

Abstract
Although multiple sclerosis (MS) has been associated with the coagulation system, the temporal and spatial regulation of coagulation activity in neuroinflammatory lesions is unknown. Using a novel molecular probe, we characterized the activity pattern of thrombin, the central protease of the coagulation cascade, in experimental autoimmune encephalomyelitis. Thrombin activity preceded onset of neurological signs, increased at disease peak, and correlated with fibrin deposition, microglial activation, demyelination, axonal damage, and clinical severity. Mice with a genetic deficit in prothrombin confirmed the specificity of the thrombin probe. Thrombin activity might be exploited for developing sensitive probes for preclinical detection and monitoring of neuroinflammation and MS progression.
Blood–brain barrier (BBB) disruption occurs before the onset of clinical symptoms and persists throughout the course of disease in multiple sclerosis (MS).1–4 As a consequence, extravasation of the plasma protein fibrinogen into the central nervous system (CNS) occurs in both human subjects and in experimental autoimmune encephalomyelitis (EAE), an MS animal model.2,3,5–7 The serine protease thrombin cleaves soluble fibrinogen to form provisional fibrin matrices that traditionally support hemostasis and tissue repair but also drive local inflammatory changes associated with neurological disease.8,9 Proteomic analysis of chronic active MS lesions identified several dysregulated coagulation factors, highlighting a potential link between the coagulation cascade and MS pathology.10 The potential pathological significance of coagulation factors in neurological disease is underscored by the finding that fibrin diminution, either genetically or using anticoagulants, significantly reduces neurological signs, inflammation, and axonal damage in EAE.5,6,10,11 Furthermore, platelets, the main cellular players in hemostasis activated by thrombin,12 are abundant within MS lesions, and their depletion ameliorates EAE.13 However, the temporal and spatial regulation of coagulation activity in neuroinflammatory lesions remains unknown. We previously developed activatable cell-penetrating peptides (ACPPs)14 for selective delivery of fluorescent and magnetic resonance imaging (MRI) agents to localized regions of high extracellular protease activity including matrix metalloproteases and elastases within tumors,14–17 and the serine protease thrombin in atherosclerotic plaques, brain ischemia, and acute blood clotting.18–20 Using a thrombin-selective ACPP, here we show that increased thrombin activity begins early and increases with progression of neuroinflammatory disease, and is specifically detected within local demyelinating lesions with prominent microglial activation and axonal damage.
Materials and Methods
EAE was induced by MOG35–55 immunization as described,6 in cohorts of Cx3cr1GFP/+ microglia reporter6 or Mx1-Cre:fIIlox/lox 21 mice, which were also injected with poly-I:C, and were all of C57BL/6 background. Our molecular probe, termed PPRSFL-ACPP, consists of the thrombin-specific cleavage sequence, PPRSFL, which links a polycationic cell-penetrating peptide tagged with Cy5 to a neutralizing polyanion. In the presence of active thrombin, the linker is proteolyzed, resulting in absorption and uptake of the Cy5-labeled peptide into cells.15 One hundred microliters of 100μM Cy5-labeled thrombin-cleavable PPRSFL or control ACPPs18,19 was injected intravenously at different stages of EAE. All animal procedures were performed under the guidelines of institutional animal care and use committees at the University of California, San Francisco and the University of Cincinnati and complied with National Institutes of Health guidelines. Imaging and quantification were performed by blinded observers with Li-COR Odyssey imager, Image J (NIH), or a customized script in MATLAB (MathWorks, Natick, MA). Statistics and correlation analyses were performed with Prism (GraphPad Software, La Jolla, CA) using appropriate tests, as indicated in the figure legends. Details of methods are available as Supplemental Materials and Methods.
Results
Specific Detection of Active Thrombin in EAE Using the PPRSFL-ACPP
To examine whether thrombin is locally activated in the CNS in neuroinflammation, we administered the thrombin-cleavable PPRSFL-ACPP18 to EAE-challenged mice with overt impairment in motor function.5,6 Thrombin activity dramatically increased at the peak of disease in the spinal cord, a major CNS site of disease pathology in EAE6 (Fig 1A). Imaging of whole excised spinal cords revealed multiple hot spots of PPRSFL-ACPP uptake in EAE, implying increased localized thrombin activity, which was ∼3.5-fold higher at disease peak compared to noninjected or healthy controls (p < 0.001) (Fig 1B). Signal was not detected in healthy mice or at peak of EAE after administration of control methoxy poly (ethylene glycol) (mPEG) mPEG-ACPP.
Figure 1.

Specific detection of thrombin activity in the experimental autoimmune encephalomyelitis (EAE) spinal cord. (A) Whole spinal cord scans at 700nm from mice at peak EAE or healthy controls, injected with Cy5-labeled thrombin-specific PPRSFL–activatable cell-penetrating peptide (ACPP) or Cy5-labeled noncleavable control methoxy poly (ethylene glycol) (mPEG)-ACPP show specific uptake (dark spots) of PPRSFL-ACPP, indicative of increased localized thrombin activity at the peak of EAE. Uninjected healthy control and EAE mice are also shown as controls (no probe). (B) Quantification of total fluorescent signal in whole spinal cord scans from A, corrected for size. Data are presented as mean ± standard error of the mean (SEM); ***p < 0.001, 2-way analysis of variance (ANOVA); n = 5 to 7 per group for no probe or PPRSFL-ACPP and 2 to 3 for mPEG-ACPP. (C) Genetic reduction or elimination of prothrombin abolishes localized thrombin activity detection in EAE. Whole spinal cord scans from 3 cohorts of mice injected with PPRSFL-ACPP and polyI:C at EAE peak: wild-type (WT; 100% prothrombin), Mx-1Cre(−)fIIlox/lox (20% prothrombin), and Mx-1Cre(+)fIIlox/lox (no prothrombin). Prior to Cre recombinase induction, homozygous Mx1-Cre:fIIlox/lox mice exhibit baseline circulating prothrombin levels that are ∼20% of normal, whereas intraperitoneal injection of poly-I:C over a 6-day period results in a rapid loss of hepatic prothrombin expression and a near-complete loss (<5%) of circulating prothrombin within 5 to 6 days. Poly-I:C was administered at the time of overt clinical disease onset. (D) Quantification of PPRSFL-ACPP signal in whole spinal cord scans from C shows significantly reduced PPRSFL-ACPP retention with lower thrombin levels. Data are presented as mean ± SEM; ***p < 0.0001, 1-way ANOVA; n = 5 to 6 per group.
The PPRSFL-ACPP was also tested for thrombin-dependent activity in the CNS in vivo in EAE mice with genetically reduced or eliminated prothrombin (Mx1-Cre:fIIlox/lox).21 Genetic reduction of prothrombin plasma levels significantly reduced PPRSFL-ACPP detection in the spinal cord of mice at EAE peak (Fig 1C). Quantification showed ∼5.6 and ∼6.8 fold reduction for mice with 20% and 0% of normal prothrombin levels, respectively, relative to wild-type controls, providing genetic confirmation that the PPRSFL-ACPP is thrombin-specific (Fig 1D). The thrombin specificity of PPRSFL-ACPP was also previously shown using thrombin inhibitors in animal models of stroke and atherosclerosis, as well as in vitro by cleavage experiments with a spectrum of other proteases, including Factor Xa and matrix metalloproteinase (MMP)-9.18–20
Thrombin Activation Occurs Early and Correlates with Disease Progression in Neuroinflammation
We further sought to examine the temporal regulation of thrombin activity and its correlation with neuroinflammatory disease progression. We administered thrombin probe in mice at 3 stages of EAE: before neurological signs, at early onset, and at disease peak (Fig 2). In whole spinal cord scans thrombin activity was detected before symptoms appeared and gradually increased at onset and peak of neurologic signs. Compared to healthy controls, thrombin activity was significantly increased both at disease onset during the appearance of the earliest neurologic signs and also at disease peak. There was a linear correlation between thrombin activity and the severity of neurological signs (R2 = 0.86, p < 0.0001, F test), suggesting that thrombin activity is detected early and is a molecular marker of disease progression in neuroinflammation.
Figure 2.
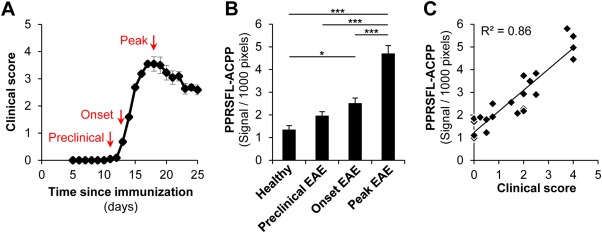
Uptake of PPRSFL–activatable cell-penetrating peptide (ACPP) correlates with disease progression and neurologic symptoms. (A) Representative experimental autoimmune encephalomyelitis (EAE) clinical score curve illustrates the different time points of PPRSFL-ACPP administration. (B) Quantification of total fluorescent signal in whole spinal cord scans from mice injected with PPRSFL-ACPP at different time points of EAE shows that detection of thrombin activity increases with EAE progression. Data are presented as mean ± standard error of the mean; *p < 0.05, ***p < 0.001, 1-way analysis of variance; n = 5 to 7 per group. (C) Scatter plot shows correlation between total thrombin activity in the entire spinal cord and clinical score of EAE (R2 = 0.86, p < 0.0001, F test). [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Thrombin Activity Is a Marker of Fibrin-Laden Inflammatory Demyelinated Lesions with Axonal Damage
We previously showed that microglia form perivascular clusters at sites of fibrin deposition prior to myelin loss or paralysis onset in EAE.6 Thrombin activity was detected in spinal cords as early as the onset of EAE within regions with microglial activation and fibrin deposition (Fig 3A). At the peak of EAE, thrombin activity was specifically detected in areas with extensive fibrin deposition and microglial clustering, but not in healthy controls. PPRSFL-ACPP thrombin cleavage product was consistently taken up within inflammatory lesions by cells including but not limited to microglia (Fig 3B). Moreover, increased thrombin activity was detected in multiple inflamed spinal cord areas at the peak of EAE (Supplemental Fig 1), whereas retention of nonspecific probe was undetectable at sites of inflammation or BBB disruption (Supplemental Fig 2).
Figure 3.

Increased thrombin activity in experimental autoimmune encephalomyelitis (EAE) spatially correlates with blood–brain barrier (BBB) disruption, microglial activation, demyelination, and axonal damage. (A) Confocal microscopy of spinal cord sections from Cx3cr1GFP/+ mice showing the spatial and temporal correlation between thrombin activity (PPRSFL–activatable cell-penetrating peptide [ACPP], red), areas of BBB disruption (fibrinogen deposition, cyan) and local inflammation (green fluorescent protein–labeled microglia, green) at onset or peak of EAE. Healthy Cx3cr1GFP/+ controls with no signs of microglial activation or fibrin deposition show no thrombin activity. (B) Single-plane analysis of image stacks acquired at high magnification with confocal microscopy showed that microglia (green) uptake a significant amount of the thrombin-sensitive PPRSFL-ACPP (red) in neuroinflammatory lesions. (C) Thrombin activity correlates with areas of demyelination in EAE lesions, at the peak of disease. Confocal images of spinal cord sections immunohistochemically stained for myelin basic protein (MBP, cyan) show that thrombin activity (PPRSFL-ACPP, red) is pronounced in white matter areas where myelin is damaged. Dotted line indicates area of parenchymal demyelination, and stars indicate perivascular demyelination sites. (D) Confocal images of spinal cord sections stained with anti–SMI-32 (cyan), a marker for axonal damage not detected in healthy spinal cords, show that thrombin activity (PPRSFL-ACPP, red) is pronounced in white matter areas with extensive signs of axonal damage. Bars represent: A, 50μm; B, 5μm; C, 50μm; D, 20μm. (E–G) Scatter plots show positive correlation between thrombin activity (PPRSFL-ACPP signal) and fibrinogen (E; R2 = 0.55, p < 0.001, F test), microglial activation (F; R2 = 0.35, p < 0.01, F test), and demyelination (G; R2 = 0.52, p < 0.01, F test) in spinal cords at the peak of EAE.
In active EAE and MS lesions, fibrin deposition correlates with axonal damage and demyelination.2,3,5–7 The thrombin inhibitor hirudin reduces clinical severity,10 microglial activation, demyelination, and axonal damage in EAE.6 Thrombin activity was detected within demyelinated lesions at the peak of EAE (Fig 3C) in areas with microglial activation (Supplemental Fig 3) and prominent axonal damage (Fig 3D). Quantification showed strong correlation between thrombin activity and fibrin deposition (R2 = 0.55, P < 0.001, F test), increased microglial activation (R2 = 0.35, P < 0.01, F test), and the size of demyelinated lesions (R2 = 0.52, P < 0.01, F test) at disease peak. Overall, these results suggest that thrombin activation occurs early in EAE and is a specific marker of inflammatory demyelinated lesions.
Discussion
The present study demonstrates that a thrombin-specific ACPP detects inflammatory demyelination in vivo. We show strong association between thrombin activity and BBB disruption, microglial activation, inflammatory demyelination, and axonal damage. Intriguingly, thrombin activation begins early at disease onset, prior to demyelination, and correlates with disease progression. Together with prior studies showing BBB disruption and deposition of fibrin early in EAE and MS,1,3,6 these results suggest that activation of the coagulation cascade occurs at the earliest signs of inflammatory activity in lesions prior to demyelination.
Association of thrombin activity with sites of increased vascular permeability might indicate that the main source of thrombin in EAE is plasma-derived. It is possible that upon BBB disruption fibrinogen together with prothrombin enter the CNS and local activation of thrombin results in fibrin deposition. However, because prothrombin RNA can be expressed by neurons and microglia,22 we cannot rule out the possibility that prothrombin might also be produced in situ in neuroinflammation. Pharmacological studies have demonstrated a pathogenic role of thrombin in EAE, as its inhibition by hirudin protects from neurological signs,10 demyelination, and axonal damage.6 Thrombin might be pathogenic by catalyzing the formation of fibrin, which is a major activator of microglia and an enabler of inflammatory demyelination.5,6 Our data show that thrombin activation can be detected at sites of fibrin deposition both at onset and at the peak of neuroinflammation. Studies using mice genetically deficient for fibrinogen or fibrin inhibitors that do not interfere with thrombin activation show protection from EAE,5,6,8 suggesting that generation of fibrin is a major pathogenic mechanism of thrombin activation. However, it is also possible that thrombin might exert fibrin-independent effects through activation of protease-activated receptors, which are expressed in several CNS cells. Future studies will determine the relative contribution of local thrombin synthesis and the mechanisms of action of thrombin in neuroinflammation.
Our work is a proof-of-principle study using a fluorescently labeled thrombin ACPP to demonstrate that detection of coagulation activity can be exploited for clinical detection of neuroinflammatory lesions. Protease-specific probes appear to be excellent sensors of disease activity, as we previously showed in cancer.14,15,17,23 In contrast to cancer, molecular probes to detect specific constituents of MS plaques have not been developed.24 ACPPs are ideal for clinical application, as they can be generated to carry a fluorescent dye, gadolinium (Gd), or both, allowing for multimodal detection of protease activity in vivo.17 For example, we showed that high levels of Gd were retained in tumors when Gd-labeled MMP-ACPPs were used, resulting in enhanced T1 contrast that lasted for several days.17 In MS, Gd-enhanced MRI remains the main clinical tool for lesion detection. More sensitive, targeted, and instructive strategies employing advanced molecular probes could significantly enhance the early detection of MS lesions, possibly even in preclinical stages. In that regard, the ACPP technology might offer an advantage over the detection of passively diffusing Gd, as it would allow local signal enhancement by cellular probe uptake, specifically in areas with increased proteolytic activity. Therefore, MRI using a Gd-fused thrombin-ACPP may improve sensitivity, as it would detect a concentrated amount of probe possibly even around a small or slowly leaking vessel, where a new lesion may initiate. Future studies will determine the pharmacokinetic properties of a Gd-labeled thrombin-specific ACPP and exploit its translation in MS. If successful, this technology could be further used for early patient diagnosis and therapeutic intervention, and also for rapid evaluation of patient response to treatments. Because BBB disruption and fibrin deposition are prominent not only in MS, but also in other CNS diseases such as Alzheimer disease, spinal cord injury, and brain trauma,4,8,9,25 sensitive molecular sensors of coagulation activity could also prove to be invaluable clinical tools for detecting early pathological manifestations in CNS injuries and neurodegenerative diseases.
Acknowledgments
This research was supported by National Multiple Sclerosis Society postdoctoral fellowships to D.D. and J.K.R, a Young Investigator Award from the Race to Erase MS to D.D., DoD W81XWH-05-1-0183 Era of Hope Innovator Award to R.Y.T., NIH National Heart, Lung, and Blood Institute grant HL096126 to J.L.D., and NIH National Institute of Neurological Diseases and Stroke grants NS027177 to R.Y.T., and NS052189 and NS066361 to K.A. M.A.P. is a fellow of the Pediatric Scientist Development Program and was supported in part by research grant No. 4-FY10-461 from the March of Dimes.
We thank K. Thorn for assistance with image analysis, M. Finucane for advice on statistical analyses, and G. Howard for editorial assistance.
Authorship
D.D. and K.M.B. generated and analyzed data, and designed research; M.A.W., E.S.M., B.F., and E.S.O. performed research, analyzed data, and assisted with experimental design; J.K.R., M.A.P., and C.B. performed research and analyzed data; D.S.S. analyzed data; J.L.D. designed experiments and analyzed data; R.Y.T. and K.A. conceived the study, designed experiments, and analyzed data; D.D., K.M.B, and K.A. wrote the manuscript with assistance from the other authors.
D.D. and K.M.B. contributed equally to this work.
Potential Conflicts of Interest
M.A.W.: equity, Avelas Biosciences; patent covering ACPPs (licensee, Avelas Biosciences; royalties paid to University of California, San Diego). E.S.O.: patent on peptides whose uptake is controllable. R.Y.T.: stock, Avelas Biosciences; patents covering ACPPs (licensee, Avelas Biosciences; royalties paid to University of California, San Diego). K.A.: grants, personal fees, Lundbeck.
Supporting Information
References
- Kermode AG, Thompson AJ, Tofts P, et al. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic and clinical implications. Brain. 1990;113:1477–1489. doi: 10.1093/brain/113.5.1477. [DOI] [PubMed] [Google Scholar]
- Kwon EE, Prineas JW. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. J Neuropathol Exp Neurol. 1994;53:625–636. doi: 10.1097/00005072-199411000-00010. [DOI] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, et al. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Adams RA, Bauer J, Flick MJ, et al. The fibrin-derived gamma377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay D, Esiri M. Blood-brain barrier damage in acute multiple sclerosis plaques. An immunocytological study. Brain. 1991;114:557–572. doi: 10.1093/brain/114.1.557. [DOI] [PubMed] [Google Scholar]
- Adams RA, Schachtrup C, Davalos D, et al. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 2007;14:2925–2936. doi: 10.2174/092986707782360015. [DOI] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Inoue A, Koh CS, Shimada K, et al. Suppression of cell-transferred experimental autoimmune encephalomyelitis in defibrinated Lewis rats. J Neuroimmunol. 1996;71:131–137. doi: 10.1016/s0165-5728(96)00150-6. [DOI] [PubMed] [Google Scholar]
- Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- Langer HF, Choi EY, Zhou H, et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res. 2012;110:1202–1210. doi: 10.1161/CIRCRESAHA.111.256370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Olson ES, Nguyen QT, et al. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ES, Aguilera TA, Jiang T, et al. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol (Camb) 2009;1:382–393. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney M, Crisp JL, Olson ES, et al. Parallel in vivo and in vitro selection using phage display identifies protease-dependent tumor-targeting peptides. J Biol Chem. 2010;285:22532–22541. doi: 10.1074/jbc.M110.138297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ES, Jiang T, Aguilera TA, et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Friedman B, Whitney MA, et al. Thrombin activity associated with neuronal damage during acute focal ischemia. J Neurosci. 2012;32:7622–7631. doi: 10.1523/JNEUROSCI.0369-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ES, Whitney MA, Friedman B, et al. In vivo fluorescence imaging of atherosclerotic plaques with activatable cell-penetrating peptides targeting thrombin activity. Integr Biol (Camb) 2012;4:595–605. doi: 10.1039/c2ib00161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney M, Savariar EN, Friedman B, et al. Ratiometric activatable cell-penetrating peptides provide rapid in vivo readout of thrombin activation. Angew Chem Int Ed Engl. 2013;52:325–330. doi: 10.1002/anie.201205721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins ES, Kombrinck KW, Talmage KE, et al. Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood. 2009;113:696–704. doi: 10.1182/blood-2008-07-169003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Miklossy J, Klegeris A, et al. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol. 2006;65:19–25. doi: 10.1097/01.jnen.0000196133.74087.cb. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Olson ES, Aguilera TA, et al. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc Natl Acad Sci U S A. 2010;107:4317–4322. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol. 2013;74:317–327. doi: 10.1002/ana.24009. [DOI] [PubMed] [Google Scholar]
- Merlini M, Davalos D, Akassoglou K. In vivo imaging of the neurovascular unit in CNS disease. Intravital. 2012;1:87–94. doi: 10.4161/intv.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.