

Abstract
Mitochondrial respiratory chain biogenesis is orchestrated by hundreds of assembly factors, many of which are yet to be discovered. Using an integrative approach based on clues from evolutionary history, protein localization and human genetics, we have identified a conserved mitochondrial protein, C1orf31/COA6, and shown its requirement for respiratory complex IV biogenesis in yeast, zebrafish and human cells. A recent next-generation sequencing study reported potential pathogenic mutations within the evolutionarily conserved Cx9CxnCx10C motif of COA6, implicating it in mitochondrial disease biology. Using yeast coa6Δ cells, we show that conserved residues in the motif, including the residue mutated in a patient with mitochondrial disease, are essential for COA6 function, thus confirming the pathogenicity of the patient mutation. Furthermore, we show that zebrafish embryos with zfcoa6 knockdown display reduced heart rate and cardiac developmental defects, recapitulating the observed pathology in the human mitochondrial disease patient who died of neonatal hypertrophic cardiomyopathy. The specific requirement of Coa6 for respiratory complex IV biogenesis, its intramitochondrial localization and the presence of the Cx9CxnCx10C motif suggested a role in mitochondrial copper metabolism. In support of this, we show that exogenous copper supplementation completely rescues respiratory and complex IV assembly defects in yeast coa6Δ cells. Taken together, our results establish an evolutionarily conserved role of Coa6 in complex IV assembly and support a causal role of the COA6 mutation in the human mitochondrial disease patient.
INTRODUCTION
Genetic mutations that perturb mitochondrial respiratory chain (MRC) function manifest clinically in mitochondrial respiratory chain disease (MRCD), one of the most common inborn errors of metabolism with an estimated prevalence of 1:5000 live births (1). MRCD is genetically heterogeneous and can be caused by mutations in both mitochondrial and nuclear genes (2,3). Known defects in the MRC have already been mapped to ∼110 genes, and this list is growing rapidly with advancements in sequencing technologies (4). In the next-generation sequencing era, it has become much faster to detect mutations in patient DNA; however, distinguishing causality from naturally occurring sequence variation remains challenging (5). The rapid molecular diagnosis of MRCD is further hampered by the fact that the full complement of genes required for MRC biogenesis is currently unknown.
To address this biomedical challenge, we have focused on identifying and assigning function to evolutionarily conserved, uncharacterized human mitochondrial proteins. Our approach is based on the premise that factors required for building a conserved pathway for mitochondrial ATP synthesis should also be highly conserved and localized to the mitochondria. The power of our approach for discovering novel MRCD genes is highlighted by a recent next-generation sequencing study that identified potentially pathogenic mutations in one of our prioritized candidates, C1orf31 (6). The causal nature of the C1orf31 mutations identified in the MRCD patient was not established because of the absence of an MRC-related phenotype in patient fibroblasts and a lack of surrogate model systems (6). C1orf31 was first identified as a putative cytochrome c oxidase (CcO, mitochondrial respiratory complex IV) assembly factor based on an iterative orthology prediction (7). Subsequently, it was found in a proteomic survey of mitochondrial intermembrane space (IMS) proteins in the yeast Saccharomyces cerevisiae and renamed Coa6 because of its requirement for CcO assembly (8). However, the molecular function of Coa6 in CcO assembly remains unknown.
CcO is the terminal enzyme of the MRC and is the main site for cellular respiration. It is a highly conserved, multi-subunit enzyme complex, consisting of 11 subunits in yeast and 13 in mammals (9). In addition to protein subunits, CcO contains several cofactors, including two copper (Cu) sites (CuA and CuB), two heme groups (heme a and a3) and a magnesium and a zinc ion (10). CcO biogenesis is an extremely complex process that requires ∼40 different assembly factors (reviewed in 9). CcO assembly in yeast and human cells is modular and begins with the independent maturation of mitochondrial DNA-encoded subunits Cox1, Cox2 and Cox3; however, important differences exist. For example, in yeast, the Cox1 containing subcomplex consists of Cox5a and Cox6 (9), whereas in humans, it consists of Cox5a and Cox4 (11). The formation of mature, catalytically active CcO requires the addition of the remaining subunits and prosthetic groups, which is facilitated by a number of assembly factors (9). Of all the MRC complexes, biogenesis of CcO is the best characterized, yet the precise molecular function of many of the so-called assembly factors, including Coa6, remains to be determined.
The assembly defects in CcO are clinically heterogeneous, affecting either an isolated organ or multiple organ-systems, with onset from infancy to adulthood (12). These disorders (mitochondrial complex IV deficiency, OMIM:220110) are typically characterized by a wide range of disease phenotypes, including cardiomyopathy, encephalomyopathy, skeletal muscle myopathy, Leigh syndrome, metabolic acidosis and occasional hepatic failure. While only a few complex IV deficiencies can be traced to the core subunits of CcO, the majority of patient mutations are found in genes encoding assembly factors, which include the leucine-rich pentatricopeptide repeat motif-containing protein LRPPRC, a mitochondrial mRNA stabilizing protein (13–18); TACO1, a translational activator of COX1 (19); and the human COX14 ortholog, C12orf62, a factor involved in the synthesis and assembly of COX1 (20). Pathogenic mutations have also been identified in COX10 and COX15, CcO assembly factors required for heme synthesis (21–23), and in SCO1 and SCO2, which are required for insertion of Cu in CcO (24–26). The exact role of other disease genes, including SURF1 (27), C2orf64/COA5 (28) and FAM36A (29), in CcO assembly is not fully defined.
In this report, we have used a combination of yeast COA6 knockout cells, an allelic series of COA6 knockdown human cell lines and coa6 knockdown zebrafish embryos to demonstrate an evolutionarily conserved role of Coa6 in the biogenesis of CcO. We show that the CcO assembly defect in Coa6 deficient cells can be completely rescued by Cu supplementation, suggesting its role in the Cu-delivery pathway to CcO. Our mutagenesis experiments show that the patient mutations, present in the conserved Cx9CxnCx10C motif of Coa6, disrupt its function. Furthermore, Coa6-silencing experiments in zebrafish embryos revealed a marked cardiac developmental defect, a phenotype that resembles the MRCD patient who died of hypertrophic cardiomyopathy. Thus, our study provides strong support for the pathogenic role of the Coa6 mutations in the MRCD patient and uncovers a novel role of Coa6 in mitochondrial Cu metabolism.
RESULTS
Coa6 is required for mitochondrial respiration and CcO assembly in the yeast Saccharomyces cerevisiae
C1orf31/Coa6 emerged as one of the top candidates in our search for MRC biogenesis factors because it is an evolutionarily conserved protein that has been previously shown to localize to the mitochondria of yeast (8) and mice (30), and is known to interact with MRC assembly factors (7). Because of the ease of performing biochemical and functional genomic experiments, we utilized S. cerevisiae as a primary model system to uncover the role of Coa6 in mitochondrial energy metabolism. We began with extensive characterization of the growth of coa6Δ cells under different carbon source and temperature conditions. The growth of coa6Δ cells in glucose-containing fermentable medium (YPD) is comparable to wildtype (WT) up to the early stationary phase (Fig. 1A). However, coa6Δ has reduced growth in the stationary phase in YPD media and a prolonged lag phase in glycerol/ethanol-containing, non-fermentable medium (YPGE), indicating an MRC defect (Fig. 1A). We also observed slower growth and a prolonged lag phase in lactate-containing non-fermentable liquid medium (data not shown). As expected, the growth of coa6Δ in solid YPD medium was comparable to WT, but the growth in solid YPGE was reduced, with the effect being more severe at higher temperature (Fig. 1B). We verified the respiratory deficient growth phenotypes arising from Coa6 deficiency by constructing a COA6 deletion in the BY4742 strain background (data not shown). The growth defect of coa6Δ cells in non-fermentable medium suggested respiratory deficiency. To directly assess respiration, we measured oxygen consumption in WT and coa6Δ cells and observed a statistically significant 50% decrease in the oxygen consumption rate (OCR) of coa6Δ cells compared with WT cells (Fig. 1C). In order to unravel the biochemical basis of reduced respiration, we performed western blot analysis of MRC proteins from WT and coa6Δ cells under denaturing and native conditions. Consistent with a recent report (8), under denaturing conditions, we observed a specific reduction in CcO (CIV) subunits Cox2 and Cox3, without alteration in subunits of any of the other MRC complexes (Fig. 1D). Blue native polyacrylamide gel electrophoresis (BNPAGE)/western blotting revealed decreased assembly in the CcO-containing supercomplexes (III2IV2 and III2IV) without affecting the steady-state levels of complex II, III and V (Fig. 1E). Together, these results demonstrate that Coa6 is required for normal respiration and CcO biogenesis.
Figure 1.

Yeast coa6Δ cells exhibit reduced respiration and diminished CcO assembly. (A) WT and coa6Δ cells were precultured in YPD and inoculated in fresh YPD or YPGE liquid media at 30°C with a starting A600 of 0.1. Absorbance was measured at the indicated times at 600 nm. Data are representative of at least three independent measurements. (B) Serial dilutions of WT and coa6Δ cells were spotted on YPD and YPGE plates at 30°C and 37°C. Pictures were taken after 2–5 days of spotting. Data are representative of three independent experiments. (C) WT and coa6Δ cells were grown overnight in YPD at 30°C. After 18 h of growth, cells were harvested, washed, counted and resuspended in ethanol-containing respiratory medium. Basal oxygen consumption rate (OCR) was measured on half a million cells using an extracellular flux analyzer. Error bars represent mean ± SD (n = 10), * denotes statistically significant differences, P < 0.001, t-test. (D) Mitochondria were isolated from WT and coa6Δ cells grown to early stationary phase in YPD medium. Mitochondrial protein was extracted and analyzed by SDSPAGE/western blot. Subunit-specific antibodies were used to detect MRC complexes II–V. Porin was used as loading control. (E) Mitochondria from (D) and shy1Δ were solubilized in 1% digitonin, followed by BNPAGE/western blot of native MRC complexes. The shy1Δ cells lack CcO and thus were used as a control for CcO assembly. The stoichiometry and molecular weights of the supercomplexes are indicated.
COA6 is required for maintenance of CcO subunit levels in human skin fibroblasts
In order to determine the role of COA6 in MRC biogenesis in mammals, we engineered stable knockdowns of COA6 in human immortalized fibroblasts (MCH58) using five different shRNA constructs (kd1 to kd5). The COA6 transcript level in knockdown cells was 5–40% of the control, as determined by qPCR (Fig. 2A), which was consistent with the COA6 protein levels in the knockdown cell lines (Fig. 2B). Western blot analyses of mitochondrial proteins isolated from COA6 knockdown cells showed a specific decrease in the COX2 subunit of CcO without alteration in subunits of other MRC complexes (Fig. 2C). Although the relationship between COA6 transcript and COX2 protein levels was not linear, we found a strong correlation between these parameters (Pearson correlation coefficient = 0.82) (Fig. 2D), indicating that COA6 becomes limiting for CcO biogenesis once its level decreases to a certain threshold. The CcO specific defect in human cells mirrors the yeast results and suggests an evolutionarily conserved role of COA6 in CcO biogenesis.
Figure 2.

Coa6 is required for the maintenance of MRC complex IV subunit (COX2) levels in human cells. (A) COA6 mRNA was quantified by qPCR on RNA extracted from MCH58 human fibroblasts infected either with an empty vector, pLKO.1 (Ctrl), or with one of the five independent shRNAs targeting COA6 (kd1–kd5). Values are reported as percent change in expression over control (Ctrl). Three replicates were used per hairpin (error bars represent S.D.). β-actin was used as an endogenous control in the qPCR. (B) Western blot detection of Coa6 protein abundance in Coa6 knockdown cell lines. (C) Western blot of individual subunits of MRC complexes (I–V) in mitochondria isolated from each of the COA6 knockdown cell lines. Western blot image is representative of three independent experiments. (D) Correlation between COA6 mRNA levels (from A) and COX2 levels (from C). COX2 levels were quantified by densitometry.
Patient mutations within the conserved Cx9CxnCx10C motif disrupt Coa6 function
COA6 is an evolutionarily conserved protein, containing a Cx9CxnCx10C motif, with homologues in mammals, fish, fly and yeast (Fig. 3). This non-canonical Cx9CxnCx10C motif is similar to the twin Cx9C motifs found in a subset of mitochondrial IMS proteins that are required for CcO assembly (31). Although the molecular function of twin Cx9C proteins remain unclear, the motif is proposed to be required for IMS import via the Mia40-Erv1 disulfide relay system (32,33). A recent next-generation sequencing study identified a missense mutation in the conserved tryptophan residue (p.W59C) and a nonsense mutation in the conserved glutamic acid residue (p.E87X) within the Cx9CxnCx10C motif in an MRCD patient (Fig. 3) (6). The patient died of hypertrophic cardiomyopathy at a young age (<1 year old) and his heart tissue exhibited a reduction in CcO enzyme activity (6). In order to test the requirement of the conserved cysteines and patient mutations for Coa6 function, we cloned yeast COA6 into the Gateway expression vector pAG423GPD-ccdB-HA and performed site-directed mutagenesis to replace each of the four cysteines of the Cx9CxnCx10C motif with alanines and replace tryptophan with cysteine (p.W26C) to mimic the patient mutation (p.W59C) (Fig. 4A). The second mutation (p.E87X) in the patient is a nonsense mutation that results in a truncated protein that is devoid of the fourth cysteine, similar to our construct with a Coa6 (p.C68A) mutation. As shown in Figure 4B, WT Coa6 rescued the respiratory growth defect of coa6Δ cells. None of the point mutations in conserved residues, including the patient mutation, could rescue the respiratory growth phenotype, suggesting their critical requirement for Coa6 function (Fig. 4B). Episomally expressed WT and mutant Coa6 were localized to mitochondria (Fig. 4C). However, the steady-state level of the Coa6 mutants in mitochondria was reduced compared with WT Coa6, with the reduction being most severe in mutant proteins that correspond to patient mutations (Fig. 4C). The decrease in the steady-state levels of the mutant proteins in mitochondria could be because of a defect in their mitochondrial import or because of their reduced stability, as we did not detect Coa6 mutants in the cytosolic fraction (Fig. 4C). This reduction could, in part, explain their temperature-sensitive growth (Fig. 4B). Taken together, these results suggest that the mutations reported in the described MRCD patient likely play a causal role in disease pathogenesis.
Figure 3.
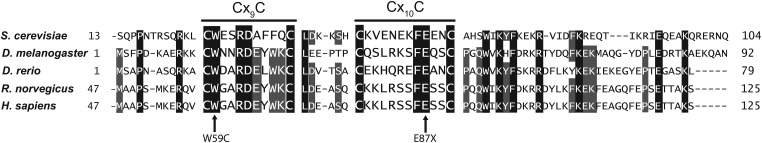
COA6 contains an evolutionarily conserved Cx9CxnCx10C motif. Sequence alignment of the conserved region of the human COA6 protein with its orthologs in yeast (Saccharomyces cerevisiae), fly (Drosophila melanogaster), fish (Danio rerio) and rat (Rattus norvegicus). The sequence alignment was performed using ClustalW. Horizontal lines above Cx9C and Cx10C residues show the conserved motif. Arrows indicate amino acid residues (tryptophan 59 and glutamic acid 87) that were mutated in a mitochondrial disease patient (6).
Figure 4.

Conserved residues in the Cx9CxnCx10C motif are essential for its function. (A) Schematic representation of the Cx9CxnCx10C motif of human COA6 and yeast Coa6 proteins showing the location of cysteine and tryptophan residues (arrows) targeted for site-directed mutagenesis. (B) WT and coa6Δ cells transformed with empty vector (EV), COA6, or one of its mutant forms were streaked onto YPGE plates and incubated at 30°C or 37°C for 5 days before imaging. (C) Western blot analysis of mitochondrial and cytosolic extract from coa6Δ cells transformed with EV, HA-tagged COA6, and its mutant forms. Pgk1 is used as a cytosolic marker, and Porin is a mitochondrial marker.
Exogenous Cu supplementation rescues respiratory and CcO assembly defects of coa6Δ cells
A subset of mitochondrial IMS twin Cx9C proteins, including Cox17, Cox19 and Cmc1, has been previously shown to be required for CcO assembly by regulating Cu delivery to the CcO subunits (34). Previous studies have also shown that Cu supplementation rescues the respiratory growth defects of cox17Δ and cmc1Δ yeast cells (35,36). Since Coa6 shares many of the features of these twin Cx9C proteins, we asked whether exogenous Cu supplementation could rescue the respiratory and CcO assembly defects of coa6Δ cells. Indeed, Cu supplementation completely rescued the respiratory growth defect of coa6Δ cells (Fig. 5A). Notably, the rescue was specific to Cu, as supplementation with the other bivalent metals, including cobalt, magnesium and zinc, failed to rescue the respiratory growth defect (Fig. 5A and B). Cu supplementation in coa6Δ cells also restored respiration and Cox2 protein levels to WT levels (Fig. 5C and D). Conversely, depletion of Cu with an extracellular copper-specific chelator, bathocuproinedisulfonic acid, completely inhibited the growth of coa6Δ cells on non-fermentable medium even at 30°C (Fig. 5E). These results implicate Coa6 in mitochondrial Cu homeostasis and transport to CcO.
Figure 5.

Cu supplementation rescues respiratory and CcO assembly defects of coa6Δ. (A) Serially diluted WT and coa6Δ cells were spotted on YPGE plates in the presence and absence of 5 µm Cu, Co, Mg and Zn bivalent salts at 30°C and 37°C and allowed to grow for 4–5 days before imaging. (B) Growth of WT and coa6Δ cells in YPGE liquid media in the presence of 5 µm Cu, Co, Mg and Zn bivalent salts at 30°C. (C) Oxygen consumption rate (OCR) of WT, coa6Δ and coa6Δ supplemented with 5 µM CuSO4. Error bars represent mean ± SD (n = 6), *P < 0.05, t-test. NS, not significant. (D) Western blot of Cox2 in mitochondria isolated from WT, coa6Δ, and coa6Δ cells supplemented with 5 µm CuSO4. Porin is used as a loading control. (E) Serial dilutions of WT and coa6Δ strains grown in synthetic medium with a non-fermentable carbon source (2% glycerol; CSG) with or without 20 μm bathocuproinedisulfonic acid (BCS). The pictures were taken 5 days after seeding.
Steady-state levels of another mitochondrial Cu-containing enzyme, superoxide dismutase (Sod1), are not altered in coa6Δ cells
Two mitochondrial proteins that are known to use Cu as a cofactor are CcO and Sod1. Both of these enzymes have been proposed to use the same mitochondrial matrix Cu pool for holoenzyme formation (31,37). How Cu is directed from the matrix to the recipient CcO and Sod1 metallochaperones in the IMS is unknown. Coa6 could be involved in trafficking Cu to both CcO and Sod1. To explore this possibility, we determined the steady-state levels of the Sod1 protein by western blotting. Our result showed that unlike CcO, Sod1 protein levels are not altered in coa6Δ cells (Fig. 6A), suggesting that Coa6 specifically functions in the Cu-delivery pathway to CcO. A previous study showed that yeast CcO mutants exhibit elevated Sod1 activity (36). Consistent with that finding, we also observed a modest increase in Sod1 activity in coa6Δ cells compared with WT (Fig. 6B and C).
Figure 6.

Mitochondrial Sod1 levels are unaltered in coa6Δ cells. (A) Mitochondria were isolated from the WT, coa6Δ and sod1Δ cells grown at 30°C in YPD to early stationary phase. Mitochondrial protein extract (2.5, 5 and 10 µg) was separated on 4–12% polyacrylamide gel and probed for Sod1 and porin by immunoblotting. Porin was used as a loading control. The blot is a representative of two independent experiments. (B) Sod activity in isolated mitochondria from the indicated strains was measured in an in-gel assay as described in Materials and Methods. Gel is representative of two independent experiments. (C) For quantification of the Sod activity in (B), the images were digitalized and densitometry performed using the ImageJ software.
Zebrafish embryos treated with zfcoa6 morpholino exhibit heart developmental defects
In order to decipher the physiological activity of Coa6 in the context of an intact developing animal, we constructed a zebrafish model of Coa6 deficiency. We chose zebrafish embryos because they are transparent and develop rapidly outside of the mother, enabling observation of organ development over time (38). Importantly, they do not require a functional cardiovascular system for the first 4–5 days after fertilization, as their small size allows sufficient passive oxygen diffusion (39), and can therefore be used to examine phenotypes that would lead to embryonic lethality in mammalian models. A zfcoa6 translation blocking (TB) and a mismatch control (MMC) morpholino were injected separately into 1–4 cell stage embryos. The TB and MMC morphants were examined at 24, 48, 72 and 96 h post fertilization (hpf). At 24 and 48 hpf, we observed mild pericardial edema, slightly smaller eyes and head, and shorter tails in TB embryos. After 48 hpf, defects in heart development became apparent, including weak heartbeat, little-to-no red blood cell circulation and a bag-like heart with two enlarged chambers and thin walls (data not shown). These cardiac phenotypes persisted through 72 and 96 hpf, with the heart failing to loop (Fig. 7A) and heart rate showing progressive decline (Fig. 7B). The most prominent defect was the presence of massive pericardial edema that extended to the yolk sac (Fig. 7A). Western blot analysis of TB morphants showed a specific decrease in the CcO subunit Cox2 (Fig. 7C). Taken together, these results show marked functional and developmental heart defects in zfcoa6 morpholino-treated zebrafish, which parallel the observed MRCD patient defect in terms of the affected organ system.
Figure 7.

Coa6 knockdown in zebrafish embryos results in cardiac defects. (A) Low- and high-resolution images of zebrafish embryos treated with mismatch control (MMC) and zfcoa6 translation blocking (TB) morpholinos at 72 and 96 h post fertilization (hpf). Arrows indicate pronounced pericardiac edema. High resolution images of the zebrafish heart in MMC and TB embryos were captured 72 and 96 hpf, where heart perimeter is outlined in MMC images to facilitate comparison to TB hearts. The images are representative of four independent experiments, where each experiment had between 150–250 embryos per morpholino. (B) Heart rate (beats/min) was measured in MMC and TB embryos at 72 and 96 hpf. Error bars represent mean ± SD and * indicates statistically significant differences, P < 0.001, t-test. (C) Western blot analysis of whole embryo protein extracts with indicated antibodies against CcO and complex V subunits. β actin is used as a loading control. The blot is representative of three independent experiments.
DISCUSSION
Next-generation sequencing technologies have greatly improved our ability to identify sequence variants associated with human disease; however, proving the pathogenicity of the identified mutation has remained challenging. One of the major obstacles in establishing the pathogenicity of disease-causing mutations is the lack of an ascribed function to the gene in question. This is most apparent in monogenic Mendelian disorders such as inborn errors of energy metabolism, where a plethora of genes are known to cause MRCD, many of which are completely uncharacterized at the time of initial identification in human patients. In this report, we demonstrate an evolutionarily conserved role of a human MRCD candidate disease gene, COA6, in the assembly of MRC complex IV in yeast, zebrafish and human cells. Using the yeast S. cerevisiae as a model system, we show that patient mutations disrupt Coa6 function, possibly by regulating Cu delivery to CcO. Furthermore, we show a critical requirement of Coa6 in heart development and function by performing morpholino-based gene-silencing experiments in zebrafish embryos. This finding parallels clinical phenotypes observed in a human MRCD patient who died of neonatal hypertrophic cardiomyopathy. Our study thus underscores the power of an integrative approach to discover MRCD candidate genes and demonstrates the utility of multiple model systems in interpreting sequence variants associated with human genetic disorders.
In our search for novel MRC assembly factors, we chose to focus on COA6 because three independent lines of evidence linked it to MRC function. First, it is an evolutionarily conserved protein that localizes to yeast and mammalian mitochondria (8,30). Second, previous genomic and proteomic studies in yeast have shown that Coa6 plays a role in CcO assembly and interacts with known MRC components (7,8). Third, potential pathogenic mutations were reported in the conserved residues of COA6 in an MRCD patient (6). The next-generation sequencing study that reported the patient mutations did not establish the pathogenicity of the mutations (6). Utilizing knockdown models in human cell lines and zebrafish embryos, as well as a knockout model in the yeast S. cerevisiae, we showed that COA6 is essential for the maintenance of steady-state levels of CcO (Figs 1D, E, 2C and 7C), and its deficiency results in reduced respiration and diminished cardiac function (Figs 1C and 7B). The patient mutations were present within the evolutionarily conserved Cx9CxnCx10C motif (Fig. 3), suggesting their essential function. We generated point mutations in the yeast Coa6 protein that were synonymous to compound heterozygous mutations reported in the human patient and showed that the mutant proteins failed to rescue the respiratory deficient growth of coa6Δ cells (Fig. 4B), thus establishing the pathogenicity of patient mutations.
The mutations in conserved residues within the Cx9CxnCx10C motif, including the patient mutations, resulted in decreased mitochondrial steady-state levels of Coa6 (Fig. 4C), implying that these residues are required either for mitochondrial import of Coa6 or for its stability. Since a previous report (8) has shown that the mitochondrial import of Coa6 is dependent on MIA import machinery, which utilizes the characteristic twin Cx9C motif, our results are consistent with the possibility that mutations in the conserved residues within the Cx9CxnCx10C motif will abrogate mitochondrial import of Coa6.
The evolutionarily conserved, non-canonical Cx9CxnCx10C motif of Coa6 is similar to the twin Cx9C motif found in a subset of mitochondrial IMS proteins involved in Cu delivery to CcO (34). The rescue of respiratory and CcO assembly defects of yeast coa6Δ cells with exogenous Cu supplementation (Fig. 5) suggests a role for Coa6 in the Cu-delivery pathway to CcO (Fig. 8). Notably, genetic mutations in three IMS proteins, including the mitochondrial Cu chaperones, Sco1 and Sco2, and a twin Cx9C protein C2orf64/COA5, have been previously reported in MRCD patients with pronounced CcO deficiency and distinct, early-onset fatal clinical phenotypes (24–26,28). Patients with SCO1 mutations suffer from neonatal hepatic failure and ketoacidotic coma (24), whereas SCO2 and COA5 mutations are associated with fatal neonatal cardiomyopathy (25,26,28). The clinical symptoms of COA6 mutations are thus similar to SCO2 and COA5 patient mutations, suggesting that these proteins partake in a common biochemical process and are particularly essential for cardiac function. Interestingly, Cu supplementation has been shown to rescue CcO activity in patient-derived SCO2 cells (40), as well as improve the clinical symptoms of patients suffering from aberrant mitochondrial Cu homeostasis (41,42). Our demonstration of Cu rescue of coa6Δ yeast cells motivates further investigation of Cu-mediated rescue of COA6 deficiency in mammalian cells.
Figure 8.
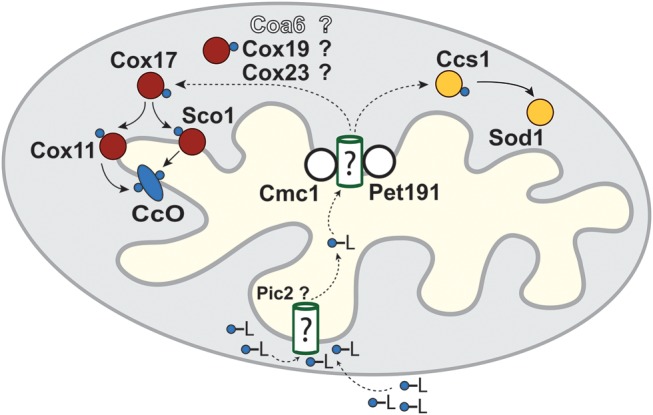
Schematic of the proposed mitochondrial copper transport pathways to CcO subunits. Cu (blue circles) enters the mitochondrial matrix bound to an unidentified ligand (-L) partially through the recently identified Pic2 protein. The Cu-L is stored in the matrix and exported to the intermembrane space to be delivered to CcO and Sod1 through a series of metallochaperones depicted in red and yellow, respectively. Only the final steps of Cu delivery mediated by Cox17, Cox11 and Sco1 are experimentally demonstrated. Dashed arrows indicate the hypothetical role of the several Cx9C containing proteins, including Coa6, in Cu transfer from a matrix Cu pool, across the inner mitochondrial membrane, to Cox17.
Although Cu supplementation offers an exciting therapeutic avenue for MRCD patients with aberrant mitochondrial Cu metabolism, many important questions regarding the mechanism of Cu-mediated rescue and the Cu-delivery pathway remain unanswered. For example, why and how is Cu supplementation able to bypass the complete absence of a Cu metallochaperone? Why are there so many IMS proteins in the Cu-delivery pathway? Despite the discovery of several IMS Cu-binding proteins over the last decade and a half, our understanding of the mitochondrial Cu-delivery pathway to CcO subunits remains incomplete. In S. cerevisiae, up to 10 proteins (Cox11, Sco1, Sco2, Cox17, Cox19, Cox23, Cmc1, Cmc2, Pet191 and Pic2) have been implicated so far in the Cu-delivery pathway to CcO (34,43). It has been suggested that in a daisy chain transfer mechanism, Cu is successively transferred from its non-protein ligand in the matrix to the IMS proteins for final delivery to Cox1 and Cox2 (34) (Fig. 8). However, only the last few steps that involve successive transfer of Cu from Cox17 to Cox1 and Cox2, via Cox11 and Sco1 respectively, are well characterized (44,45). We predict Coa6 to be either a regulator or a bona fide member of this Cu-delivery pathway because of (i) the presence of a highly conserved cysteine-rich Cx9CxnCx10C motif, (ii) IMS localization, (iii) small size (14 kDa) and (iv) Cu rescue of a CcO-specific MRC defect. The identification of patient mutations through human genetics and our construction of multiple model systems have provided the necessary tools that will guide future experiments to unravel the precise biochemical functions of Coa6 in the Cu-delivery pathway for CcO assembly.
MATERIALS AND METHODS
Yeast strains, plasmids and culture conditions
Saccharomyces cerevisiae strains used in this study were obtained from Open Biosystems. These include the haploid BY4741 WT (Mat-a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0), and the isogenic coa6Δ (Mat-a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa6::KanMX4), shy1Δ (Mat-a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, shy1::KanMX4) and sod1Δ (Mat-a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sod1::KanMX4). All strains were confirmed by PCR as well as by replica plating on dropout plates. For growth in liquid medium, yeast cells were precultured in YPD (1% yeast extract, 2% peptone and 2% glucose) and then inoculated into medium containing either 2% glucose (YPD) or 3% glycerol + 1% ethanol (YPGE) and grown to early stationary phase. Solid YPD and YPGE media were prepared by addition of 2% agar. For metal supplementation experiments, growth medium was supplemented with 5 µm divalent chloride salts of Cu, Co, Mg or Zn. A Gateway cloning vector pAG423-GPD-ccdB-HA (Addgene) was used to express yeast COA6 with a C-terminal HA tag. The same construct was used as a template to perform site-directed mutagenesis (Agilent Technologies QuikChange Lightning) for generating all the point mutants. All constructs were confirmed by DNA sequencing. Yeast cells transformed with the pAG423GPD-ccdB-HA vector were pre-cultured in 2% glucose-containing synthetic media lacking histidine before plating on YPGE.
Yeast oxygen consumption and isolation of subcellular fractions
BY4741 WT and coa6Δ cells were grown in YPD media to late log phase and then washed, counted and resuspended in the assay medium (0.176% yeast nitrogen base, 0.5% ammonium sulfate and 2% ethanol) before seeding in XF24-well microplates (Seahorse Bioscience) at 5 × 105 cells/well. After seeding, cells were centrifuged at 100g for 2 min and incubated at 30°C for 30 min in the assay medium prior to measurements. The cellular OCR was measured using an XF24 extracellular flux analyzer (Seahorse Biosciences) at 30°C. Mitochondria were isolated from yeast cells grown to late log phase in YPD by the previously described method (46) and protein concentrations were determined by the BCA assay (Thermo Scientific). Mitochondrial samples were stored at −80°C before performing any protein analysis. Cytosolic fractions were prepared from yeast cells grown to late log phase using acid-washed glass beads as described previously (47). Briefly, 2 g of yeast cells were taken and washed in 25 ml prechilled PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] lysis buffer containing 20 mm PIPES/KOH (pH 6.8), 250 mm sorbitol, 100 mm potassium acetate, 50 mm KCl, 5 mm MgCl2, 2 mm DTT, 1 mm PMSF and protease inhibitor cocktail (Roche Diagnostic). Cell pellets were suspended in 2 ml prechilled PIPES lysis buffer and 3 g of glass beads and lysed by vortexing for 15 min at 4°C. Cell debris and glass beads were cleared at 3000g for 10 min and the supernatant was centrifuged at 150 000g for 1 h. The final supernatant was then used as the cytosolic fraction for protein analysis.
Human cell culture
MCH58 immortalized human skin fibroblasts (obtained from Dr Eric Shoubridge) were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Sigma) and 1 mm sodium pyruvate (Life Technologies). The COA6 knockdown cells were cultured in DMEM medium supplemented with 2 μg/ml puromycin. Mitochondria from human MCH58 cells were isolated as per the manufacturer's protocol (Abcam) and protein concentrations were determined by the BCA assay (Thermo Scientific).
shRNA lentiviral production and infection
The lentiviral vector (pLKO.1) for expressing shRNA was purchased from the Mission shRNA collection (Sigma). Lentiviral particle production and infection was performed as described previously (14). For constructing stable knockdown cell lines, 100 000 cells were seeded in a six-well dish and 100 μl of viral supernatant was added to cells to a final volume of 2 ml of medium containing 8 μg/ml polybrene. The plates were spun at a relative centrifugal force of 805g for 30 min at 30°C, returned to a 37°C incubator, and selected for infection after 24 h with 2 μg/ml puromycin-containing medium.
The shRNA sequences used for COA6 knockdown are shown below.
.
| COA6 | Sequence (5′→3′) |
| kd1 | CTGGAAGTGTTTAGATGAGAACTCGAGTTCTCATCTAAACACTTCCAG |
| kd2 | GATGAGTACTGGAAGTGTTTACTCGAGTAAACACTTCCAGTACTCATC |
| kd3 | GCAAGAAGTTAAGAAGCTCTTCTCGAGAAGAGCTTCTTAACTTCTTGC |
| kd4 | CTATGGAACAGTAATAGTTTGCTCGAGCAAACTATTACTGTTCCATAG |
| kd5 | GAAGCTCTTTCGAATCAAGTTCTCGAGAACTTGATTCGAAAGAGCTTC |
Quantitative PCR
RNA was isolated from cells using the RNeasy kit (Qiagen). An amount of 500 ng of RNA was used as starting material to synthesize first-strand cDNA using Superscript III (Life Technologies). The qPCR was performed on cDNA samples in a Stratagene Mx30005P RT-PCR system in a 96-well plate. 20 μl of PCR reactions were prepared with 2X mastermix and 20X COA6-specific TaqMan assay from Applied Biosystems (assay ID Hs01372973_m1). COA6 mRNA levels were normalized to β-actin expression levels.
BNPAGE, SDSPAGE and western blotting
BNPAGE and denaturing sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDSPAGE) were performed to separate intact and denatured MRC protein complexes, respectively (48). For BNPAGE sample preparation, yeast mitochondria (10–20 μg) were solubilized in 1% digitonin, and soluble lysate was resolved on a 3–12% gradient native PAGE Bis–Tris gel (Invitrogen). SDSPAGE was performed on mitochondrial samples solubilized in RIPA lysis buffer (150 mm NaCl, 1 mm EDTA, 50 mm Tris–HCl, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with protease inhibitor cocktail (Roche Diagnostic). Western blot was performed using a Trans–Blot transfer cell (Bio-Rad). Membranes were blocked in 5% fatty acid-free BSA dissolved in Tris–buffered saline with 0.1% Tween 20 (TBST–BSA) and probed with antibodies as indicated. The separated proteins were blotted onto a polyvinylidene difluoride membrane and blocked for 1 h at room temperature in TBST–BSA. Membranes were incubated with primary antibody in TBST–BSA overnight at 4°C. Primary antibodies for human proteins were used at the following dilutions: COX2, 1:4000 (Abcam 110 258); NDUFB8, 1:2000 (Abcam 110 242); SDHB, 1:1000 (Abcam 14 714); UQCRC2, 1:1000 (Abcam 14 745); ATP5A, 1:10 000 (Abcam 14 748); Porin, 1:1000 (Abcam 14 734); and COA6/C1orf31, 1:500 (Proteintech 24 209–1-AP). The primary antibodies for yeast were used at the following dilutions: Cox2, 1:1000 (Abcam 110 271); Cox3, 1:1000 (Abcam 110 259); Sdh2, 1:5000 (from Dr Dennis Winge); Rip1, 1:50 000 (from Dr Vincenzo Zara); Atp2, 1:40 000 (from Dr Sharon Ackerman); Porin, 1:1000 (Abcam 110 326); Sod1, 1:5000 (from Dr Valeria Culotta); and Pgk1, 1:2500 (from Dr Craig Kaplan). The primary antibodies for zebrafish were used at the following dilutions: COX2, 1:4000 (Abcam 110 258); ATP5A, 1:10 000 (Abcam 14 748); and β actin, 1:1000 (Sigma A2228). Secondary antibodies were used at 1:5000 dilution for 1 h at room temperature. Membranes were developed using WesternLightning Plus-ECL (PerkinElmer), or SuperSignal West Femto (Thermo Scientific) for the human COA6/C1orf31 antibody.
In-gel Sod activity
To measure the Sod1 activity in isolated mitochondria, we used an in-gel assay as described previously by Horn et al. (36). Briefly, 2 mg of isolated mitochondria were solubilized in 300 μl of 20 mm potassium phosphate buffer, pH 7.4, containing 1 mm EDTA, 0.1% Triton X-100, 4 mm PMSF with protease inhibitor cocktail (Roche Diagnostic) and incubated on ice for 10 min. The protein concentration in the supernatant was estimated by BCA assay and 35 μg of protein supplemented with loading dye (50% glycerol, 0.2% bromophenol blue) were separated onto a 4–16% native PAGE gel. The gel was stained for Sod activity as described (49). To quantify Sod1 activity, the images were digitalized and densitometric analyses performed using the ImageJ software.
Heart development and cardiac function in zebrafish embryos
Wild-type zebrafish (AB strain) were obtained from the Zebrafish International Resource Center. Zebrafish were maintained and crossed according to standard methods. Fertilized eggs were collected and placed in E3 embryo medium, and maintained in an incubator set at 28.5°C with a 14/10 h light/dark cycle. Embryos were staged using the criteria of Kimmel et al. (50). All animal studies were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (AR #2850) and performed in accordance with the guidelines. Zebrafish Coa6 (si:ch211–258f14.5) TB and 5 bp MMC morpholinos were designed with Gene Tools, LLC (TB sequence: CGCTCATCTCTGTCTCTCCTCACTC, MMC sequence: CGgTgATCTgTGTCTgTgCTCACTC). Morpholinos were titrated to determine the optimal concentration required for knockdown without overt toxicity, and 0.7 ng final concentrations of TB and MMC morpholino were used for injection. An aliquot of 3 nL of TB or MMC morpholino was injected into the 1–4 cell stage embryo (1–2 hpf). Embryonic development was monitored at 24, 48, 72 and 96 hpf by taking low- and high-magnification images (51). Heart rate was measured at 72 and 96 hpf by counting the number of atrial contractions in embryos using a Zeiss Axio Observer A.1 inverted microscope. Beats per min were calculated and expressed as the mean ± standard deviation. Protein extracts for western blotting were prepared from deyolked embryos as described previously (51). Briefly, 20 embryos were pooled for each time point and quickly frozen and stored at −80°C. Protein lysates were prepared by homogenization in 35 μl RIPA buffer using microfuge pestles. Samples were centrifuged at 12 000g for 10 min at 4°C, and protein concentration in the supernatant was measured by BCA assay.
FUNDING
This work was supported by scientist development grant (#13SDG17060112) from the American Heart Association and startup funds from Texas A&M University to V.M.G., National Institutes of Health awards 5P20RR024485-02, 8 P20 GM103542-02 and startup funds from the Medical University of South Carolina to S.S.L.C. S.A.T. was supported by a Research Experiences for Undergraduates (REU) fellowship from the Department of Biochemistry and Biophysics, Texas A&M University.
ACKNOWLEDGEMENTS
We thank Eric Shoubridge for the MCH58 cell line; Sharon Ackerman, Valeria Culotta, Dennis Winge, Vincenzo Zara, and Craig Kaplan for their generous gift of antibodies. We also thank members of the Gohil lab, including Connor McBroom, Sarah Theriault, Charli Baker and Morgan Thompson, for valuable comments and technical support, and Krista Stackley from the Chan lab for zebrafish husbandry.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Skladal D., Halliday J., Thorburn D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 2.DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 3.Fernández-Vizarra E., Tiranti V., Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim. Biophys. Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 4.Vafai S.B., Mootha V.K. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- 5.Chakravarti A., Clark A.G., Mootha V.K. Distilling pathophysiology from complex disease genetics. Cell. 2013;155:21–26. doi: 10.1016/j.cell.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calvo S.E., Compton A.G., Hershman S.G., Lim S.C., Lieber D.S., Tucker E.J., Laskowski A., Garone C., Liu S., Jaffe D.B., et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003310. 118ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szklarczyk R., Wanschers B.F., Cuypers T.D., Esseling J.J., Riemersma M., van den Brand M.A., Gloerich J., Lasonder E., van den Heuvel L.P., Nijtmans L.G., et al. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012;13:R12. doi: 10.1186/gb-2012-13-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vögtle F.N., Burkhart J.M., Rao S., Gerbeth C., Hinrichs J., Martinou J.C., Chacinska A., Sickmann A., Zahedi R.P., Meisinger C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteomics. 2012;11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soto I.C., Fontanesi F., Liu J., Barrientos A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta. 2012;1817:883–897. doi: 10.1016/j.bbabio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 11.Fornuskova D., Stiburek L., Wenchich L., Vinsova K., Hansikova H., Zeman J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem. J. 2010;428:363–374. doi: 10.1042/BJ20091714. [DOI] [PubMed] [Google Scholar]
- 12.Shoubridge E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- 13.Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F., et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gohil V.M., Nilsson R., Belcher-Timme C.A., Luo B., Root D.E., Mootha V.K. Mitochondrial and nuclear genomic responses to loss of LRPPRC expression. J. Biol. Chem. 2010;285:13742–13747. doi: 10.1074/jbc.M109.098400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A. LSFC Consortium. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chujo T., Ohira T., Sakaguchi Y., Goshima N., Nomura N., Nagao A., Suzuki T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012;40:8033–8047. doi: 10.1093/nar/gks506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruzzenente B., Metodiev M.D., Wredenberg A., Bratic A., Park C.B., Cámara Y., Milenkovic D., Zickermann V., Wibom R., Hultenby K., et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012;31:443–456. doi: 10.1038/emboj.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu F., Addis J.B., Cameron J.M., Robinson B.H. LRPPRC mutation suppresses cytochrome oxidase activity by altering mitochondrial RNA transcript stability in a mouse model. Biochem. J. 2012;441:275–283. doi: 10.1042/BJ20110985. [DOI] [PubMed] [Google Scholar]
- 19.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 20.Weraarpachai W., Sasarman F., Nishimura T., Antonicka H., Aure K., Rotig A., Lombes A., Shoubridge E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valnot I., von Kleist-Retzow J.C., Barrientos A., Gorbatyuk M., Taanman J.W., Mehaye B., Rustin P., Tzagoloff A., Munnich A., Rotig A. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- 22.Antonicka H., Leary S.C., Guercin G.H., Agar J.N., Horvath R., Kennaway N.G., Harding C.O., Jaksch M., Shoubridge E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 23.Antonicka H., Mattman A., Carlson C.G., Glerum D.M., Hoffbuhr K.C., Leary S.C., Kennaway N.G., Shoubridge E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valnot I., Osmond S., Gigarel N., Mehaye B., Amiel J., Cormier-Daire V., Munnich A., Bonnefont J.P., Rustin P., Rotig A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000;67:1104–1109. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papadopoulou L.C., Sue C.M., Davidson M.M., Tanji K., Nishino I., Sadlock J.E., Krishna S., Walker W., Selby J., Glerum D.M., et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 26.Jaksch M., Ogilvie I., Yao J., Kortenhaus G., Bresser H.G., Gerbitz K.D., Shoubridge E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000;9:795–801. doi: 10.1093/hmg/9.5.795. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M., et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 28.Huigsloot M., Nijtmans L.G., Szklarczyk R., Baars M.J., van den Brand M.A., Hendriksfranssen M.G., van den Heuvel L.P., Smeitink J.A., Huynen M.A., Rodenburg R.J. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet., 2011;88:488–493. doi: 10.1016/j.ajhg.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szklarczyk R., Wanschers B.F., Nijtmans L.G., Rodenburg R.J., Zschocke J., Dikow N., van den Brand M.A., Hendriks-Franssen M.G., Gilissen C., Veltman J.A., et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 2013;22:656–667. doi: 10.1093/hmg/dds473. [DOI] [PubMed] [Google Scholar]
- 30.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K., et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson N.J., Winge D.R. Copper metallochaperones. Annu. Rev. Biochem. 2010;79:537–562. doi: 10.1146/annurev-biochem-030409-143539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chacinska A., Koehler C.M., Milenkovic D., Lithgow T., Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longen S., Bien M., Bihlmaier K., Kloeppel C., Kauff F., Hammermeister M., Westermann B., Herrmann J.M., Riemer J. Systematic analysis of the twin cx(9)c protein family. J. Mol. Biol. 2009;393:356–368. doi: 10.1016/j.jmb.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 34.Horn D., Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life. 2008;60:421–429. doi: 10.1002/iub.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glerum D.M., Shtanko A., Tzagoloff A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 36.Horn D., Al-Ali H., Barrientos A. Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol. Cell Biol. 2008;28:4354–4364. doi: 10.1128/MCB.01920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobine P.A., Ojeda L.D., Rigby K.M., Winge D.R. Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem. 2004;279:14447–14455. doi: 10.1074/jbc.M312693200. [DOI] [PubMed] [Google Scholar]
- 38.Liu J., Stainier D.Y. Zebrafish in the study of early cardiac development. Circ. Res. 2012;110:870–874. doi: 10.1161/CIRCRESAHA.111.246504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelster B., Burggren W.W. Disruption of hemoglobin oxygen transport does not impact oxygen-dependent physiological processes in developing embryos of zebra fish (Danio rerio) Circ. Res. 1996;79:358–362. doi: 10.1161/01.res.79.2.358. [DOI] [PubMed] [Google Scholar]
- 40.Jaksch M., Paret C., Stucka R., Horn N., Müller-Höcker J., Horvath R., Trepesch N., Stecker G., Freisinger P., Thirion C., et al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum. Mol. Genet. 2001;10:3025–3035. doi: 10.1093/hmg/10.26.3025. [DOI] [PubMed] [Google Scholar]
- 41.Freisinger P., Horvath R., Macmillan C., Peters J., Jaksch M. Reversion of hypertrophic cardiomyopathy in a patient with deficiency of the mitochondrial copper binding protein Sco2: is there a potential effect of copper? J. Inherit. Metab. Dis. 2004;27:67–79. doi: 10.1023/B:BOLI.0000016614.47380.2f. [DOI] [PubMed] [Google Scholar]
- 42.Horváth R., Freisinger P., Rubio R., Merl T., Bax R., Mayr J.A., Shawan, Müller-Höcker J., Pongratz D., Moller L.B., et al. Congenital cataract, muscular hypotonia, developmental delay and sensorineural hearing loss associated with a defect in copper metabolism. J. Inherit. Metab. Dis. 2005;28:479–492. doi: 10.1007/s10545-005-0479-x. [DOI] [PubMed] [Google Scholar]
- 43.Vest K.E., Leary S.C., Winge D.R., Cobine P.A. Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 2013;288:23884–23892. doi: 10.1074/jbc.M113.470674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiser L., Di Valentin M., Hamer A.G., Hosler J.P. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000;275:619–623. doi: 10.1074/jbc.275.1.619. [DOI] [PubMed] [Google Scholar]
- 45.Rigby K., Cobine P.A., Khalimonchuk O., Winge D.R. Mapping the functional interaction of Sco1 and Cox2 in cytochrome oxidase biogenesis. J. Biol. Chem. 2008;283:15015–15022. doi: 10.1074/jbc.M710072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meisinger C., Pfanner N., Truscott K.N. Isolation of yeast mitochondria. Methods Mol. Biol. 2006;313:33–39. doi: 10.1385/1-59259-958-3:033. [DOI] [PubMed] [Google Scholar]
- 47.Haas A. A quantitative assay to measure homotypic vacuole fusion in vitro. Methods Cell Sci. 1995;17:283–294. [Google Scholar]
- 48.Wittig I., Braun H.P., Schägger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 49.Flohe L., Otting F. Superoxide dismutase assays. Methods Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 50.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 51.Rahn J.J., Stackley K.D., Chan S.S. Opa1 is required for proper mitochondrial metabolism in early development. PLoS One. 2013;8:e59218. doi: 10.1371/journal.pone.0059218. [DOI] [PMC free article] [PubMed] [Google Scholar]