

Abstract
Although coding variants in THAP1 have been causally associated with primary dystonia, the contribution of noncoding variants remains uncertain. Herein, we examine a previously identified Intron 1 variant (c.71+9C>A, rs200209986). Among 1672 subjects with mainly adult-onset primary dystonia, 12 harbored the variant in contrast to 1/1574 controls (P < 0.01). Dystonia classification included cervical dystonia (N = 3), laryngeal dystonia (adductor subtype, N = 3), jaw-opening oromandibular dystonia (N = 1), blepharospasm (N = 2), and unclassified (N = 3). Age of dystonia onset ranged from 25 to 69 years (mean = 54 years). In comparison to controls with no identified THAP1 sequence variants, the c.71+9C>A variant was associated with an elevated ratio of Isoform 1 (NM_018105) to Isoform 2 (NM_199003) in leukocytes. In silico and minigene analyses indicated that c.71+9C>A alters THAP1 splicing. Lymphoblastoid cells harboring the c.71+9C>A variant showed extensive apoptosis with relatively fewer cells in the G2 phase of the cell cycle. Differentially expressed genes from lymphoblastoid cells revealed that the c.71+9C>A variant exerts effects on DNA synthesis, cell growth and proliferation, cell survival, and cytotoxicity. In aggregate, these data indicate that THAP1 c.71+9C>A is a risk factor for adult-onset primary dystonia.
Keywords: Dystonia, DYT6, intronic variant, minigene assay, THAP1
Introduction
Dystonia is a neurological disorder characterized by sustained or intermittent involuntary muscle contractions leading to abnormal postures and movements that may be tremulous (Fahn et al. 1998; Albanese et al. 2013). Physiologically, dystonia may be associated with atypical cocontractions of agonist and antagonist muscle groups, impaired control of somatotopically contiguous muscles, and reduced inhibition of spinal and brainstem reflexes (LeDoux 2012a). Dystonia is classified along two axes: clinical characteristics and etiological categories. Clinical characteristics include age of onset, body distribution, temporal pattern, coexistence of other movement disorders, and other neurological manifestations (Fahn et al. 1998; LeDoux 2012b; Albanese et al. 2013). Etiological categories include nervous system pathology and inherited. In general, adult-onset dystonia without evidence of overt degeneration or structural lesions of the nervous system is referred to as primary or isolated dystonia and inherited in autosomal-dominant fashion with reduced penetrance (Fahn et al. 1998; LeDoux 2012b). To date, coding mutations in TOR1A, THAP1, CIZ1, GNAL, and ANO3 have been causally associated with primary dystonia (Ozelius et al. 1997; Fuchs et al. 2009, 2012; Xiao, et al., 2010; Xiao et al. 2012; Vemula et al. 2013). The role of large structural and noncoding variants in the etiopathogenesis of primary dystonia remains largely unexplored (Moscovich et al. 2013).
Among the known genetic causes of adult-onset primary dystonia, THAP1 dystonia appears to be associated with the most diverse ages of onset and anatomical distributions (Fuchs et al. 2009; Xiao et al. 2010; LeDoux et al. 2012). THAP1 encodes Thanatos-associated THAP domain containing, apoptosis-associated protein 1, encoding DNA-binding transcription factor THAP1. Located on Chr.8p11.21 (42,691,817–42,698,474: 6658 bp), the major isoform of this gene (NM_018105) contains three exons. THAP1 harbors a zinc-binding THAP domain, nuclear localization signal, and coiled-coiled domain. This zinc-binding domain is present in other proteins that are involved in cell-cycle control, apoptosis, and transcriptional regulation. THAP proteins are absent from plants and bacteria, but found in Drosophila, Caenorhabditis elegans, Xenopus, zebrafish, and mammals (Roussigne et al. 2003). Monomeric THAP1 shows low affinity for DNA (Campagne et al. 2010), and amino acid residues 154–166 are critical for dimerization (Sengel et al. 2011). THAP1 is widely expressed in brain and extraneural tissues including whole blood (Zhao et al. 2013).
THAP1 binds to specific DNA sequences, regulating cell proliferation and the G1/S checkpoint through pRB/E2F cell-cycle target genes (Clouaire et al. 2005; Cayrol et al. 2007; Sabogal et al. 2010; LeDoux et al. 2012, 2013). For instance, RRM1 is a direct transcriptional target of THAP1 (Cayrol et al. 2007). Both overexpression and knockdown of THAP1 in endothelial cells (ECs) inhibits proliferation.
To date, over 80 disease-specific sequence variants have been localized to the coding regions of THAP1 and associated with focal, segmental, multifocal, and generalized dystonia with age of onset ranging from 3 to over 60 years (LeDoux et al. 2012). Consistent with the characteristically reduced penetrance of adult-onset dystonia, several asymptomatic carriers have been identified in the relatives of probands. The broad clinical heterogeneity of THAP1 dystonia and low penetrance of known coding variants suggest that noncoding variants in THAP1 could contribute to the risk of developing adult-onset primary dystonia. In 2010, we identified a sequence variant (c.71+9C>A) (rs200209986) near the donor site of Intron 1 that might increase the risk of developing primary dystonia (Xiao, et al. 2010). The variant was found in 7/1210 subjects with dystonia but only 1/600 controls. Although the control did not manifest dystonia, she did report trismus and had experienced seizures as a child. Herein, we present the results of a larger definitive case–control study of THAP1 c.71+9C>A in 1672 subjects with primary, mainly adult-onset, dystonia and 1574 neurologically normal Caucasian controls. Moreover, we characterize the relative effects of THAP1 c.71+9C>A on cell-cycle control, splicing, and gene expression.
Materials and Methods
Human subjects
All human studies were conducted in accordance with the Declaration of Helsinki with formal approval from the institutional review boards at each participating study site. All subjects gave written informed consent. Recruitment of patients with primary dystonia and neurologically normal controls has been described (Xiao, et al. 2010; LeDoux et al. 2012; Vemula et al. 2013). Demographics and phenotypes for dystonia and control subjects are presented in Table 1 and Table S1. The control subject with trismus reported earlier (Xiao, et al. 2010) was excluded from the analyses reported herein because of uncertainty regarding how her phenotype should be classified. Of note, the data contained in Table 1 do not include the family members of probands. In addition, all neurologically normal controls and all subjects classified as primary dystonia were Caucasians of European descent. Minor allele frequencies (MAFs) were compared with those reported in 1000 Genomes (http://www.1000genomes.org) and Exome Variant Server (EVS) (http://evs.gs.washington.edu) databases using Fisher's exact test or Chi-square with Yates correction.
Table 1.
Clinical diagnoses and demographics
| Gender | |||||
|---|---|---|---|---|---|
| Clinical diagnosis | Number (age of onset)1 | Family history2 | Male | Female | Number of subjects with c.71+9C>A |
| Laryngeal dystonia | 472 (45.4 ± 15.7, 7–85) | 8.3% | 110 | 362 | 3 |
| Cervical dystonia | 509 (44.6 ± 13.6, 4–76) | 10.0% | 118 | 391 | 3 |
| Blepharospasm | 198 (58.1 ± 9.4, 20–73) | 10.1% | 61 | 137 | 2 |
| Hand–forearm dystonia | 52 (34.9 ± 13.0, 7–60) | 9.6% | 23 | 29 | 0 |
| Oromandibular dystonia | 18 (52.5 ± 12.3, 20–70) | 11.1% | 4 | 14 | 1 |
| Other primary focal dystonia | 38 (43.3 ± 18.1, 10–74) | 13.2% | 15 | 23 | 0 |
| Segmental dystonia | 143 (48.0 ± 12.2, 12–74) | 12.6% | 47 | 96 | 0 |
| Multifocal dystonia | 24 (33.3 ± 16.1, 7–67) | 20.8% | 8 | 16 | 0 |
| Generalized dystonia | 26 (23.3 ± 17.8, 1–57) | 15.4% | 12 | 14 | 0 |
| Classified dystonias | 1480 | 398 | 1082 | 9 (P = 0.0095)3 | |
| Unclassified dystonias4 | 192 | NA | NA | NA | 3 |
| Dystonia total | 1672 | 12 (P = 0.0035)3 | |||
| Neurologically normal controls | 1574 (61.2 ± 14.4, 23–95) | NA | 688 | 886 | 1 |
NA, not available or applicable.
Mean age at study enrollment ± standard error, range (years).
First- or second-degree relative with dystonia.
Fisher's exact test for case–control analysis.
Subjects with unclassified dystonia were obtained from Athena Diagnostics.
HRM and genotyping
High-resolution melting (HRM) was performed with the LightCycler® 480 real-time polymerase chain reaction (PCR) system and High Resolution Master Mix (Roche; Indianapolis, IN) in accordance with manufacturer instructions and our laboratory protocol (Xiao, et al. 2010). Melting curves and difference plots were analyzed using Gene Scanning Software. For samples with shifted melting curves, PCR products were cleaned using ExoSAP-IT® (United States Biochemical, Santa Clara, CA) and sequenced in the forward and reverse directions on an Applied Biosystems (Grand Island, NY) 3130XL Genetic Analyzer (Table S2).
Extraction of RNA from leukocytes and cell lines
Ambion's (Grand Island, NY) LeukoLOCK™ Total RNA Isolation System and TRI Reagent® were used to isolate RNA from peripheral blood leukocytes of subjects with dystonia and controls. Using relative quantitative real-time reverse transcription PCR (QRT-PCR), we examined the effects of c.71+9C>A on THAP1 gene expression in dystonia subjects harboring c.71+9C>A (n = 6) in comparison to controls without the variant (n = 12). For microarray, whole-genome gene expression studies, RNA was extracted from lymphoblastoid cell lines derived from six dystonia patients with c.71+9C>A and nine normal controls without this variant. Sanger sequencing was used to exclude coding, splice site, 5' untranslated region, and other previously reported intronic sequence variants (e.g., c.71+126T>C) from the controls.
To establish lymphoblastoid cell lines, peripheral blood mononuclear cells were separated by centrifugation on a sodium diatrizoate polysucrose gradient and transformed with Epstein–Barr virus (Coriell, Camden, NJ). The lymphoblastoid cell lines were propagated in Roswell Park Memorial Institute-1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 15% fetal bovine serum (Sigma-Aldrich) and 2 mmol/L l-glutamine (Invitrogen, Grand Island, NY) at 37°C in 5% CO2. Confluent cells (1 × 106 cells/mL) were harvested for RNA using TRI Reagent® from Ambion. The quality of total RNA derived from leukocytes and lymphoblastoid cell lines was accessed with a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Chip kit. Subsequent downstream analyses were limited to samples with RNA Integrity Numbers greater than 8.0.
Relative quantitative reverse transcription PCR
Reverse transcription was performed with Ambion's RETROscript™ kit using 500 ng of total RNA as template. The reaction mixture was incubated at 44°C for 1 hr followed by 10 min at 92°C. QRT-PCR was performed using the Roche LightCycler® 480 with specific primers for THAP1 and TOR1A (Table S2), along with Universal ProbeLibrary (Roche) probes for human THAP1, TOR1A, and the endogenous control (cyclophilin D). Four endogenous controls (encoded proteins: cyclophilin D, β-tubulin, TATA-binding protein, hypoxanthine-guanine phoshoribosyltransferase) were examined. Cyclophilin D showed the smallest sample-to-sample variance. Student's t-test was used to compare RNA expression between dystonia and control samples.
Whole-genome gene expression analysis
For microarray analyses, we used the Illumina® HumanHT-12 v.4 expression microarray platform (Illumina, San Diego, CA) to assess the expression levels in each individual sample. These arrays investigate whole-genome expression, providing coverage for more than 47,000 transcripts and known splice variants across the human transcriptome. In total, 15 samples, six from patient lymphoblastoid cell lines and nine from normal controls were processed on two chips. Total RNA (200 ng) was processed with the Illumina® TotalPrep™ RNA Amplification Kit (Applied Biosystems) to produce biotinylated complementary RNAs (cRNAs). This procedure included the reverse transcription of RNA to synthesize the first strand complementary DNA (cDNA), second-strand cDNA synthesis, cDNA purification, in vitro transcription to synthesize cRNA, cRNA amplification, and purification. The concentration of the cRNA solution was determined by measuring the absorbance at 260 nm/280 nm using the NanoDrop 1000A spectrophotometer. The biotinylated cRNAs (750 ng/sample) were hybridized to the arrays for 20 h at 58°C. These direct hybridized microarrays were scanned with the BeadArray® reader (Illumina, San Diego, CA). The raw data were processed for errors and quality checks using Illumina's proprietary GenomeStudio® software. Gene expression data were quartile normalized and summarized further with GeneSpring GX® 12.1 software (Agilent® Technologies, Santa Clara, CA). An unpaired t-test was done to filter probes that were significant at P ≤ 0.05 with a mean fold change ≥1.5. Filtered probes were further investigated for biological significance using WebGestalt Gene Set Analysis Toolkit 2.0 (http://bioinfo.vanderbilt.edu/webgestalt/) (Zhang et al. 2005). The differentially expressed gene set was compared to the human genome using the hypergeometric test followed by correction for multiple testing using the Benjamini and Hochberg (1995) (BH) method at a significance level of 0.05 (False Discovery Rate < 0.05). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were accessed from WebGestalt (Ashburner et al. 2000; Kanehisa and Goto 2000). Dysregulated cellular networks were examined with Ingenuity Pathway Analysis (IPA; Ingenuity, Redwood City, CA).
Minigene assay and in silico analysis of splicing
To test the effect of c.71+9C>A variant on splicing of THAP1, we performed a minigene assay. The wild-type minigene construct was produced by cloning into the exon trap vector pET01 (MoBiTec, Göttingen, Germany) in two steps. First, we introduced genomic DNA encompassing ∼400 bp 5′ to Exon 1, Exon 1, and ∼400 bp of Intron 1 into the pET01 vector. Genomic DNA was amplified with the SequalPrep™ Long PCR Kit from Invitrogen, followed by digestion of the PCR product and pET01 with XhoI and BamHI at 37°C for 2 h and then at 65°C for 20 min. This was followed by ligation of cohesive-ended gel-purified digested PCR products into pET01 with the LigaFast™ Rapid DNA Ligation System from Promega (Madison, WI). After sequence confirmation, the second part of the gene, which included ∼400 bp of Intron 1, Exon 2, Intron 2, and ∼600 bp of Exon 3, was introduced into the minigene construct with BamHI and NotI digestion (Table S3). After sequence confirmation, the variant c.71+9C>A was introduced into the wild-type minigene with the QuikChange® II XL Site-Directed Mutagenesis Kit from Stratagene (La Jolla, CA) to form the mutant minigene.
Human embryonic kidney (HEK293) cells were transfected in triplicates and total RNA was extracted after 72 h with TRI Reagent®. DNA was removed with DNase I (Ambion) and total RNA quality was examined with both agarose gel electrophoresis and a NanoDrop® ND-1000 spectrophotometer. Reverse transcription was performed with Ambion's RETROscript™ kit using 500 ng of total RNA as template. PCR products were examined on a 1% agarose gel, followed by quantifying the bands with ImageJ (http://rsbweb.nih.gov/ig/). Effects of genotype were evaluated with a Student's t-test.
The c.71+9C>A Intron 1 variant was further evaluated for its effects on splicing using a broad array of in silico programs which examine splicing, enhancer sites, and silencer sites. The programs used included Relative Enhancer and Silencer Classification by Unanimous Enrichment exonic enhancer site (RESCUE-ESE) (Fairbrother et al. 2002), Putative Exonic Splicing Enhancers/Silencers (PESX) (Zhang and Chasin 2004), Human Splicing Finder (HSF) (Desmet et al. 2009), NetGene2 (Brunak et al. 1991), Splice Site Prediction by Neural Network (NNSplice) (Reese et al. 1997), MaxEntScan (MES) (Yeo and Burge 2004), ESEfinder (Cartegni et al. 2003), and Automated Splice Site and Exon Definition Analyses (ASSAED) (Rogan et al. 1998).
Flow cytometry cell-cycle analysis
For flow cytometry, 10 × 106 lymphoblastoid cells were washed with phosphate-buffered saline (PBS) and fixed overnight with 90% ice-chilled ethanol at −20°C. Fixed cells were then washed with 3× PBS followed by incubation with propidium iodide staining buffer (PBS with RNaseA 100 μg/mL and propidium iodide 50 μg/mL) at 37°C for 30 min, and then overnight at 4°C in the dark. The analysis was performed with a BD Biosciences LSR II Flow Cytometer (San Jose, CA).
Results
Case–control analysis of THAP1 c.71+9C>A frequency
The THAP1 c.71+9C>A variant (rs200209986) was found in neurologically normal Caucasian controls with a MAF of 0.0318% (1/3148). This is comparable to reports from the 1000 Genomes project (MAF = 0.0916%, 2/2184) and EVS database (MAF = 0.199%, 26/13006). In subjects with dystonia, the MAF was significantly higher (P < 0.05) at 0.359% (12/3344) when compared with the 1000 Genomes project and neurologically normal Caucasian controls genotyped in this study. However, the difference in MAF did not reach statistical significance when compared with the EVS database (P = 0.13).
Effects of c.71+9C>A on THAP1 and TOR1A expression
In comparison to controls with no identified THAP1 sequence variants, leukocytes that harbored the c.71+9C>A variant were associated with an elevated ratio of Isoform 1 (NM_018105, 3-exon variant) to Isoform 2 (NM_199003, 2-exon variant) (Table 2). However, the c.71+9C>A variant had no effect on overall expression of THAP1. In addition, the c.71+9C>A variant had no effect on expression of TOR1A in either lymphoblastoid cell lines or leukocytes (Table S4).
Table 2.
THAP1 mRNA expression in wild-type and mutant cells
| Isoform 1 | Isoform 2 | Isoform 1/Isoform 2 | ||||
|---|---|---|---|---|---|---|
| Genotype/phenotype | Mean ± SEM | P1 | Mean ± SEM | P1 | Mean ± SEM | P1 |
| Leukocytes | ||||||
| Normal controls (n = 12) | 14.99 ± 4.68 | 0.96 | 1.00 ± 0.33 | 0.29 | 14.99 ± 3.03 | 0.028 |
| c.71+9C>A dystonia (n = 6) | 14.93 ± 5.10 | 0.70 ± 0.27 | 21.25 ± 3.90 | |||
| Lymphoblastoid cell lines | ||||||
| Normal controls (n = 12) | 10.14 ± 0.41 | 0.19 | 1.06 ± 0.05 | 0.24 | 9.63 ± 0.30 | 0.99 |
| c.71+9C>A dystonia (n = 6) | 8.82 ± 0.83 | 0.93 ± 0.10 | 9.62 ± 0.34 | |||
P-value of t-test statistic.
THAP1 c.71+9C>A is associated with transcriptional dysregulation
Microarray gene expression analysis revealed significant upregulation of 234 genes; 277 genes were downregulated (Tables S5, S6). Gene set enrichment analysis using WebGestalt identified 29 significant KEGG pathways (Table S7). Upregulated pathways included cytokine–cytokine receptor interaction (LIF, IL4R, CCL5, TGFBR2, LIFR, CCR2, and CXCL6), chemokine signaling (VAV3, CCL5, PLCB2, CCR2, and CXCL6), natural killer cell-mediated cytotoxicity (PPP3CA, VAV3, SH2D1A, and RAET1E), WNT signaling (CTBP2, PPP3CA, PLCB2, and WNT7A), phophatidylinositol signaling (PI4KA, DGKD, and PLCB2), and JAK-STAT signaling (LIF, LIFR, IL4R, and SOCS5). Downregulated pathways included MAPK signaling (ELK4, FGFR3, RAC1, CACNG6, MAPK8IP2, and PLA2G6), endocytosis (FGFR3, FOLR4, RNF41, and ADRBK2), high-affinity receptor for the Fc region of immunoglobulin E and vascular endothelial growth factor (VEGF) signaling (RAC1, PLA2G2F, and PLA2G6) (Table S8). IPA interactome analysis of upregulated genes identified CDKN1A, MDM2, and MYC as hubs (Fig. 1). ERK1/2 was a major hub for downregulated genes (Fig. 2).
Figure 1.

Ingenuity interactome analysis of upregulated genes.
Figure 2.

Ingenuity interactome analysis of downregulated genes.
Top canonical pathways upregulated, as indicated by IPA, included PI3K signaling. Top downregulated networks were involved in l-3, 4-dihydroxyphenylalanine degradation, glutamine degradation, and 1,25-dihydroxyvitamin D3 biosynthesis (Table S9).
c.71+9C>A alters THAP1 splicing
Minigene results from three wild-type and three c.71+9C>A transfections showed that the c.71+9C>A variant exerted significant effects on splicing patterns (P < 0.05) (Fig. 3). Overall, mutant cells had reduced incorporation of Exon 2 and increased incorporation of Exon 3.
Figure 3.
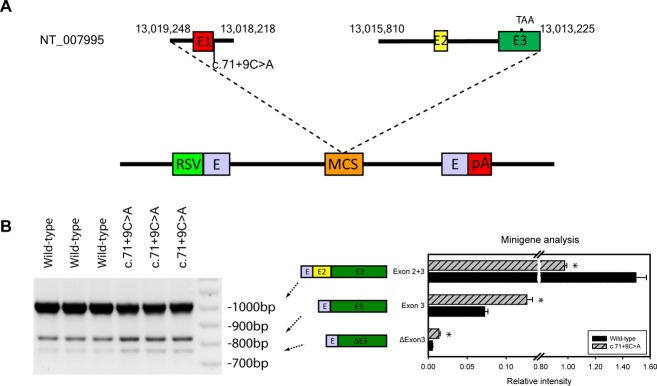
Minigene assay of THAP1 splicing. (A) Segments of THAP1 were cloned into the multiple cloning site (MCS) of the pET01 exon trap vector, which contains a short stretch of a eukaryotic phosphatase gene (E) 3′ to the eukaryotic strong long terminal repeater promoter of the Rous Sarcoma Virus (RSV). A second segment of E is followed by a polyadenylation site (pA). (B) The results of three wild-type and three c.71+9C>A transfections showed that the C>A mutation exerted significant effects on splicing patterns in HEK293 cells (*P < 0.05, for all).
In silico analysis suggested that the c.71+9C>A variant may exert deleterious effects on transcription of THAP1. In aggregate, in silico analysis did not predict overt effects on splicing although the c.71+9C>A variant did alter enhancer locations. More specifically, HSF, NNSPLICE, MES, NetGene2 predicted no changes in splicing due to the mutant allele. On the other hand, ESEfinder, RESCUE-ESE, PESX, and ASSAED indicated that ESEs are altered by c.71+9C>A. RESCUE-ESE identified a new site, whereas PESX showed the wild-type ESE site as broken. ESEfinder indicated a drop in score from 93.79 to 90.90 for serine/arginine rich (SR) protein SC35. Other SR proteins, splicing factor 2/alternative splicing factor (SF2/ASF) and SRp40, were predicted to bind to new sites. ASSAED incurred changes in SC35 (6.7 bits to 6.0 bits), SRp40 (−1.0 bits to 5.3 bits), SF2/ASF (from 0.4 bits to 2.1 bits). All programs reported that exonic silencer sites (ESSs) remained unchanged by the c.71+9C>A variant.
THAP1 c.71+9C>A exerts effects on cell-cycle control
Cell-cycle analysis of control and c.71+9C>A lymphoblastoid cell lines showed overt differences (Fig. 4). Mutant variant cells, when compared with the normal wild-type genotypes, showed an increased percentage of apoptosis and decreased activity in G2 phase (Table S10).
Figure 4.
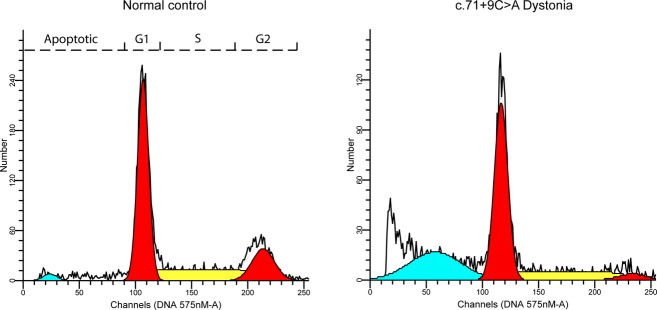
Cell-cycle analysis of lymphoblastoid cell lines. Data from representative normal and c.71+9C>A cells are shown. In comparison to normal cells, the proportion of c.71+9C>A cells in apoptotic phase is increased and G2 phase is decreased.
Discussion
Our study investigated the effects of a sequence variant (c.71+9C>A, rs200209986) located within Intron 1 of THAP1 on gene splicing and gene expression in subjects with primary dystonia. The c.71+9C>A variant is rare in Caucasian populations with a MAF at less than 0.1% (1/3148) in our control cohort. This MAF is similar to data from the 1000 Genomes but lower than the MAF reported by the EVS database. In dystonia patients, the MAF (c.71+9C>A) was significantly higher than the controls of the 1000 Genomes project but not significantly higher than subjects in the EVS database. In other THAP1 screening series, this variant was reported in 3/455 subjects with primary dystonia and 1/185 controls (Groen et al. 2010), 1/567 dystonia subjects and 1/365 controls (Lohmann et al. 2012), 0/109 dystonia subjects and 1/185 controls (Groen et al. 2011). In reports from other groups, it is not entirely clear if population controls had normal neurological examinations or a family history of any movement disorder including dystonia. Moreover, subjects recruited into EVS were not carefully examined to exclude dystonia, the diagnosis of which can be difficult even for an experienced general neurologist, often requiring the expertise of a movement disorders specialist. In addition, the EVS dataset is comprised of 2203 African-Americans and 4300 European-Americans, whereas our cohort was limited to Caucasians. The EVS cohort also included subjects with the extremes of specific traits and diseases including lipid disorders, hypertension, and coronary artery disease. Finally, sequence variants reported in the EVS dataset have not been validated with Sanger sequencing and some may be next-gen read errors (LeDoux 2012c). Therefore, case–control comparisons using the EVS cohort as a “control” group is problematic.
In vitro studies have identified a possible role for THAP1 in regulating the expression of TOR1A by binding to its promoter region (Gavarini et al. 2010; Kaiser et al. 2010). However, we did not detect an effect of variant THAP1 on expression of TOR1A in either leukocytes or lymphoblastoid cell lines. Similarly, in previous work, RNAi knockdown of THAP1 in fibroblasts showed no effect on expression levels of TOR1A (Gavarini et al. 2010). Overexpression of THAP1 in ECs has been used as an indirect means of identifying THAP1 targets – TOR1A was not among the 16 genes that were significantly upregulated or the 80 genes were downregulated (Cayrol et al. 2007). Consistent with these results, THAP1 variants do not act as disease modifiers in DYT1 dystonia (Kamm et al. 2011).
The effects of c.71+9C>A on THAP1 were studied with an integrated collection of in silico and in vitro approaches. Our in silico analysis showed that c.71+9C>A exerted significant effects on the exon splicing enhancer site, but not the splicing site. This finding is compatible with recent work showing that intronic variants can exert important effects on splicing enhancer sites (Fukao et al. 2010; Oeffner et al. 2011; Cohen et al. 2012; Jakubauskiene et al. 2012). Our in silico analysis was supported by a minigene assay which showed significant alterations in incorporation of Exons 2 and 3 in HEK293 cells transfected with the c.71+9C>A variant. In addition, leukocytes harboring the c.71+9C>A variant showed an elevated ratio of Isoform 1 (3-exon variant) to Isoform 2 (2-exon variant). This could represent a false positive, however, as we did not detect an effect of c.71+9C>A on splicing in lymphoblastoid cell lines. Although not entirely concordant, the minigene assay in HEK293 cells, in silico analyses, and QRT-PCR results in lymphoblastoid cell lines and leukocytes suggest that the c.71+9C>A variant may exert deleterious effects on splicing in human brain. In this regard, it is well-established that splicing shows tissue type and developmental specificity (Heinzen et al. 2008; Mondal et al. 2010).
Flow cytometry showed evidence of increased apoptosis and cell-cycle disruption in the variant lymphoblastoid cell lines. Previous studies defined a role for THAP1 as a transcription factor binding to specific DNA sequences and regulating cell proliferation through pRB/E2F cell-cycle target genes (Clouaire et al. 2005; Cayrol et al. 2007). Furthermore, the pRB/E2F G1/S cell-cycle pathway appears to play a central role in the pathogenesis of dystonia, in general (LeDoux et al. 2013), and contributes to the apoptotic dopaminergic cell death associated with Parkinson's disease (Hoglinger et al. 2007).
The limitations of our functional analysis are well-recognized. Leukocytes and lymphoblastoid cell lines are not ideal surrogates for neural tissues. In this regard, the effects of genomic variants on splicing may be developmentally regulated and show tissue-type specificity. Ideally, the relative roles of the major and minor THAP1 isoforms should be studied in a mammalian system via transgenic expression of the respective cDNAs on a THAP1-null background.
Gene expression data from lymphoblastoid cell lines provided evidence of significant transcriptional dysregulation associated with the c.71+9C>A variant. Upregulated pathways included cytokine–cytokine receptor interaction, chemokine signaling, cytotoxicity, Wnt signaling, and Jak-STAT signaling. Two genes that play a central role in the G1/S checkpoint pathway (CDKN1A and MDM2) were hubs in the interactome for upregulated genes. Conversely, downregulated pathways included MAPK signaling, endocytosis, and VEGF signaling. A critical element of MAPK signaling (extracellular signal-regulated kinase [ERK]1/2) was identified as a hub in the interactome for downregulated genes.
The differentially expressed individual genes identified in this study were mainly involved in cell division, communication, migration, and survival. The mitotic cell cycle is accomplished through a sequence of events that include DNA replication (S phase) and mitosis (M phase), separated by phases G1 and G2. This progression is regulated by cyclin-dependent kinases (CDKs). Any damage to the genome during these steps is corrected by repair pathways or arrest of progression from one stage to the next. Details from flow cytometry of c.71+9C>A lymphoblastoid cells indicate that apoptosis occurred prior to G1 with cells showing impairments in moving through the G1/S checkpoint and, as a result, a smaller percentage of cells were in the G2 phase. In a study by Cayrol et al. (2007), the overexpression of THAP1 was associated with inhibition of EC proliferation. Cell-cycle analysis of ECs transfected with THAP1 siRNAs showed a reduction in both the S-phase and G2/M-phase cell populations and an increase in the number of cells in G1 thereby indicating the role of endogenous THAP1 in G1/S cell-cycle progression (Cayrol et al. 2007). Their data from both microarray and qPCR revealed that overexpression of THAP1 in primary HUVECs (human umbilical vein ECs) inhibits cell proliferation through coordinated repression of critical cell-cycle regulators and pRb/E2F target genes.
Upon phosphorylation, pRB is released, allowing the E2F transcription factor to interact with the transcriptional machinery of the cell, transcribing proteins crucial for G1/S progression and the S phase of the cell cycle. Phosphorylation of pRB is initiated by the cyclin D/Cdk4 and Cdk6 complex and continued by the cyclin E/Cdk2 complex. MAPK signaling, which was an enriched downregulated KEGG pathway in variant lymphoblastoid cells, activates Cyclin D/Cdk4 and Cdk6.
The MAPK signaling family plays an important role in complex cellular programs like proliferation, differentiation, development, transformation, and apoptosis. At least three MAPK pathways have been characterized: ERK, Jun kinase (JNK/SAPK) and p38 MAPK (Zhang and Liu 2002). ERK, when activated, will translocate to nucleus and change gene expression promoting differentiation or mitosis. ERK1/2 is involved in activating the microtubule-organizing center which controls the assembly of cytosolic microtubules in interphase cells and the mitotic spindle of dividing cells (Verlhac et al. 1993). ERK can translocate to the nucleus and phosphorylate transcription factors including ELK1, ELK4, and Tal. The ERK pathway can also link G0/G1 mitogenic signals to the immediate early response. The ERK pathway is known to be an intracellular checkpoint for cellular mitogenesis. The ERK cascade is a key in controlling the cell-cycle progression (Buchkovich and Ziff 1994). The Raf-MEK-ERK cascade regulates the posttranslational regulation of the assembly of cyclin D-Cdk4/6 complexes, which phosphorylate Rb protein causing activation of the E2F transcription factors that regulate the transcription of genes needed for G1/S transition (Blanchard et al. 2000).
Mean age of onset for THAP1-associated dystonia is 16.8 years and ranges from 3 to over 60 years (LeDoux et al. 2012). Although DYT1 dystonia is widely viewed as a neurodevelopmental circuit disorder (Xiao et al. 2004; Zhao et al. 2011), neurodegenerative processes may contribute to THAP1 and other mainly adult-onset dystonias (LeDoux et al. 2013). In recent work, the c.71+9C>A variant was associated with a significantly lower linear density of Purkinje cells in postmortem cerebellum from subjects with craniocervical dystonia and normal controls (Prudente et al. 2013). Other findings in subjects with craniocervical dystonia included ubiquitin-positive Marinesco bodies in the substantia nigra and areas of focal gliosis in cerebellar cortex. Transcriptional dysregulation in lymphoblastoid cells lines from subjects harboring the c.71+9C>A are compatible with these pathological findings and current themes in dystonia research which point to cerebellar Purkinje cells and nigrostriatal signaling as sites of functional abnormalities.
In the context of relative neurodevelopmental and neurodegenerative contributions to the pathogenesis of mainly adult-onset primary dystonia, Wnt, Jak-STAT, PI3K, and G1/S checkpoint pathways showed evidence of dysregulation in c.71+9C>A cells. Wnt pathways contribute to cell polarity, transcriptional regulation, and intracellular calcium homeostasis, all of which have been implicated in dystonia (LeDoux 2011; LeDoux et al. 2013). The JAK-STAT system is a cell-surface to nucleus signaling pathway leading to activation of genes that contribute to cell growth and differentiation. PI3K activates AKT which activates mammalian target of rapamycin (mTOR). As such the PI3K pathway is often referred to as the PI3K/AKT/mTOR pathway. The PI3K/AKT/mTOR pathway is central to the biology of glial tumors but also contributes to neural development (Wong 2013).
Given the apparent histological changes associated with c.71+9C>A postmortem human tissue and the results of IPA interactome analyses, the G1/S checkpoint pathway is a potential focal point of cellular pathology (Fig. 1). CDKN1A directly interacts with the dystonia-associated protein CIZ1 (Copeland et al. 2010). Moreover, other dystonia-associated proteins including THAP1, TAF1, ATM, and Gα(olf), contribute to the G1/S checkpoint pathway (LeDoux et al. 2013). MDM2 is an E3 ubiquitin-protein ligase and negative regulator of p53. In theory, upregulation of CDKN1A and MDM2 could dampen the effects of THAP1 loss-of-function variants on G1/S cell-cycle control. Disturbances of the G1/S checkpoint could manifest as ultrastructural neurodevelopmental defects, apoptosis or both in terminally differentiated neurons such as Purkinje cells (Florio et al. 2012; Jurk et al. 2012).
Acknowledgments
The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services; nor does mention by trade names, commercial practices, or organizations imply endorsement by the U.S. Government.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Phenotypes associated with the c.71+9C>A sequence variant.
Table S2. Primers used for HRM, Sanger sequencing, and quantitative RT-PCR.
Table S3. PCR primers used for the minigene assay.
Table S4. TOR1A mRNA expression in leukocyte and lymphoblastoid cell lines.
Table S5. Genes significantly upregulated in mutant cell lymphoblastoid lines.
Table S6. Genes significantly downregulated in mutant cell lymphoblastoid lines.
Table S7. Enriched KEGG pathways.
Table S8. IPA top upregulated biological functions and canonical pathways.
Table S9. IPA top downregulated biological functions and canonical pathways.
Table S10. Cell-cycle analysis with propidium iodide.
References
- Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov. Disord. 2013;28:863–873. doi: 10.1002/mds.25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate – a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995;57:289–300. [Google Scholar]
- Blanchard DA, Mouhamad S, Auffredou MT, Pesty A, Bertoglio J, Leca G, et al. Cdk2 associates with MAP kinase in vivo and its nuclear translocation is dependent on MAP kinase activation in IL-2-dependent Kit 225 T lymphocytes. Oncogene. 2000;19:4184–4189. doi: 10.1038/sj.onc.1203761. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Buchkovich KJ, Ziff EB. Nerve growth-factor regulates the expression and activity of P33(Cdk2) and P34(Cdc2) kinases in PC12 pheochromocytoma cells. Mol. Biol. Cell. 1994;5:1225–1241. doi: 10.1091/mbc.5.11.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagne S, Saurel O, Gervais V, Milon A. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res. 2010;38:3466–3476. doi: 10.1093/nar/gkq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang JH, Zhu ZW, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C, Lacroix C, Mathe C, Ecochard V, Ceribelli M, Loreau E, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood. 2007;109:584–594. doi: 10.1182/blood-2006-03-012013. [DOI] [PubMed] [Google Scholar]
- Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Girard JP. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc. Natl. Acad. Sci. USA. 2005;102:6907–6912. doi: 10.1073/pnas.0406882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen YH, Bargal R, Zeigler M, Markus-Eidlitz T, Zuri V, Zeharia A. Hyperargininemia: a family with a novel mutation in an unexpected site. JIMD Rep. 2012;5:83–88. doi: 10.1007/8904_2011_101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NA, Sercombe HE, Ainscough JF, Coverley D. Ciz1 cooperates with cyclin-A-CDK2 to activate mammalian DNA replication in vitro. J. Cell Sci. 2010;123:1108–1115. doi: 10.1242/jcs.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, Bressman SB, Marsden CD. Classification of dystonia. Adv. Neurol. 1998;78:1–10. [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Florio M, Leto K, Muzio L, Tinterri A, Badaloni A, Croci L, et al. Neurogenin 2 regulates progenitor cell-cycle progression and Purkinje cell dendritogenesis in cerebellar development. Development. 2012;139:2308–2320. doi: 10.1242/dev.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat. Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat. Genet. 2012;45:88–92. doi: 10.1038/ng.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao T, Horikawa R, Naiki Y, Tanaka T, Takayanagi M, Yamaguchi S, et al. A novel mutation (c.951C > T) in an exonic splicing enhancer results in exon 10 skipping in the human mitochondrial acetoacetyl-CoA thiolase gene. Mol. Genet. Metab. 2010;100:339–344. doi: 10.1016/j.ymgme.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard JP, et al. Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann. Neurol. 2010;68:549–553. doi: 10.1002/ana.22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen JL, Ritz K, Contarino MF, van de Warrenburg BP, Aramideh M, Foncke EM, et al. DYT6 dystonia: mutation screening, phenotype, and response to deep brain stimulation. Mov. Disord. 2010;25:2420–2427. doi: 10.1002/mds.23285. [DOI] [PubMed] [Google Scholar]
- Groen JL, Yildirim E, Ritz K, Baas F, van Hilten JJ, van der Meulen FW, et al. THAP1 mutations are infrequent in spasmodic dysphonia. Mov. Disord. 2011;26:1952–1954. doi: 10.1002/mds.23682. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Ge D, Cronin KD, Maia JM, Shianna KV, Gabriel WN, et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglinger GU, Breunig JJ, Depboylu C, Rouaux C, Michel PP, Alvarez-Fischer D, et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2007;104:3585–3590. doi: 10.1073/pnas.0611671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubauskiene E, Janaviciute V, Peciuliene I, Soderkvist P, Kanopka A. G/A polymorphism in intronic sequence affects the processing of MAO-B gene in patients with Parkinson disease. FEBS Lett. 2012;586:3698–3704. doi: 10.1016/j.febslet.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. doi: 10.1111/j.1474-9726.2012.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser FJ, Osmanoric A, Rakovic A, Erogullari A, Uflacker N, Braunholz D, et al. The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6) Ann. Neurol. 2010;68:554–559. doi: 10.1002/ana.22157. [DOI] [PubMed] [Google Scholar]
- Kamm C, Uflacker N, Asmus F, Schrader C, Wolters A, Wittstock M, et al. No evidence for THAP1/DYT6 variants as disease modifiers in DYT1 dystonia. Mov. Disord. 2011;26:2136–2137. doi: 10.1002/mds.23777. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS. Animal models of dystonia: lessons from a mutant rat. Neurobiol. Dis. 2011;42:152–161. doi: 10.1016/j.nbd.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS. Dystonia: phenomenology. Parkinsonism Relat. Disord. 2012a;18(Suppl. 1):S162–S164. doi: 10.1016/S1353-8020(11)70050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS. The genetics of dystonias. Adv. Genet. 2012b;79:35–85. doi: 10.1016/B978-0-12-394395-8.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS. Exome sequencing for gene discovery: time does not stand still. Ann. Neurol. 2012c;72:628–629. doi: 10.1002/ana.23660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Xiao J, Rudzinska M, Bastian RW, Wszolek ZK, Van Gerpen JA, et al. Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat. Disord. 2012;18:414–425. doi: 10.1016/j.parkreldis.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Dauer WT, Warner TT. Emerging common molecular pathways for primary dystonia. Mov. Disord. 2013;28:968–981. doi: 10.1002/mds.25547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann K, Uflacker N, Erogullari A, Lohnau T, Winkler S, Dendorfer A, et al. Identification and functional analysis of novel THAP1 mutations. Eur. J. Hum. Genet. 2012;20:171–175. doi: 10.1038/ejhg.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal AK, Das SK, Baldini G, Chu WS, Sharma NK, Hackney OG, et al. Genotype and tissue-specific effects on alternative splicing of the transcription factor 7-like 2 gene in humans. J. Clin. Endocrinol. Metab. 2010;95:1450–1457. doi: 10.1210/jc.2009-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovich M, LeDoux MS, Xiao J, Rampon GL, Vemula SR, Rodriguez RL, et al. Dystonia, facial dysmorphism, intellectual disability and breast cancer associated with a chromosome 13q34 duplication and overexpression of TFDP1: case report. BMC Med. Genet. 2013;14:70. doi: 10.1186/1471-2350-14-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffner F, Martinez F, Schaffer J, Salhi A, Monfort S, Oltra S, et al. Intronic mutations affecting splicing of MBTPS2 cause ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome. Exp. Dermatol. 2011;20:448–449. doi: 10.1111/j.1600-0625.2010.01238.x. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Prudente CN, Pardo CA, Xiao J, Hanfelt J, Hess EJ, LeDoux MS, et al. Neuropathology of cervical dystonia. Exp. Neurol. 2013;241:95–104. doi: 10.1016/j.expneurol.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum. Mutat. 1998;12:153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Roussigne M, Cayrol C, Clouaire T, Amalric F, Girard JP. THAP1 is a nuclear proapoptotic factor that links prostate-apoptosis-response-4 (Par-4) to PML nuclear bodies. Oncogene. 2003;22:2432–2442. doi: 10.1038/sj.onc.1206271. [DOI] [PubMed] [Google Scholar]
- Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat. Struct. Mol. Biol. 2010;17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengel C, Gavarini S, Sharma N, Ozelius LJ, Bragg DC. Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain. J. Neurochem. 2011;118:1087–1100. doi: 10.1111/j.1471-4159.2011.07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemula SR, Puschmann A, Xiao J, Zhao Y, Rudzinska M, Frei KP, et al. Role of Galpha(olf) in familial and sporadic adult-onset primary dystonia. Hum. Mol. Genet. 2013;22:2510–2519. doi: 10.1093/hmg/ddt102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac MH, de Pennart H, Maro B, Cobb MH, Clarke HJ. MAP kinase becomes stably activated at metaphase and is associated with microtubule-organizing centers during meiotic maturation of mouse oocytes. Dev. Biol. 1993;158:330–340. doi: 10.1006/dbio.1993.1192. [DOI] [PubMed] [Google Scholar]
- Wong M. Mammalian target of rapamycin (mTOR) activation in focal cortical dysplasia and related focal cortical malformations. Exp. Neurol. 2013;244:22–26. doi: 10.1016/j.expneurol.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Gong S, Zhao Y, LeDoux MS. Developmental expression of rat torsinA transcript and protein. Brain Res. Dev. Brain Res. 2004;152:47–60. doi: 10.1016/j.devbrainres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, et al. Novel THAP1 sequence variants in primary dystonia. Neurology. 2010;74:229–238. doi: 10.1212/WNL.0b013e3181ca00ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Uitti RJ, Zhao Y, Vemula SR, Perlmutter JS, Wszolek ZK, et al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann. Neurol. 2012;71:458–469. doi: 10.1002/ana.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- Zhang XHF, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sharma N, LeDoux MS. The DYT1 carrier state increases energy demand in the olivocerebellar network. Neuroscience. 2011;177:183–194. doi: 10.1016/j.neuroscience.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Xiao J, Gong S, Clara JA, LeDoux MS. Neural expression of the transcription factor THAP1 during development in rat. Neuroscience. 2013;231:282–295. doi: 10.1016/j.neuroscience.2012.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Phenotypes associated with the c.71+9C>A sequence variant.
Table S2. Primers used for HRM, Sanger sequencing, and quantitative RT-PCR.
Table S3. PCR primers used for the minigene assay.
Table S4. TOR1A mRNA expression in leukocyte and lymphoblastoid cell lines.
Table S5. Genes significantly upregulated in mutant cell lymphoblastoid lines.
Table S6. Genes significantly downregulated in mutant cell lymphoblastoid lines.
Table S7. Enriched KEGG pathways.
Table S8. IPA top upregulated biological functions and canonical pathways.
Table S9. IPA top downregulated biological functions and canonical pathways.
Table S10. Cell-cycle analysis with propidium iodide.