

Abstract
Deoxycytidine kinase (dCK) and human antigen R (HuR) have been associated with response to gemcitabine in small studies. The present study investigates the prognostic and predictive value of dCK and HuR expression levels for sensitivity to gemcitabine and 5-fluorouracil (5-FU) in a large phase III adjuvant trial with chemoradiation backbone in pancreatic ductal adenocarcinoma (PDA). The dCK and HuR expression levels were determined by immunohistochemistry on a tissue microarray of 165 resected PDAs from the Radiation Therapy Oncology Group (RTOG) 9704 trial. Association with overall survival (OS) and disease-free survival (DFS) status were analyzed using the log-rank test and the Cox proportional hazards model. Experiments with cultured PDA cells were performed to explore mechanisms linking dCK and HuR expression to drug sensitivity. dCK expression levels were associated with improved OS for all patients analyzed from RTOG 9704 (HR: 0.66, 95% CI [0.47–0.93], P = 0.015). In a subset analysis based on treatment arm, the effect was restricted to patients receiving 5-FU (HR: 0.53, 95% CI [0.33–0.85], P = 0.0078). Studies in cultured cells confirmed that dCK expression rendered cells more sensitive to 5-FU. HuR cytoplasmic expression was neither prognostic nor predictive of treatment response. Previous studies along with drug sensitivity and biochemical studies demonstrate that radiation interferes with HuR’s regulatory effects on dCK, and could account for the negative findings herein based on the clinical study design (i.e., inclusion of radiation). Finally, we demonstrate that 5-FU can increase HuR function by enhancing HuR translocation from the nucleus to the cytoplasm, similar to the effect of gemcitabine in PDA cells. For the first time, in the pre-treatment tumor samples, dCK and HuR cytoplasmic expression were strongly correlated (chi-square P = 0.015). This dual-institutional follow up study, in a multi-institutional PDA randomized clinical trial, observed that dCK expression levels were prognostic and had predictive value for sensitivity to 5-FU.
Keywords: pancreatic cancer, gemcitabine, 5-fluorouracil, HuR, dCK
Introduction
Pancreatic ductal adenocarcinoma (PDA) is one of the most aggressive cancers. In patients with advanced PDA, survival is typically less than 6 mo.1 Multi-agent regimens are superior to gemcitabine monotherapy in the metastatic setting, but the benefit is limited to just a few months at the expense of greater toxicities.2,3 Patients with localized disease who undergo resection have a slightly better prognosis, but the vast majority of patients recur and succumb to their disease.4 The median survival after resection is approximately 18 mo, and the 5-y overall survival rate is just 15%. Based on prospective and randomized data from Europe5,6 and the United States,7,8 the standard of care for adjuvant treatment remains either gemcitabine (2′,2′-difluorodeoxycytidine) or 5-fluorouracil (5-FU) monotherapy, with or without chemoradiation. In practice, gemcitabine tends to be favored over 5-FU by many oncologists since the drug has a slightly better toxicity profile.6 Nevertheless, gemcitabine and 5-FU are both considered acceptable options in the adjuvant setting, especially in regards to new standard of care therapies that have recently emerged (e.g., gemcitabine/nab-paclitaxel and FOLFIRINOX).3,9
Gemcitabine is a prodrug antimetabolite that functions as a cell cycle-dependent deoxycytidine analog, once activated within cancer cells.10,11 The drug enters the cell via the human equilibrative nucleoside transporter (hENT1).12 Under normal conditions (i.e., no treatment), deoxycytidine kinase (dCK) functions primarily as the rate limiting enzyme in the deoxyribonucleoside salvage pathway, working to recycle DNA degradation products. However, dCK also phosphorylates gemcitabine into a monophosphate derivative, which is further phosphorylated by additional nucleotide kinases to the active di- and triphosphate forms. Cytotoxicity occurs when the active metabolites incorporate into DNA, resulting in masked-chain termination.13,14 Deficiency of dCK, detectable by immunohistochemistry (IHC), has been associated with gemcitabine resistance (i.e., impaired intratumoral activation) and worse overall survival in patients.15 Recently, the RNA binding protein human antigen R (HuR) was found to bind and stabilize the dCK transcript, resulting in higher cytoplasmic dCK levels and improved gemcitabine efficacy.16 Concordant with these findings, increased cytoplasmic HuR expression was associated with improved survival in patients who received gemcitabine after pancreatectomy for pancreatic cancer.16,17 Thus, posttranscriptional regulation of dCK by activated HuR in PDAs has been hypothesized as an important factor determining gemcitabine efficacy.16,17
Like gemcitabine, 5-FU is an anti-metabolite that interferes with DNA synthesis and repair. The drug is a uracil analog with a fluorine atom in place of a hydrogen in the pyrimidine ring.18 Intracellularly, 5-FU is metabolized to FdUMP through a series of steps, which covalently binds and inhibits thymidylate synthase (TS), thereby impairing the formation of thymidine monophosphate. However, it is still unclear whether TS is the main target of 5-FU.19,20 Additionally, the 5-FU metabolite FdUTP is misincorporated into DNA. These events lead to DNA damage and cell death, particularly in proliferating cells.21,22
While gemcitabine and 5-FU have comparable activity in the treatment of PDA, they likely affect different patient groups differently, due to the different mechanism of a prodrug activation and drug catabolism. In support of this concept, different molecular markers have been proposed to predict tumor response for the different drugs. TS, dihydropyrimidine dehydrogenase (DPD),23 and mismatch repair enzymes are examples of putative predictive markers with 5-FU treatment20; hENT1,12,24,25 ribonucleotide reductase M1 (RRM1),26 excision repair cross complementation group 1 (ERCC1),26 dCK,15 and HuR16,17 are examples for gemcitabine. Clearly, the discovery and application of reliable predictive biomarkers to exploit non-overlapping pharmacogenomic profiles between the two principal backbone agents to treat PDA represents perhaps the “lowest-hanging fruit” in the effort to improve outcomes of this devastating disease, yet such biomarkers remain elusive.
While dozens of studies have explored predictive markers for gemcitabine and 5-FU efficacy, these primarily were based on retrospective cohorts with a small sample size. As a result, no predictive biomarkers have been validated and integrated into routine clinical care, particularly in patients with pancreatic cancer. Prospective and randomized adjuvant trials are invaluable for investigations of candidate predictive markers for several reasons: (1) the large sample size; (2) excellent patient follow-up; (3) tissue availability; (4) uniform treatment plans; and (5) diminished confounding or patient selection bias, as a result of the randomized study design.
There has been only a single phase III adjuvant study performed in the United States in the past 25 y for pancreatic cancer,7 which limits the opportunities to optimally explore predictive markers for this disease. The Radiation Therapy Oncology Group (RTOG) 9704 trial was a phase III trial in patients with resected adenocarcinoma of the pancreas treated with 5-FU-based chemoradiation, sandwiched between 5-FU or gemcitabine chemotherapy. To our knowledge, three prior biomarker studies have been published using the tissue samples from this trial. Low histone modification was associated with poor survival with 5-FU,27 hENT1 expression was associated with a possible benefit from gemcitabine-based therapy,24 and a variant single nucleotide polymorphism in cytidine deaminase was associated with less gemcitabine toxicity.28
We were granted access to tissue microarrays of specimens from the RTOG 9704 trial to test the value of dCK and HuR expression levels in both study arms.15-17 While both proteins were previously associated with improved outcomes in patients receiving gemcitabine,15-17 we observed improved survival with increased dCK expression in patients receiving 5-FU. Independent of the survival outcomes, we tested for an association between dCK and HuR expression as evidence of a regulatory interaction between the proteins in human tumors. We provide experimental data in cultured PDA cells supporting the role of dCK as a predictive marker of 5-FU efficacy in PDA. Additionally, we provide intriguing mechanistic data, based on previous work,29 that could explain the discrepancy between prior findings linking dCK and HuR to gemcitabine response, with the results reported herein.
Results
Study population
RTOG 9704 began accruing patients on July 20, 1998 and closed on July 26, 2002 with a total of 538 patients. Of the 279 patients with analyzable tissue, 114 patients were excluded from the present study; 93 did not have sufficient tissue and 21 failed to meet eligibility requirements for the clinical trial. A total of 165 patients therefore were analyzed for dCK expression, including 76 in the gemcitabine arm and 89 in the 5-FU arm. A summary of included and excluded patients are provided in Figure S1. The median dCK score across the entire cohort was 2.67 (range, 0–3). Based on this finding, patients were categorized into low- (<2.67) and high-dCK groups (≥2.67).
Patient demographics, pathologic features, and treatment allocation were similar between patients with high and low dCK expression (Table S1), except that a higher proportion of tumors with high dCK were located in the pancreatic head, relative to tumors with low dCK. Low- and high-dCK expression groups were similar in the subgroup analysis stratified by treatment arms with two exceptions. Within the gemcitabine arm, a higher proportion of patients with high dCK expression had cancers in the pancreatic head (93% vs. 67%, P = 0.003). Additionally, a higher proportion of the patients in this group had a poor performance status (53% vs. 24%, P = 0.01), relative to patients harboring tumors with low dCK expression (Table S2).
Prognostic value of dCK expression (analysis of the entire cohort)
The disease-free survival was similar between patients with high and low dCK protein expression levels in the total cohort (Fig. 1A). However, patients with high dCK had improved OS (Fig. 1B), with a hazard ratio of 0.66 on univariate analysis (95% CI: [0.47–0.93], P = 0.015). The hazard ratio was slightly better (0.61, 95% CI [0.44, 0.86], P = 0.0044) in the multivariate analysis after adjusting for regional lymph node metastases and Karnofsky Performance Status (KPS) (Table 1).

Figure 1. Survival of patients enrolled in RTOG 9704 stratified by dCK expression level. Disease-free and overall survival for all evaluable patients (A and B), patients on the gemcitabine arm (C and D) and patients on the 5-FU arm (E and F).
Table 1. Hazard ratios for OS and DFS based upon dCK expression (high vs. low [reference level]) patients treated with gemcitabine or 5-FU.
| Total patients analyzed (n = 165) | Gemcitabine arm (n = 76) | 5-FU arm (n = 89) | |||||
|---|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | ||
| Univariable analysis | DFS | 0.78 (0.57, 1.08) | P = 0.13 | 0.96 (0.60, 1.54) | P = 0.87 | 0.66 (0.42, 1.03) | P = 0.065 |
| OS | 0.66 (0.47, 0.93) | P = 0.016 | 0.83 (0.51, 1.36) | P = 0.45 | 0.56 (0.35, 0.88) | P = 0.012 | |
| Multivariable analysis | DFS | 0.75 (0.54–1.04) | P = 0.08 | 0.96 (0.60, 1.54) | P = 0.87 | 0.60 (0.38, 0.95) | P = 0.027 |
| OS | 0.61 (0.44, 0.86) | P = 0.0044 | 0.79 (0.48, 1.30) | P = 0.35 | 0.53 (0.33, 0.85) | P = 0.0078 | |
Predictive value of dCK expression for gemcitabine and 5-FU efficacy (stratified by treatment)
In a subgroup univariate analysis of the gemcitabine arm, OS and DFS were similar between patients with low and high dCK expression (Fig. 1C and D). The similarity was maintained in the multivariate analysis (Table 1). The DFS was also similar between low and high dCK groups in the 5-FU arm, although a trend toward superior DFS in the high dCK group was noted (HR: 0.66, 95% CI [0.42–1.03], P = 0.065). However, patients with high dCK expression had significantly improved OS in the 5-FU arm (HR: 0.56, 95% CI [0.35–0.88], P = 0.012) (Fig. 1E and F). The median OS for patients with high and low dCK levels in the 5-FU arm were 21 mo (95% CI: 15.1, 30.7) and 12.9 mo (95% CI: 11.1, 17.6), respectively (log-rank P value = 0.012) (Table S3). In the multivariate analysis of the 5-FU arm, after adjusting for KPS and lymph node metastases, an increase in DFS and OS were observed in patients with high dCK expression; hazard ratios were 0.60 (95% CI [0.38, 0.95], P = 0.027) and 0.53 (95% CI [0.33, 0.85], P = 0.0078), respectively (Table 1).
dCK expression impacts 5-FU efficacy in pancreatic cancer cells
MiaPaCa2 pancreatic cancer cells were transfected with siRNA oligos designed to inhibit dCK mRNA and with control siRNA oligos. The efficacy of silencing was confirmed by RT-qPCR analysis of dCK mRNA levels (Fig. 2A). Cells were left untreated or treated with 5 or 10 µM 5-FU. At the higher dose, survival was greater in the cells that showed lower dCK expression levels (Fig. 2B). These results are consistent with the above human studies, and demonstrate that dCK expression sensitizes cells to 5-FU treatment (i.e., improved cell killing in cell culture or improved patient survival in the RTOG-9704 study).
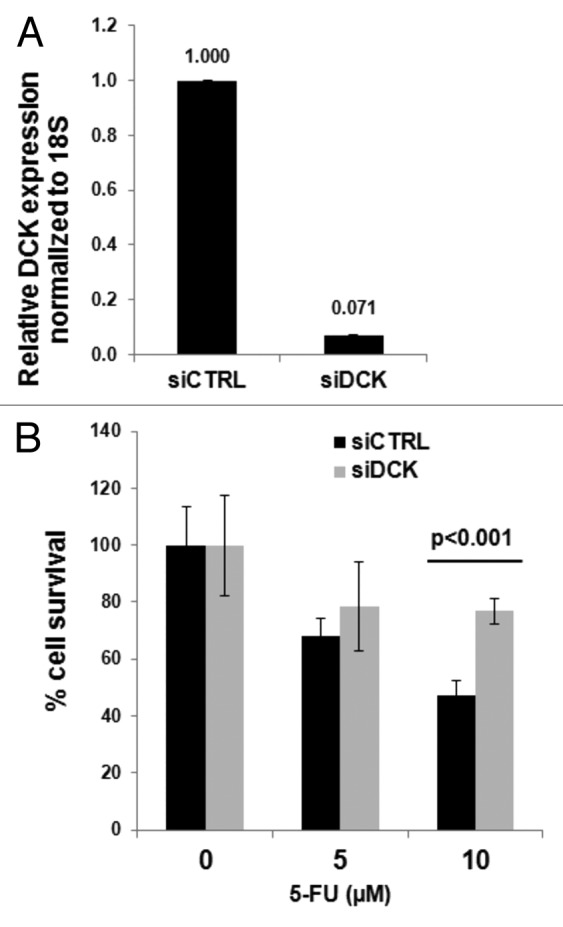
Figure 2. Inhibition of dCK in pancreatic cancer cells impairs 5-FU efficacy. MiaPaCa2 cells. (A) RT-qPCR validation of dCK silencing using siRNA oligos. (B) Improved cell survival after 5-FU treatment in cells with decreased dCK expression.
Prognostic and predictive value of HuR
In the context of previous retrospective studies demonstrating an association between cytoplasmic HuR expression and prolonged survival in patients receiving gemcitabine, we assessed the predictive value of HuR abundance in the RTOG 9704 cohort. A total of 116 patients had available tissue for immunohistochemical analyses. There were no differences between patients with low- or high HuR-expressing tumors in the total cohort with respect to DFS (HR 1.6, 95% CI [0.74–1.53], P = 0.73) and OS (HR 1.18, 95% CI [0.81–1.72], P = 0.37) (Fig. S2A and B). No differences were identified in subgroup analyses for either treatment arm (Fig. S2C–F).
HuR is co-expressed with dCK in human PDA
We tested for an association between HuR cytoplasmic expression and dCK expression in patients with tissue available for immunolabeling with both biomarkers. Out of 116 informative samples, 62 (53%) had high dCK expression. However, among samples with low cytoplasmic HuR expression, the proportion of samples with high dCK expression dropped to 46%; the proportion in the high HuR cytoplasmic expression group was 69%. These data provide the first evidence in human tumors that HuR and dCK are co-expressed (P = 0.015), likely due to post-transcriptional regulation of the dCK transcript by HuR (Table 2).
Table 2. Association between dCK status and HuR levels.
| HuR status | |||
|---|---|---|---|
| dCK | Low | High | Total |
| Low (<2.67) | 42 (55%) | 12 (32%) | 54 (47%) |
| High (≥2.67) | 35 (46%) | 27 (69%) | 62 (53%) |
| Total | 77 (66%) | 39 (34%) | 116 (100%) |
chi-square P = 0.015
Radiation interferes with HuR biology
To our surprise, HuR and dCK were not predictive of outcome in patients receiving gemcitabine. These data conflict with previous clinical studies and in vitro experiments which suggest that HuR stabilization of the dCK transcript enhances gemcitabine metabolism and efficacy.15-17 We hypothesized that the use of radiation in the RTOG 9704 interferes with HuR biology to the extent that HuR or its target transcripts (e.g., dCK) are no longer reliable as predictive markers. This hypothesis is founded on convincing molecular data demonstrating that ionizing radiation induces the dissociation of all mRNAs bound to HuR, thereby preventing post-transcriptional regulation of these target genes (such as dCK) by HuR.29
To explore this possibility, we first examined the importance of sequencing order of gemcitabine and radiation on pancreatic cancer cell survival. If radiation causes HuR protein and dCK mRNA to dissociate, then cancer cells should be less sensitive to gemcitabine administered as the second treatment. MiaPaCa2 cells were plated in soft agar and treated with a combination of 1 µM gemcitabine and 2 Gy of radiation (varied sequence order) and negative controls. The impact of treatment on anchorage-independent growth at 3 wk was compared. As anticipated, combination chemoradiation was only effective when gemcitabine preceded radiation treatment. (Fig. 3A). These data were validated with a PicoGreen survival assay analyzed at 7 d. There was 70% more cell death in MiaPaCa2 cells treated with gemcitabine followed by radiation, as compared with the opposite sequence (Fig. 3B). Importantly, there were no survival differences between the two sequenced chemoradiation protocols when HuR was removed from the system using siRNA oligos (Fig. 3D). Efficacy of HuR siRNA treatment was confirmed by RT-qPCR on the transfected MiaPaCa2 cells (Fig. 3C). These data suggest that HuR regulatory functions contributed to the observed differences in the parental cells (Fig. 3A and B). Immunoblots in Figure 3E provide mechanistic evidence of HuR’s regulation of dCK. MiaPaCa2 cells were treated with gemcitabine or radiation alone, as well as in combination. For the latter scenario, both treatment sequences were tested. Cytoplasmic HuR was increased relative to the untreated sample for each treatment, except when radiation preceded gemcitabine (Fig. 3E, lane 5). Similarly, no dCK expression was detected in the cells with this treatment sequence (Fig. 3E, lane 5). The greatest amount of dCK was apparent with chemoradiation, sequenced with gemcitabine first, and followed by radiation (Fig. 3E, lane 3). Accordingly, we reproduced previous findings by our group29 in pancreatic cancer cells (MiaPaCa2 cells). In brief, we validated and demonstrated in ribonucleoprotein–immunoprecipitation assays that dCK mRNA bound to HuR (80-fold compared to IgG control), and that when gemcitabine treated PDA cells were irradiated, there was a ~80% reduction in dCK mRNA bound to HuR protein (data not shown). Taken together, these findings support a model where gemcitabine given prior to radiation increases cell killing, while the opposite sequence negates gemcitabine efficacy. Mechanistically, gemcitabine efficacy is contingent on HuR’s ability to stabilize dCK.

Figure 3. Gemcitabine prior to radiation (XRT) is more efficacious than gemcitabine (GEM) alone or XRT prior to GEM due to differential modulation of HuR and dCK proteins. (A) Anchorage-independent colony formation assays were performed for quantification of colonies formation in MiaPaCa2 cells treated with gemcitabine (1 uM) alone or with gemcitabine preceded or followed by radiation (2 Gy). (B) PicoGreen cell survival assay on cells treated with radiation or with gemcitabine preceded or followed by radiation. (C) PicoGreen cell survival assay after HuR silencing with siRNA oligos. (D) RT-qPCR validation of HuR siRNA treatment in MiaPaCa2 cells. (E) Western blot for dCK, HuR, and α-tubulin (loading control) on lysates obtained from cells treated with gemcitabine alone, radiation alone, gemcitabine prior to radiation and radiation prior to gemcitabine.
5-FU activated HuR
We have previously shown that gemcitabine induces HuR translocation from the nucleus to the cytoplasm,16 a step necessary for HuR function in the stabilization and translational control of target mRNAs. Recently, we also published that other chemotherapy agents used to treat PDA similarly induce the cytoplasmic export of HuR.30 Therefore, we evaluated whether PDA cells treated with 5-FU induced HuR translocation from the nucleus to the cytoplasm. Indeed, when MiaPaCa2 cells are treated with ~IC50 doses of 5-FU, HuR translocates to the cytoplasm (Fig. S3). Ongoing studies are aimed at identifying the HuR target mRNAs that are regulated (likely stabilized) when PDA cells are exposed to 5-FU.
Discussion
RTOG 9704 compared chemoradiation and gemcitabine (pre- and post-chemoradiation) to chemoradiation and 5-FU (pre- and post- chemoradiation).7 An updated analysis reported that the two treatments were equivalent with respect to OS. These results were consistent with the results of a European trial that compared gemcitabine and 5-FU, without adjuvant chemoradiation.6 As one of just a handful of adjuvant randomized trials with available pancreatic cancer tissue for biomarker studies, and the only randomized American adjuvant trial performed in the past 25 y, we identified the RTOG 9704 biorepository as particularly appealing to test HuR and dCK as possible predictive markers for chemotherapeutic efficacy. Metastatic pancreatic cancer, in theory, offers a far better model to study predictive markers since treatment response can be directly measured. In contrast, patients enrolled in adjuvant trials typically are disease-free at randomization, and treatment efficacy must be extrapolated from survival data (DFS or OS). Unfortunately, sufficient tissue for biomarker studies is a major limitation of palliative chemotherapy trials, and often precludes such correlative studies in patients with advanced disease. Adjuvant trials are less challenged with regards to tissue procurement, but face other limitations. For instance, the predictive value of a given biomarker is much more difficult to discern than prognostic information, but may be inferred when an association with survival varies between treatment groups.
Retrospective analyses of specimens from multi-institution and randomized adjuvant chemotherapy trials are particularly important for biomarker studies for reasons already stated, yet only a small number have ever been published. Table 3 provides a list of predictive biomarker studies with “positive” results, using specimens from any tumor type obtained from randomized adjuvant trials that tested either gemcitabine or 5-FU. Only 2 studies based on randomized trials to-date have identified predictive markers (hENT1 and cytidine deaminase) for gemcitabine.12,24,28 Including the present one, a dozen studies have identified predictive markers (microsatellite instability, thymidylate synthase, HER2, serine threonine receptor-associated protein, MYC, p53, decoy receptor 3, KRAS, and histone modification) for 5-FU.27,31-40 We provide the first demonstration in human samples that dCK levels are associated with survival in patients receiving 5-FU.
Table 3. Prior studies of predictive markers of 5-fluoruracil and gemcitabine efficacy from randomized adjuvant trials.
| First author | Year | Tumor | Treatment | Marker | Result summary |
|---|---|---|---|---|---|
| Ohrling39 | 2013 | Colorectal | 5-FU | MSI and TS | 5-FU beneficial for ↓MSI and ↑TS |
| Farrell29,* | 2012 | Pancreas | Gem | CDA | Gem less toxic with CDA variant SNP |
| Sinicrope41 | 2011 | Colorectal | 5-FU | MSI | 5-FU beneficial for ↑MSI |
| Manayakorn28,* | 2010 | Pancreas | 5-FU | H3K4me2, H3K9me2 | 5-FU ineffective with low histone modification |
| Farrell25,* | 2009 | Pancreas | Gem | hENT1 | Gem beneficial for ↑hENT1 |
| Colozza36 | 2005 | Breast | C, M, 5-FU | HER2 | 5-FU ineffective for ↑HER2 |
| Buess35 | 2004 | Colorectal | 5-FU, MMC | STRAP | Chemotherapy ineffective with STRAP amplification |
| Ribic40 | 2003 | Colorectal | 5-FU | MSI | 5-FU beneficial for ↓MSI |
| Edler37 | 2002 | Colorectal | 5-FU | TS | 5-FU beneficial for ↑TS |
| Mild38 | 2002 | Colorectal | 5-FU, MMC | DCR3 | 5-FU ineffective for DCR3 amplification |
| Ahnen32 | 1998 | Colorectal | 5-FU | KRAS and P53 | 5-FU beneficial for WT KRAS and ↓P53 |
| Augenlicht34 | 1997 | Colorectal | 5-FU | MYC | 5-FU beneficial for low MYC amplification |
*From RTOG 9704. Additional abbs: 5-FU, 5-fluorouracil; C, cyclophosphamide; CDA, cytidine deaminase; DCR3, decoy receptor 3; Gem, gemcitabine; hENT1, human equilibrative nucleoside transporter; H3K4me2, H3K9me2, H3K18ac, modified histones; M, methotrexate; MMC, mitomycin C; MSI, microsatellite instability; STRAP, serine/threonine kinase receptor associated protein; TS, thymidylate synthase). Pubmed search used to populate table: (A) randomized[title/abstract] AND fluorouracil[title/abstract] AND adjuvant[title/abstract] AND marker[title/abstract]; (B) randomized[title/abstract] AND 5-fluorouracil[title/abstract] AND adjuvant[title/abstract] AND expression[title/abstract AND survival[title/abstract]; (C) RTOG 9704.
This association was previously hinted at in a prior study using a gemcitabine resistant and dCK deficient ovarian cancer cell line, which was also noted to be resistant to 5-FU and a host of other drugs.41 We validated the importance of dCK on 5-FU efficacy in a pancreatic cancer cell line model. The experimental data support the finding that elevated dCK levels may identify tumors sensitive to 5-FU, but perhaps even more important, highlight dCK as a potential synthetic lethal target to complement 5-FU. Future studies are needed to validate dCK as a bona fide predictive marker of 5-FU efficacy, and to elucidate a mechanistic link between dCK expression and 5-FU response. We suspect that the observed interaction in vivo and in vitro reflects an increased susceptibility to 5-FU in proliferating cells (i.e., dCK is an important enzyme for DNA synthesis).
In prior work, we demonstrated that dCK (normally an important enzyme in DNA synthesis) is posttranscriptionally regulated by a cancer-promoting protein, HuR.16,42 HuR is overexpressed in a wide variety of tumor types,43,44 and increased cytoplasmic HuR expression (a key step regulating HuR function) is associated with aggressive pathologic features and worse survival.45 In the present study, we observed a correlation between HuR and dCK expression in patient samples, providing evidence that HuR regulates dCK in vivo. Similar analyses have been performed for other validated HuR binding targets in PDA, including DR5,46 COX2, and VEGF.16,17
Despite this observed association, and previous studies suggesting that dCK and HuR are predictive of gemcitabine efficacy in patients with PDA,15-17 these biomarkers were not informative in patients from the RTOG 9704 trial who received gemcitabine. Based on new insights into the impact of radiation on HuR biology29 and data presented herein, it is entirely plausible that the use of radiation in the RTOG 9704 trial decouples HuR and dCK expression in residual cancer cells (after treatment, as the samples we found a correlation between dCK and HuR were in naively treated). This in turn would diminish gemcitabine efficacy. We acknowledge that the in vitro studies testing the importance of chemotherapy and radiation sequencing performed in both the previous29 and the present studies are not a completely accurate treatment model for the RTOG 9704 regimen. It would be very difficult to simulate the complex treatment schedule (pre-treatment chemotherapy for 1 mo, chemoradiation for 5 wk, post-treatment chemotherapy for 3 mo) in cell culture, and vice versa. Nevertheless, when one considers the gemcitabine treatment arm in the RTOG 9704 study in the context of our new understanding of HuR biology, the results are in fact quite predictable. Patients did not begin their main course of gemcitabine until 5 mo after surgery (10% of patients will have already recurred clinically by then6); moreover, this treatment course begins on the heels of radiation which likely disrupts the HuR–dCK axis. Finally, we should also note that another limitation of the study was the fact that only ~30% of the patients enrolled in the study had tissue available.
Based on the present results, we are currently performing a similar analysis with tissues from the ESPAC3 trial,47 comparing adjuvant gemcitabine and 5-FU, without chemoradiation. Based on our new observations that 5-FU activates HuR and dCK is a predictive marker of 5-FU, we are eager to validate these data in an independent clinical setting (e.g., the retrospective setting of the ESPAC3 trial). This study demonstrates the importance of mechanistically understanding how and why a specific biomarker (e.g., HuR) may or may not be valuable in certain clinical settings (e.g., gemcitabine in combination with radiation). Based on our collective past work from two institutions,15,16 we followed up on our biomarker studies with additional experimentation and validation work, and we believe, these studies (with further rigorous validation) could lay the groundwork for setting up a future, prospective, and personalized trial for PDA patients.
Materials and Methods
Patient specimens
Paraffin-embedded tissues were collected from patients enrolled into the RTOG 9704 clinical trial. All patients consented to specimen submission and tissue was collected and stored at the RTOG Biospecimen Resource. Full details of the RTOG 9704 clinical protocol and the IRB approval are available through the online Supplementary Materials. For simplicity, the treatment arms consisting of gemcitabine plus chemoradiation (using 5-FU as a radiosensitizer) or 5-FU plus chemoradiation (using 5-FU as a radiosensitizer) will be designated gemcitabine and 5-FU arms, respectively.
Immunohistochemistry for dCK and HuR
Unstained 4-μm sections were deparaffinized and placed in Retrieval Solution (Dako) for 20 min at 100 °C. The slides were quenched with 3% H2O2 for 5 min and incubated at room temperature (RT) with polyclonal rabbit anti-dCK antibody (1:200 dilution) for 1 h (generously provided by Dr Iannis Talianidis). The antibody was generated against amino acids 246 to 260 of dCK corresponding to the COOH terminal portion of the protein.48 Labeling with a secondary antibody alone (anti-rabbit) was used as a negative control. IHC labeling was detected with the Dako Envision system and scored on an intensity scale of 0 to 3; 0 for no staining, 1 for weak positive labeling of neoplastic epithelium (weaker in intensity to lymphocytes present in the same section), 2 for positive labeling of epithelial cells (equal in intensity to lymphocytes), and 3 for intense positive labeling (greater in intensity to lymphocytes). A final score was calculated for each patient as an average obtained from replicate samples (a total of 2 or 3 replicates per patient). Patients with only a single score were excluded from the analysis. IHC scoring was performed by two authors who remained blinded to corresponding patient clinical information. There was concordance between pathologists. HuR labeling (antibody purchased from Santa Cruz Biotechnology, clone sc-5261) and scoring were based on the percentage of cells with positive cytoplasmic labeling, as previously described.16,17 Expression scores for HuR were categorized into 2 groups: low (scores of 0 and 1) and high (scores of >2).16
Cell culture, transfection, treatment, and RT-qPCR
The human PDA cell line MiaPaCa2 (American Type Culture Collection) was maintained in Dulbecco’s modified essential medium (DMEM), supplemented with 10% fetal bovine serum, l-glutamine, and penicillin/streptomycin (Invitrogen). Gemcitabine was obtained from Eli-Lilly as a powder and reconstituted in phosphate-buffered saline for delivery in our cell culture model. Radiation was delivered at a dose of 2 or 10 Gy. Silencing siRNA oligos targeting HuR and a scrambled control were obtained from Santa Cruz Biotechnology (sequence available upon request), siRNA were used at a final concentration of 15 nM. Transfection of cells was performed using Oligofectamine and Optimem per manufacturers’ instructions (Invitrogen). RT-qPCR for HuR and dCK mRNA expression were performed to confirm the lack of RNA amplification after siRNA treatment.
Soft agar assay
Single-cell suspensions of MiaPaCa2 cells in 3 mL of standard DMEM supplemented with 0.36% agar were created after treatment with gemcitabine, radiation or both. Suspensions were plated in 60-mm dishes overlying a 5 mL layer of 0.75% agar/DMEM and grown for three weeks. Colonies were counted and morphology was noted under the light microscope.
Cell survival assay
MiaPaCa2 cells were plated in 96-well plates at a density of 1000 cells per well. Plates were then treated with gemcitabine in varying doses with or without radiation. At seven days, the cells were lysed using deionized water and stained with Quant-iT PicoGreen dsDNA reagent (Life Technologies). Double-stranded DNA was then counted using a Tecan GENios Fluorescence, Absorbance and Luminescence Reader (Tecan Group).
Ribonucleoprotein–immunoprecipitation analysis (data not shown)
Ribonucleoprotein–immunoprecipitation (RIP) assays were performed as previously described.16,49
Protein analysis
Cytoplasmic, nuclear, and whole cell extracts
MiaPaCa2 PDA cells were plated at 60–70% confluence. Cells were treated with 5 µM 5-fluorouracil (USB) for 6 h. Cytoplasmic and nuclear extracts were prepared using the NE-PER kit (ThermoScientific) as previously described.30 Whole cell extracts were lysed using RIPA buffer (Invitrogen) supplemented with 10 μL/mL PMSF, 10 μL/mL sodium orthovanadate, and 10 μL/mL protease and phosphate inhibitors (Pierce) as previously described.30
SDS-PAGE/western blotting
Samples were mixed 1:1 with 2× Laemmli buffer, boiled for 5 min at 95 °C. Approximately 10–20 µg of protein was separated using a 10% Bis-Trispolyacrylimide gel (Gibco/Invitrogen) and transferred to PVDF membrane (Invitrogen).
The membrane was preincubated in 5% milk with 1× TBST for 1 h at RT before incubating with primary antibodies overnight at 4 °C. Antibodies included HuR (Santa Cruz, sc-5621, 1:1000), α-tubulin (Santa Cruz, 1:1000), GAPDH (Cell Signaling), and dCK (gift from Dr Talianidis, 1:5000). Blots were then washed 5 times with 1× TBST 5 min each followed by secondary antibody incubation at 1:10 000 dilution with HRP conjugated antibody for 1 h at RT. Protein complexes were visualized with ECL (Thermo Scientific).
Immunofluorescence
MiaPaCa2 cells were plated onto chamber slides at 1000 cells per chamber cell confluency and treated with 5 µM 5-flurouracil (USB) for 12 h. Cells were then washed 3 times with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton-X, blocked with 5% goat serum for 1 h at RT and incubated with the HuR (3A2, 1:200) antibody overnight at 4 °C. Cell nuclei were mounted on chamber slides with ProLong® Gold antifade reagent with DAPI (Invitrogen) for analysis with a Zeiss LSM-510 Confocal Laser Microscope.
Statistical analyses
Statistical comparisons to assess potential associations between baseline characteristics and dCK or HuR expression were performed using the Chi-square test. Median overall survival (OS) and disease-free survival (DFS) were estimated by the Kaplan–Meier method50 and survival between groups analyzed using the log-rank test. A Cox proportional hazards model51 was used for multivariable survival analyses; patients harboring tumors with low dCK expression served as the reference group. The following variables were included in the multivariable model: dCK expression, treatment arm (5-FU vs. gemcitabine), primary tumor location (head vs. everything else), nodal involvement (no vs. yes), tumor diameter (<3 vs. ≥3 cm), Karnofsky Performance Status (KPS) (90–100 vs. 60–80), and resection margin status (negative vs. positive). Backward selection process was used to build the multivariable models, and variables with a P value < 0.05 were kept in the models. However, dCK expression was retained in models regardless of the P value. Definitions have been defined elsewhere.7 In brief, an overall survival event was defined as death due to any cause and was measured from the date of randomization to the date of death, or the date of the last follow-up for censored patients. Failure for disease-free survival was defined as local, regional, or distant relapse, appearance of a second primary, or death due to any cause.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
F.M. was supported by NIGMS T32GMO66691, 2012 Pancreatic Cancer Action Network-AACR Fellowship, in memory of Samuel Stroum, Grant Number 12-40-25-MCAL and Conquer Cancer Foundation Young Investigator Award 2012. J.R.B. was in part supported by resources provided by AACR-Pancreatic Cancer Action Network and A Research Scholar Grant from the American Cancer Society. M.G. and K.A. were supported by the NIA-IRP, NIH. This project was supported by the RTOG Translational Research Program (seed grant #78), RTOG grant U10 CA21661, CCOP grant U10 CA37422 from the National Cancer Institute (NCI), and the Pennsylvania Department of Health Formula Grant 4100057652. No part of this manuscript has been submitted to any other journal.
Glossary
Abbreviations:
- 5-FU
5-fluorouracil
- Gem
gemcitabine
- HR
hazard ratio
- CI
confidence interval
- OS
overall survival
- DFS
disease-free survival
- y
years
- XRT
radiation therapy
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff DD, Ervin T, Arena FP, Chiorean G, Infante JR, Moore MJ, Seay TE, Tjulandin S, Ma WW, Saleh MN, et al. Randomized phase III study of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone in patients with metastatic adenocarcinoma of the pancreas (MPACT). GI ASCO, 2012. [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. Groupe Tumeurs Digestives of Unicancer. PRODIGE Intergroup FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J, Hodgin MB, Sauter PK, Hruban RH, Riall TS, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J Gastrointest Surg. 2006;10:1199–210, discussion 1210-1. doi: 10.1016/j.gassur.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 6.Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, Goldstein D, Padbury R, Moore MJ, Gallinger S, Mariette C, et al. European Study Group for Pancreatic Cancer Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304:1073–81. doi: 10.1001/jama.2010.1275. [DOI] [PubMed] [Google Scholar]
- 7.Regine WF, Winter KA, Abrams RA, Safran H, Hoffman JP, Konski A, Benson AB, Macdonald JS, Kudrimoti MR, Fromm ML, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA. 2008;299:1019–26. doi: 10.1001/jama.299.9.1019. [DOI] [PubMed] [Google Scholar]
- 8.Kalser MH, Ellenberg SS. Pancreatic cancer. Adjuvant combined radiation and chemotherapy following curative resection. Arch Surg. 1985;120:899–903. doi: 10.1001/archsurg.1985.01390320023003. [DOI] [PubMed] [Google Scholar]
- 9.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galmarini CM, Clarke ML, Jordheim L, Santos CL, Cros E, Mackey JR, Dumontet C. Resistance to gemcitabine in a human follicular lymphoma cell line is due to partial deletion of the deoxycytidine kinase gene. BMC Pharmacol. 2004;4:8. doi: 10.1186/1471-2210-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergman AM, Pinedo HM, Jongsma AP, Brouwer M, Ruiz van Haperen VW, Veerman G, Leyva A, Eriksson S, Peters GJ. Decreased resistance to gemcitabine (2′,2′-difluorodeoxycitidine) of cytosine arabinoside-resistant myeloblastic murine and rat leukemia cell lines: role of altered activity and substrate specificity of deoxycytidine kinase. Biochem Pharmacol. 1999;57:397–406. doi: 10.1016/S0006-2952(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 12.Poplin E, Wasan H, Rolfe L, Raponi M, Ikdahl T, Bondarenko I, Davidenko I, Bondar V, Garin A, Boeck S, et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J Clin Oncol. 2013;31:4453–61. doi: 10.1200/JCO.2013.51.0826. [DOI] [PubMed] [Google Scholar]
- 13.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–57. [PubMed] [Google Scholar]
- 14.Bouffard DY, Laliberté J, Momparler RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45:1857–61. doi: 10.1016/0006-2952(93)90444-2. [DOI] [PubMed] [Google Scholar]
- 15.Sebastiani V, Ricci F, Rubio-Viqueira B, Kulesza P, Yeo CJ, Hidalgo M, Klein A, Laheru D, Iacobuzio-Donahue CA. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: relationship to molecular mechanisms of gemcitabine resistance and survival. Clin Cancer Res. 2006;12:2492–7. doi: 10.1158/1078-0432.CCR-05-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A, Keen JC, Yeo CJ, Gorospe M, Brody JR. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009;69:4567–72. doi: 10.1158/0008-5472.CAN-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards NG, Rittenhouse DW, Freydin B, Cozzitorto JA, Grenda D, Rui H, Gonye G, Kennedy EP, Yeo CJ, Brody JR, et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann Surg. 2010;252:499–505, discussion 505-6. doi: 10.1097/SLA.0b013e3181f1fd44. [DOI] [PubMed] [Google Scholar]
- 18.Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, Scheiner J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179:663–6. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 19.Brody JR, Hucl T, Costantino CL, Eshleman JR, Gallmeier E, Zhu H, van der Heijden MS, Winter JM, Wikiewicz AK, Yeo CJ, et al. Limits to thymidylate synthase and TP53 genes as predictive determinants for fluoropyrimidine sensitivity and further evidence for RNA-based toxicity as a major influence. Cancer Res. 2009;69:984–91. doi: 10.1158/0008-5472.CAN-08-3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Showalter SL, Showalter TN, Witkiewicz A, Havens R, Kennedy EP, Hucl T, Kern SE, Yeo CJ, Brody JR. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol Ther. 2008;7:986–94. doi: 10.4161/cbt.7.7.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 22.Pinedo HM, Peters GF. Fluorouracil: biochemistry and pharmacology. J Clin Oncol. 1988;6:1653–64. doi: 10.1200/JCO.1988.6.10.1653. [DOI] [PubMed] [Google Scholar]
- 23.Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL, Smid K, Lunec J, Calvert AH, Marsh S, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta. 2002;1587:194–205. doi: 10.1016/S0925-4439(02)00082-0. [DOI] [PubMed] [Google Scholar]
- 24.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, Abrams R, Benson AB, Macdonald J, Cass CE, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187–95. doi: 10.1053/j.gastro.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 25.Pérez-Torras S, García-Manteiga J, Mercadé E, Casado FJ, Carbó N, Pastor-Anglada M, Mazo A. Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochem Pharmacol. 2008;76:322–9. doi: 10.1016/j.bcp.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Akita H, Zheng Z, Takeda Y, Kim C, Kittaka N, Kobayashi S, Marubashi S, Takemasa I, Nagano H, Dono K, et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. 2009;28:2903–9. doi: 10.1038/onc.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, Hines OJ, Reber H, Seligson DB, Horvath S, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. 2010;28:1358–65. doi: 10.1200/JCO.2009.24.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrell JJ, Bae K, Wong J, Guha C, Dicker AP, Elsaleh H. Cytidine deaminase single-nucleotide polymorphism is predictive of toxicity from gemcitabine in patients with pancreatic cancer: RTOG 9704. Pharmacogenomics J. 2012;12:395–403. doi: 10.1038/tpj.2011.22. [DOI] [PubMed] [Google Scholar]
- 29.Masuda K, Abdelmohsen K, Kim MM, Srikantan S, Lee EK, Tominaga K, Selimyan R, Martindale JL, Yang X, Lehrmann E, et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011;30:1040–53. doi: 10.1038/emboj.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, Romeo C, Jimbo M, Norris ZA, Yeo CJ, et al. HuR Posttranscriptionally Regulates WEE1: Implications for the DNA Damage Response in Pancreatic Cancer Cells. Cancer Res. 2014;74:1128–40. doi: 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahnen DJ, Feigl P, Quan G, Fenoglio-Preiser C, Lovato LC, Bunn PA, Jr., Stemmerman G, Wells JD, Macdonald JS, Meyskens FL., Jr. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res. 1998;58:1149–58. [PubMed] [Google Scholar]
- 32.Askmalm MS, Carstensen J, Nordenskjöld B, Olsson B, Rutqvist LE, Skoog L, Stål O. Mutation and accumulation of p53 related to results of adjuvant therapy of postmenopausal breast cancer patients. Acta Oncol. 2004;43:235–44. doi: 10.1080/02841860410029474. [DOI] [PubMed] [Google Scholar]
- 33.Augenlicht LH, Wadler S, Corner G, Richards C, Ryan L, Multani AS, Pathak S, Benson A, Haller D, Heerdt BG. Low-level c-myc amplification in human colonic carcinoma cell lines and tumors: a frequent, p53-independent mutation associated with improved outcome in a randomized multi-institutional trial. Cancer Res. 1997;57:1769–75. [PubMed] [Google Scholar]
- 34.Buess M, Terracciano L, Reuter J, Ballabeni P, Boulay JL, Laffer U, Metzger U, Herrmann R, Rochlitz C. STRAP is a strong predictive marker of adjuvant chemotherapy benefit in colorectal cancer. Neoplasia. 2004;6:813–20. doi: 10.1593/neo.04307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colozza M, Sidoni A, Mosconi AM, Cavaliere A, Bisagni G, Gori S, De Angelis V, Frassoldati A, Cherubini R, Bian AR, et al. Italian Oncology Group for Clincal Research HER2 overexpression as a predictive marker in a randomized trial comparing adjuvant cyclophosphamide/methotrexate/5-fluorouracil with epirubicin in patients with stage I/II breast cancer: long-term results. Clin Breast Cancer. 2005;6:253–9. doi: 10.3816/CBC.2005.n.028. [DOI] [PubMed] [Google Scholar]
- 36.Edler D, Glimelius B, Hallström M, Jakobsen A, Johnston PG, Magnusson I, Ragnhammar P, Blomgren H. Thymidylate synthase expression in colorectal cancer: a prognostic and predictive marker of benefit from adjuvant fluorouracil-based chemotherapy. J Clin Oncol. 2002;20:1721–8. doi: 10.1200/JCO.2002.07.039. [DOI] [PubMed] [Google Scholar]
- 37.Mild G, Bachmann F, Boulay JL, Glatz K, Laffer U, Lowy A, Metzger U, Reuter J, Terracciano L, Herrmann R, et al. DCR3 locus is a predictive marker for 5-fluorouracil-based adjuvant chemotherapy in colorectal cancer. Int J Cancer. 2002;102:254–7. doi: 10.1002/ijc.10711. [DOI] [PubMed] [Google Scholar]
- 38.Öhrling K, Karlberg M, Edler D, Hallström M, Ragnhammar P. A combined analysis of mismatch repair status and thymidylate synthase expression in stage II and III colon cancer. Clin Colorectal Cancer. 2013;12:128–35. doi: 10.1016/j.clcc.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinicrope FA, Foster NR, Thibodeau SN, Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C, Moore MJ, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011;103:863–75. doi: 10.1093/jnci/djr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergman AM, Giaccone G, van Moorsel CJ, Mauritz R, Noordhuis P, Pinedo HM, Peters GJ. Cross-resistance in the 2′,2′-difluorodeoxycytidine (gemcitabine)-resistant human ovarian cancer cell line AG6000 to standard and investigational drugs. Eur J Cancer. 2000;36:1974–83. doi: 10.1016/S0959-8049(00)00246-X. [DOI] [PubMed] [Google Scholar]
- 42.Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, Cozzitorto JA, Rigoutsos I, Yeo CJ, Brody JR, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013;10:1312–23. doi: 10.4161/rna.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.López de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2:11–3. doi: 10.4161/rna.2.1.1552. [DOI] [PubMed] [Google Scholar]
- 44.Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci U S A. 2008;105:20297–302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winter JM, Dixon DA, Brody JR. HuR. Springer, 2012. [Google Scholar]
- 46.Pineda DM, Rittenhouse DW, Valley CC, Cozzitorto JA, Burkhart RA, Leiby B, Winter JM, Weber MC, Londin ER, Rigoutsos I, et al. HuR’s post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer Biol Ther. 2012;13:946–55. doi: 10.4161/cbt.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neoptolemos JP, Moore MJ, Cox TF, Valle JW, Palmer DH, McDonald AC, Carter R, Tebbutt NC, Dervenis C, Smith D, et al. European Study Group for Pancreatic Cancer Effect of adjuvant chemotherapy with fluorouracil plus folinic acid or gemcitabine vs observation on survival in patients with resected periampullary adenocarcinoma: the ESPAC-3 periampullary cancer randomized trial. JAMA. 2012;308:147–56. doi: 10.1001/jama.2012.7352. [DOI] [PubMed] [Google Scholar]
- 48.Hatzis P, Al-Madhoon AS, Jüllig M, Petrakis TG, Eriksson S, Talianidis I. The intracellular localization of deoxycytidine kinase. J Biol Chem. 1998;273:30239–43. doi: 10.1074/jbc.273.46.30239. [DOI] [PubMed] [Google Scholar]
- 49.Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Jr., Martindale JL, Yang X, Gorospe M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol. 2008;28:4562–75. doi: 10.1128/MCB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaplan EL, Meier P. Nonparameteric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. doi: 10.1080/01621459.1958.10501452. [DOI] [Google Scholar]
- 51.Cox DR. Regression models and life-tables. J Roy Stat Soc B. 1972;34:187–220. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.