Kimura N., Inoue M., Okabayashi S., Ono F., Negishi T. Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases: a potential mechanism underlying age-dependent endocytic dysfunction. J. Biol. Chem. 2009;284:31291–31302. doi: 10.1074/jbc.M109.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sundaramoorthy V., Walker A.K., Yerbury J., Soo K.Y., Farg M.A., Hoang V., Zeineddine R., Spencer D., Atkin J.D. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 2013;70:4181–4195. doi: 10.1007/s00018-013-1385-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011;70:349–359. doi: 10.1097/NEN.0b013e3182160690. [DOI] [PubMed] [Google Scholar]
- 46.Ferraiuolo L., Kirby J., Grierson A.J., Sendtner M., Shaw P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 47.Otomo A., Pan L., Hadano S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012;2012:498428. doi: 10.1155/2012/498428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguilera M.O., Beron W., Colombo M.I. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8:1590–1603. doi: 10.4161/auto.21459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee J.Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y.S., Pandey U.B., Kaushik S., Tresse E., Lu J., et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu C.H., Fallini C., Ticozzi N., Keagle P.J., Sapp P.C., Piotrowska K., Lowe P., Koppers M., McKenna-Yasek D., Baron D.M., et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snowden J.S., Rollinson S., Thompson J.C., Harris J.M., Stopford C.L., Richardson A.M., Jones M., Gerhard A., Davidson Y.S., Robinson A., et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guil S., Long J.C., Caceres J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006;26:5744–5758. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bosco D.A., Morfini G., Karabacak N.M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B.A., Lemay N., McKenna-Yasek D., et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janssens J., Wils H., Kleinberger G., Joris G., Cuijt I., Ceuterick-de Groote C., Van Broeckhoven C., Kumar-Singh S. Overexpression of ALS-associated P.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol. Neurobiol. 2013;48:22–35. doi: 10.1007/s12035-013-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armakola M., Higgins M.J., Figley M.D., Barmada S.J., Scarborough E.A., Diaz Z., Fang X., Shorter J., Krogan N.J., Finkbeiner S., et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012;44:1302–1309. doi: 10.1038/ng.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Latonen L., Moore H.M., Bai B., Jaamaa S., Laiho M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene. 2011;30:790–805. doi: 10.1038/onc.2010.469. [DOI] [PubMed] [Google Scholar]
- 57.Tsuiji H., Iguchi Y., Furuya A., Kataoka A., Hatsuta H., Atsuta N., Tanaka F., Hashizume Y., Akatsu H., Murayama S., et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boeve B.F., Boylan K.B., Graff-Radford N.R., DeJesus-Hernandez M., Knopman D.S., Pedraza O., Vemuri P., Jones D., Lowe V., Murray M.E., et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsiung G.Y., DeJesus-Hernandez M., Feldman H.H., Sengdy P., Bouchard-Kerr P., Dwosh E., Butler R., Leung B., Fok A., Rutherford N.J., et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135:709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Derby M.C., Lieu Z.Z., Brown D., Stow J.L., Goud B., Gleeson P.A. The trans-Golgi network golgin, GCC185, is required for endosome-to-Golgi transport and maintenance of Golgi structure. Traffic. 2007;8:758–773. doi: 10.1111/j.1600-0854.2007.00563.x. [DOI] [PubMed] [Google Scholar]
- 61.Tung Y.T., Hsu W.M., Lee H., Huang W.P., Liao Y.F. The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell. Mol. Neurobiol. 2010;30:795–806. doi: 10.1007/s10571-010-9507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hosie K.A., King A.E., Blizzard C.A., Vickers J.C., Dickson T.C. Chronic excitotoxin-induced axon degeneration in a compartmented neuronal culture model. ASN Neuro. 2012:4. doi: 10.1042/AN20110031. pii: e00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.King A.E., Chung R.S., Vickers J.C., Dickson T.C. Localization of glutamate receptors in developing cortical neurons in culture and relationship to susceptibility to excitotoxicity. J. Comp. Neurol. 2006;498:277–294. doi: 10.1002/cne.21053. [DOI] [PubMed] [Google Scholar]
- 64.Chen Z.Y., Ieraci A., Tanowitz M., Lee F.S. A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol. Biol. Cell. 2005;16:5761–5772. doi: 10.1091/mbc.E05-07-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rajagopal R., Chen Z.Y., Lee F.S., Chao M.V. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J. Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arancibia-Carcamo I.L., Fairfax B.P., Moss S.J., Kittler J.T. Chapter 6. Frontiers in Neuroscience. In: Kittler J.T., Moss S.J., editors. The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology. Boca Raton, FL: CRC Press; 2006. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.