

Abstract
Adult human pancreatic β-cells are primarily quiescent (G0) yet the mechanisms controlling their quiescence are poorly understood. Here, we demonstrate, by immunofluorescence and confocal microscopy, abundant levels of the critical negative cell cycle regulators, p27(Kip1) and p18(Ink4c), 2 key members of cyclin-dependent kinase (CDK) inhibitor family, and glycogen synthase kinase-3 (GSK-3), a serine-threonine protein kinase, in islet β-cells of adult human pancreatic tissue. Our data show that p27(Kip1) localizes primarily in β-cell nuclei, whereas, p18(Ink4c) is mostly present in β-cell cytosol. Additionally, p-p27(S10), a phosphorylated form of p27(Kip1), which was shown to interact with and to sequester cyclinD-CDK4/6 in the cytoplasm, is present in substantial amounts in β-cell cytosol. Our immunofluorescence analysis displays similar distribution pattern of p27(Kip1), p-p27(S10), p18(Ink4c) and GSK-3 in islet β-cells of adult mouse pancreatic tissue. We demonstrate marked interaction of p27(Kip1) with cyclin D3, an abundant D-type cyclin in adult human islets, and vice versa as well as with its cognate kinase partners, CDK4 and CDK6. Likewise, we show marked interaction of p18(Ink4c) with CDK4. The data collectively suggest that inhibition of CDK function by p27(Kip1) and p18(Ink4c) contributes to human β-cell quiescence. Consistent with this, we have found by BrdU incorporation assay that combined treatments of small molecule GSK-3 inhibitor and mitogen/s lead to elevated proliferation of human β-cells, which is caused partly due to p27(Kip1) downregulation. The results altogether suggest that ex vivo expansion of human β-cells is achievable via increased proliferation for β-cell replacement therapy in diabetes.
Keywords: CDK inhibitors, GSK-3, adult human islets, adult pancreatic β-cell, p18(Ink4c), p27(Kip1), proliferation, quiescence
Introduction
Normal adult human pancreatic β-cells are mostly quiescent (G0) and generally do not enter into the G1/S-phase of the cell cycle. However, the mechanisms regulating such quiescence are not well understood. In order to expand human β-cells for future therapeutic intervention of diabetes, such knowledge is critical since it will contribute to their elevated entry into the cell cycle by overcoming quiescence leading to increased proliferation. Diabetes is primarily a disease of reduced β-cell mass. In type 1 diabetes, β-cell deficit is almost complete, whereas, in type 2, such deficit is partial. Therefore, in principle, replenishment of lost/reduced β-cell mass, either by β-cell replacement/transplantation or via β-cell expansion in vivo, should ameliorate hyperglycemia and correct diabetes. As proof of principle, clinical studies show that restoration of β-cell mass via islet transplantation can treat diabetes-related symptoms for a certain period of time and allow temporal insulin independence in type 1 diabetic patients.1 Additionally, studies using rodent models of β-cell ablation (type 1 diabetes) and insulin resistance (type 2 diabetes) display that restoration of lost/reduced β-cell mass by increased proliferation of pre-existing β-cells results in normoglycemia and correction of diabetes.2-5
It was first reported in 2009 that many members of the mammalian cell cycle machinery, particularly of the G1/S proteome, are expressed in adult human islets isolated from cadaveric donors.6 The critical role of positive cell cycle regulators, such as, cyclin D1, D3, and CDK6, individually or in combination, in promoting ex vivo proliferation of adult human β-cells was also revealed.6-8 However, from the point of clinical application, these studies6-8 may have significant limitations due to the use of virus-mediated overexpression systems for cyclin and/or CDK to elevate human β-cell replication. Nonetheless, such studies documented the clear potential of adult human β-cells to proliferate ex vivo. We showed, using isolated adult human islets, marked levels of several critical cell cycle regulators, including p27(Kip1) (a cyclin-dependent kinase [CDK] inhibitor), glycogen synthase kinase-3 (GSK-3) (a serine-threnine protein kinase), cyclin D3 (a member of D-type cyclins) and retinoblastoma (Rb) protein (a tumor suppressor).9 Substantial levels of both p27(Kip1) and cyclin D3 in β-cells of adult human pancreatic tissue were also reported.10 An old study indicated that in human pregnancy, maternal β-cell mass expands via β-cell hyperplasia for maintaining normal glucose homeostasis.11 A recent report has revealed about 50% increase in β-cell mass due to elevated β-cell number in obese individuals for compensating high insulin demand.12 Studies of β-cell turnover in donors displayed the presence of replicating β-cells primarily in the first 3 decades of life.13 Additionally, studies using cadaveric donors revealed strong evidence that residual β-cells in type 1 diabetic patients are in a steady-state of proliferation and apoptosis, even after 50 y of diabetes duration.14 Moreover, pancreatic β-cell ablation in very old mice (1–2 y old) demonstrated that β-cells retain the capacity for compensatory proliferation to maintain normal glucose homeostasis.15 These studies collectively indicate that irrespective of the quiescent nature of adult human and rodent β-cells, they possess an intrinsic ability to respond to growth stimuli to overcome their quiescence state and enter into the cell cycle for self-duplication/proliferation via modulation of the levels and/or function of cell cycle regulators (negative and positive).
p27(Kip1), an important member of the Cip/Kip protein family, is a primary negative regulator of cell proliferation and controls the G1/S-phase transition by binding to and regulating the activity of cyclin-dependent kinases (CDKs) that include cyclinD (D1, D2, and D3)-CDK4/6 and cyclin E-CDK2 complexes.16,17 In normal quiescent (G0) cells, protein stability of p27(Kip1) is maximal but, upon mitogenic stimulation, p27(Kip1) levels decrease rapidly to ensure entry of cells into the G1 phase as well as progression through the S phase of the cell cycle.18 The 2 major pathways that result in p27(Kip1) degradation upon mitogenic stimulation include KPC (Kip1 ubiquitylation-promoting complex)-mediated cytosolic degradation of p27(Kip1) after its nuclear export and Skp2 (S-phase kinase-associated protein 2)-mediated nuclear degradation following its Thr187 (T187) phosphorylation.18,19 Decrease in p27(Kip1) levels allows cyclin-CDK holoenzyme complexes to phosphorylate retinoblastoma (Rb) protein and its family members, p107 and p130, resulting in the release of E2F family of transcription factors and subsequently transitioning of cells from the G0 to G1/S phase of the cell cycle.18,20 Additionally, Ser10 (S10) phosphorylation of p27(Kip1) acts as an important determinant of the stability of p27(Kip1) in the G0 phase and plays critical role in regulating cytosolic assembly of p27(Kip1) with cyclin D-CDK4/6 and in turn, prevent nuclear import of cyclinD-CDK4/6 from cytosol.18 The 2 post-translational events, phosphorylation and protein-protein interaction, are thus important in controlling p27(Kip1) sub-cellular localization and stability, which, in turn, modulate non-proliferative (quiescent) vs. proliferative status of cells.
P18(Ink4c) belongs to Ink4 (inhibitors of CDK4/6) protein family that includes 3 more members, p15(Ink4b), p16(Ink4a), and p19(Ink4d).20,21 The members of this group are closely related ankyrin repeat containing proteins, which selectively form binary complexes with CDK4 or CDK6 and thus prevent CDK4/6 from binding with D-type cyclins (D1, D2, and D3) leading to disruption of D cyclin-CDK4/6 complex activation followed by cancellation of phosphorylation/hyperphosphorylation of Rb.20,21
Glycogen synthase kinase-3 (GSK-3), an important serine-threonine kinase, is constitutively active in resting cells and has 2 mammalian isoforms, GSK-3α (51 kDa protein), and GSK-3β (47 kDa protein).22,23 GSK-3 is located in the nucleus besides cytoplasm24 and its activity is primarily controlled through inhibition by various mitogenic and hormonal signaling pathways. It is a key regulator of various important signaling pathways, including receptor tyrosine kinases, Wnt and G-protein-coupled receptors, and is implicated in a variety of important cellular processes, including cell-cycle regulation and proliferation.22,23 Several independent studies demonstrated that GSK-3 acts as an important negative regulator of proliferation because of its ability to facilitate degradation of critical positive cell cycle regulators, such as, D1, E, and D3 cyclins,25-27 and stabilization of the levels of a key negative cell cycle regulator, p27(Kip1).28
In this report, we have demonstrated abundant levels of the critical negative cell-cycle regulators, p27(Kip1), its phosphorylated form, p-p27(S10), p18Ink4c, and GSK-3, in β-cells of both adult human and mouse pancreatic islets, which contribute to maintenance of β-cell quiescence. We have also shown that treatments with pharmacological inhibitor of GSK-3 and mitogen/s cause increased proliferation of human β-cells, suggesting a road map to promote their ex vivo expansion for β-cell replacement therapy in diabetes.
Results
Prevalence of β-cells than α-cells in adult human islets
We have examined the abundance of insulin (Ins) positive β-cells vs glucagon (Glu) positive α-cells in normal adult human pancreatic tissue sections. Analyses by immunofluorescence and confocal microscopy reveal the prevalence of β-cells than α-cells in majority of adult human islets (~80%) that we have examined (Fig. 1A, green [Ins] vs. white [Glu]). We have found relatively higher α-cell numbers in only about 20% islets (Fig. 1B, white [Glu] vs. A, white [Glu]), suggesting an overall majority of β-cells than α-cells in adult human islets as also demonstrated by independent studies.29 Likewise, when we examined the abundance of β- and α-cells in adult mouse islets, we found a typical β-cell core (Fig. 1C, green [Ins]) and a periphery containing α-cells (Fig. 1C, red [Glu]), indicating much higher abundance of β- than α-cells in adult mouse islets. Additionally, corresponding to other studies,29 we also have found that α-cells are generally interspersed with β-cells in human islets (Fig. 1A and B, merge data), which is contrary to mouse islet architecture where β- and α-cells remain in the core and periphery, respectively (Fig. 1C, merge data). Furthermore, few somatostatin positive δ-cells are present in majority of adult human islets (Fig. 1A and B, red [somatostatin]) that usually remain intermingled with β- and α-cells (Fig. 1A and B, merge data).

Figure 1. Prevalence of β-cells in adult human pancreatic islets. Analysis of the abundance of β-, α-, and δ- cells in normal adult pancreatic tissue sections, obtained from donors (age: 30–50 y) (A and B) and mice (age: 9 mo) (C), by immunoflluorescence and confocal microscopy. The tissue sections were incubated with the specific primary antibodies against insulin, somatostatin and glucagon followed by incubation with the appropriate secondary antibodies conjugated with either FITC (for insulin) or Cy3 (for somatostatin) or Dylight/Texas Red (TR) (for glucagon). For (A and B), insulin, somatostatin and glucagon are represented by green, red, and white, respectively and for (C), insulin and glucagon are indicated by green and red, respectively. For (A–C), DAPI-stained nuclei are represented by blue. Almost 200 individual islets (10–15 islets/section) for (A and B) were analyzed to assess the abundance of β-, α-, and δ-cells. (A) represents the distribution pattern of β-, α-, and δ-cells found in majority (80%) of adult human islets, whereas, (B) represents the distribution pattern of these cell types found only in 20% of adult human islets. (C) shows the typical distribution of β- and α-cells in adult mouse islets. In this and subsequent figures, insulin, somatostatin, and glucagon are abbreviated as Ins, Somat, and Glu.
Abundant levels of the negative cell cycle regulators, p27(Kip1), p18(Ink4c), and GSK-3, in adult human β-cells
We have found the prevalence of β-cells than α-cells in adult human islets as described above (see Fig. 1A and B). Also, our published data using lysates from isolated adult human islets showed substantial levels of the 2 important negative cell cycle regulators, p27(Kip1) and GSK-3.9 Therefore, we wanted to examine the levels and sub-cellular localization of p27(Kip1) and its 2 important phosphorylated forms, p-p27(S10) and p-p27(T187), as well as GSK-3 and also, p18(Ink4c), a critical CDK inhibitor and negative cell cycle regulator, in β-cells using adult human and mouse pancreatic tissue sections. Our immunofluorescence studies demonstrate marked levels of p27(Kip1), primarily in nuclei of adult human islet β-cells (Fig. 2A, merge data, red dots), and also, in islet β-cell cytosol (Fig. 2A, merge data, yellow color). We show substantial amounts of p-p27(S10), specifically in adult human islet β-cell cytosol (Fig. 2B, merge data, bright yellow color). However, the levels of p-p27(T187) are relatively lower and present in both nuclei and cytosol of human islet β-cells (Fig. 2C, merge data, red dots and light yellow color). Likewise, we examined the levels and sub-cellular localization of p27(Kip1) and its 2 phosphorylated forms in adult mouse β- cells. We demonstrate marked amounts of p27(Kip1), almost exclusively in mouse islet β-cell nuclei (Fig. 2D, merge data, red dots), which is contrary to the distribution pattern found in human β- cells (Fig. 2D vs. A, merge data). Additionally, we show marked levels of p-p27(S10) in adult mouse islet β-cell cytosol (Fig. 2E, merge data, bright yellow color), whereas, the amounts of p-p27(T187) are relatively lower and located in both nuclei and cytosol (Fig. 2F, merge data, red dots and light yellow color), indicating a distribution pattern similar to human β-cells (Fig. 2E and F vs. Fig. 2B and C, merge data). Our data demonstrating substantial levels of p27(Kip1) in both adult human and mouse β-cell nuclei suggest that p27(Kip1), through protein-protein interactions, likely inhibits nuclear cyclinE-CDK2 activity leading to maintenance of adult β-cell quiescence. It is worth pointing out that in our previous studies using isolated adult human islet lysates,9 we displayed robust binding of p27(Kip1) with cyclinE and its cognate kinase partner, CDK2. Additionally, we hypothesize based on our findings of the abundance and sub-cellular localization of p-p27(S10) in human and mouse β-cells and also, based on the studies using non-β-cell model systems18,30 that p-p27(S10) likely interacts with D-type cyclins and their cognate kinase partners, cyclin-dependent kinase 4/6 (CDK4/6), in cytosol of adult β-cells and in turn, prevent their entry into the nuclei. Such interactions lead to cancellation of the positive cell cycle regulatory function of cyclinD-CDK/4 complexes. We and others showed substantial levels of cyclin D3 in adult human islets/β-cells and also, found low/undetectable amounts of the other 2 D-type cyclins, D1 and D2.9,10 Next, we examined the abundance and sub-cellular localization of p27(Kip1), p-p27(S10), and p-p27(T187) in α-cells of adult human and mouse pancreatic tissue. Our immunofluorescence analyses show that both human and mouse islet α-cells primarily contain the p-p27(S10) form, which is localized almost exclusively in α-cell cytosol (Fig. 3A and B, merge data, yellow color). We didn’t find any appreciable levels of either p27(Kip1) or p-p27(T187) in adult human and mouse α-cells (data not shown). Moreover, we demonstrate substantial presence of p-p27(S10) in adult human islet δ-cells (Fig. 3C, merge data, red color in somatostatin positive [white] cells), suggesting a wider presence of this particular phophorylated form of p27(Kip1) in islet endocrine cell types, including β-, α-, and δ-cells. Similarly, we examined the levels and sub-cellular localization of GSK-3, a serine-threonine kinase that remains constitutively active in resting cells. Our immunofluorescence data show substantial amounts of GSK-3β isoform in adult human islet β-cell cytosol (Fig. 4A, merge data, bright yellow color), whereas, α-cells contain either undetectable or low amounts of this kinase (Fig. 4B, merge data, glucagon positive [white] cells). We found a similar pattern of abundance and localization of GSK-3 in adult mouse islet β-cells (data not shown). Our current and previous data,9 as well as independent studies,28 indicate that p27(Kip1) levels are likely stabilized following binding with GSK-3 in adult human and mouse β-cells resulting in maintenance of their quiescence. It’s worth pointing out that GSK-3 can also be present in the nuclei of cells via its nuclear localization signal.24 Moreover, we examined the levels and sub-cellular localization of p18(Ink4c) that is implicated in controlling islet β-cell proliferation.31 Our immunofluorescence analyses demonstrate marked levels of this negative cell cycle regulator in adult human islet β-cell cytosol (Fig. 5A and B, merge data, bright yellow color). We were unable to find any appreciable changes in the sub-cellular localization of p18(Ink4c) when we compared human islets having lower and higher numbers of α-cells (Fig. 5A vs. B, merge data). The levels of p18(Ink4c) were almost undetectable in both α-and δ-cells of adult human pancreatic tissue (Fig. 5C and D, merge data). Likewise, our analyses of this CDK inhibitor in adult mouse pancreatic islets show its substantial presence in islet β-cell cytosol (Fig. 6A, merge data, bright yellow color), whereas, its levels are undetectable in α- (Fig. 6B, merge data) and also, in δ-cells (data not shown), suggesting a high similarity with adult human β-cells (Fig. 5A–D vs. Fig. 6A–B). The findings altogether confirm the abundant levels of p27(Kip1), p18(Ink4c) and GSK-3, the 3 critical negative regulators of proliferation, particularly in adult human and mouse islet β-cells, and also, demonstrate a wider distribution of the p-p27(S10) form, including in β, α, and δ-cells, specifically in their cytosol.
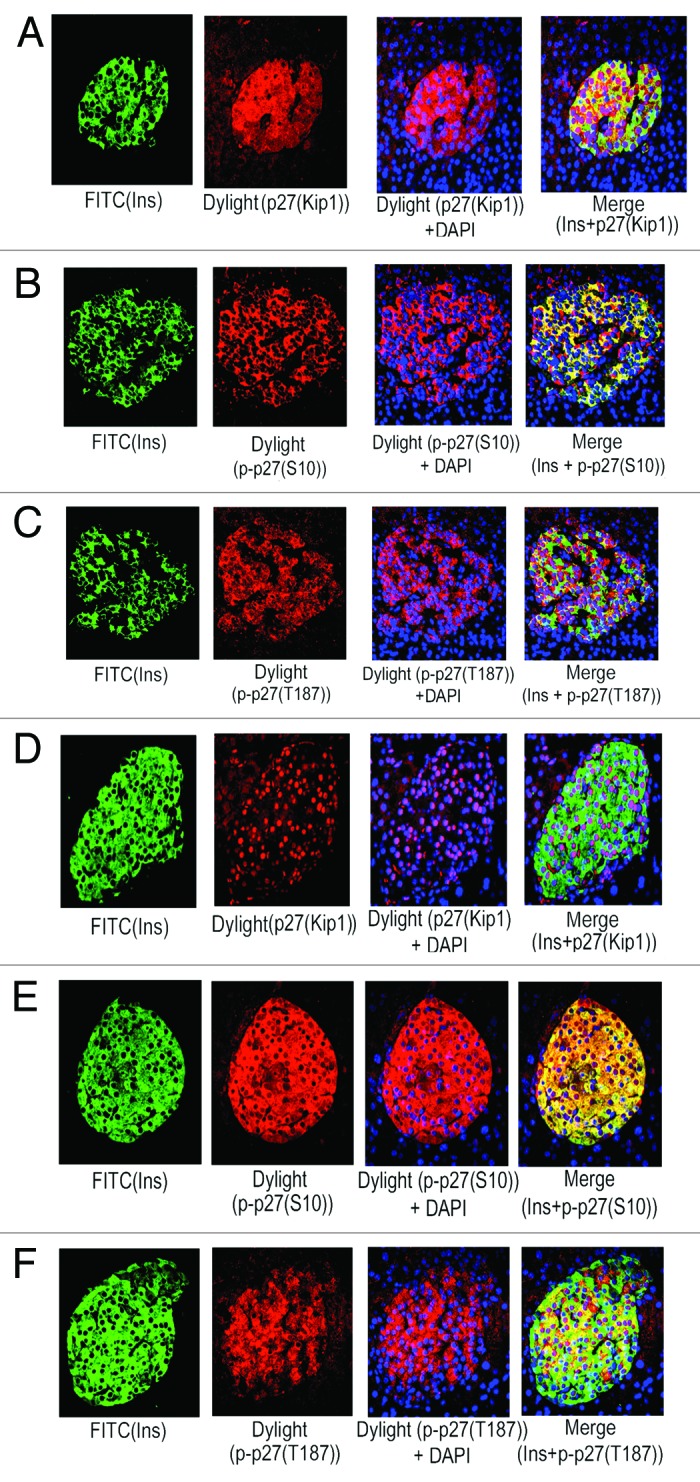
Figure 2. Adult human and mouse pancreatic β-cells contain marked levels of p27(Kip1) and its phosphorylated forms, specifically p-p27(S10). Levels and sub-cellular localization of p27(Kip1) and its 2 important phosphorylated forms, p-p27(S10) and p-p27(T187), in β-cells of adult pancreatic tissue sections, obtained from donors (A–C) and mice (D–F), were assessed by immunoflluorescence and confocal microscopy. Analyses were performed using the specific primary antibodies against insulin, p27(Kip1), p-p27(S10), and p-p27(T187) followed by incubation with the appropriate secondary antibodies conjugated with either FITC (for insulin) or Dylight (for p27[Kip1], p-p27[S10], and p-p27[T187]). In (A–F), insulin is represented by green, whereas, all of p27(Kip1), p-p27(S10), and p-p27(T187) are marked by red. Also, DAPI-stained nuclei are represented by blue. In the merge panels, (A and C; human islets), and (D and F; mouse islets), β-cell nuclear localization of p27(Kip1) and p-p27(T187) is denoted by red dots. In the merge panels, (A–C; human islets) and (E–F; mouse islets), yellow color of varying intensities indicate the levels and localization of p27(Kip1), p-p27(S10), and p-p27(T187) in β-cell cytosol. For (A–C) and (E–F), 10–15 individual islets were assessed each time (n = 3–5 independent experiments) following incubation with the specific primary and secondary antibodies as mentioned above.

Figure 3. p-p27(S10) is substantially present in adult human and mouse pancreatic α- and δ- cells. Levels and sub-cellular localization of p-p27(S10) were examined using immunofluorescence and confocal microscopy. Adult pancreatic tissue sections obtained from donors (A and C) and mice (B) were subjected to immunohistochemical analysis using the specific primary antibodies against glucagon (A and B), insulin (C), somatostatin (C) and p-p27(S10) (A–C). Glucagon is represented by either green (A) or red (B), p-p27(S10) is marked by either red (A and C) or green (B) and insulin and somatostatin by green (C) and white (C), respectively. In the merge panels, (A–C), yellow color of varying intensities refer to the levels and localization of p-p27(S10) in α-cell cytosol of adult human (A) and mouse islets (B) and also, in β-cells of adult human islets (C). The merge panel in (C) shows the abundance of p-p27(S10) (red) in somatostatin positive δ-cells (white). For (A–C), 10–15 individual islets were assessed each time (n = 4 independent experiments).

Figure 4. Abundant levels of GSK-3β in adult human pancreatic β-cells. Analysis of GSK-3β levels in β- and α-cells of adult pancreatic tissue sections obtained from donors using immunofluorescence and confocal microscopy. Pancreatic tissue sections were incubated with the specific primary antibodies against insulin, glucagon and GSK-3β followed by incubation with the appropriate secondary antibodies conjugated with either FITC (for insulin) or Dylight (for GSK-β) or TR (for glucagon). In (A), green and red represent insulin and GSK-3β, respectively, and in (B), white and red refer to glucagon and GSK-3β, respectively. In the merge panel, (A), yellow color indicates the levels and localization of GSK-3β in adult β-cell cytosol, whereas, in the merge panel, (B), GSK-3β levels are almost undetectable in α-cells. For (A and B), 10–15 individual islets were assessed each time (n = 4 independent experiments).

Figure 5. p18(Ink4c) is present in marked amounts in adult human β-cells, not in α- or δ-cells. The levels and sub-cellular localization of p18(Ink4c) were analyzed in β-, α-, and δ-cells of adult pancreatic tissue sections obtained from donors using immunofluorescence and confocal microscopy. For such studies, tissue sections were incubated with the specific primary antibodies against insulin, glucagon, somatostatin and p18(Ink4c) followed by incubation with the appropriate secondary antibodies conjugated with either FITC (for insulin and glucagon - green) or Dylight (for p18[Ink4c] - red) or Cy3 (for glucagon and somatostatin - white). In the merge panels, (A, B, and D), yellow color represents the amounts and localization of p18(Ink4c) in β-cell cytosol. In the merge panels, (C and D), no detectable levels of p18[Ink4c]are found in either α- (green) or δ-cells (white). For (A–D), 10–15 individual islets were assessed each time (n = 3–4 independent experiments).

Figure 6. Adult mouse β-cells contain substantial levels of p18(Ink4c).The amounts and sub-cellular localization of p18(Ink4c) in β- and α-cells of pancreatic tissue sections obtained from adult mice were examined by immunofluorescence and confocal microscopy. Pancreatic tissue sections were incubated with the specific antibodies against insulin, glucagon and p18(Ink4c) followed by incubation with the appropriate secondary antibodies conjugated with either FITC (for insulin and glucagon–green) or Dylight (for p18[Ink4c]-red). In the merge panel, (A), yellow color represents the levels and localization of p18(Ink4c) in β-cell cytosol, whereas, in the merge panel, (B), no detectable amounts of this protein are found in α-cells (green). For (A and B), 10–15 individual islets were assessed each time (n = 4 independent experiments).
Interaction of p27(Kip1) and p18(Ink4c) with positive cell cycle regulators in adult human islets
We and others previously showed that cyclin D3 is a major D-type cyclin in adult human islets and β-cells.7,9,10 D-type cyclins, including cyclin D3, are known to interact with their cognate kinase partners, CDK4/6, and form cyclinD-CDK4/6 holoenzyme complexes that inactivate retinoblastoma (Rb) protein via phosphorylation/hyper-phosphorylation in order to ensure the G1/S-phase progression of the cell cycle.20 It is known that p27(Kip1) has cyclin and CDK binding sites at its N-terminal30 and following binding, p27(Kip1) inhibits the positive cell cycle regulatory function of cyclin-CDK complexes. We thus wanted to assess if p27(Kip1) could interact with cyclin D3 and vice versa and also, whether p27(Kip1) could bind with CDK4/6. Our IP+WB analyses using lysates from isolated adult human islets demonstrate that each of cyclin D3, CDK4 and CDK6 is capable of binding with p27(Kip1) and such interactions are relatively much stronger for both cyclin D3 and CDK4 compared with CDK6 (Fig. 7A, lanes, αCD3 [αcyclinD3] and αCDK4 vs. C, lane, αCDK6). Additionally, by reciprocal IP and WB studies, we show that p27(Kip1) binds with cyclinD3 (Fig. 7B, lane, αp27), indicating specificities of such protein-protein interactions (Fig. 7A–C). Similarly, we examined the levels of p18(Ink4c) and also, its binding ability with CDK4/6 since independent studies demonstrated interactions of this CDK inhibitor with CDK4/6, not with any cyclins.32 Our IP and WB analyses using lysates from isolated adult human islets reveal its modest levels (Fig. 7D, lanes, αp18) as well as its ability to interact, particularly with CDK4 (Fig. 7E, lanes, αp18). We found relatively reduced binding of p18(Ink4c) with CDK6 (data not shown). Additionally, our preliminary data (unpublished) indicate an ability of p18(Ink4c) to interact with GSK-3, similar to the binding of p27(Kip1) with GSK-3 as we showed before. Altogether, the data indicate that both p27(Kip1) and p18Ink4c likely inhibit the positive cell cycle regulatory function of cyclinD3 and CDK4/6 through protein-protein interactions and eventually contribute to maintenance of adult β-cell quiescence.

Figure 7. p27(Kip1) and p18(Ink4c) interact with the positive cell cycle regulators in adult human islets. Binding abilities of p27(Kip1) and p18(Ink4c) with the positive cell cycle regulators were assessed by IP and WB assays using lysates from isolated adult human islets (islet purity: 80–90% and age of donors: 40–70 y). For each experiment (n = 3 independent experiments), total protein extracts from isolated human islets were prepared following the procedure as described before9 and equal amounts (40–50 μg) of such lysates for each sample were IP using the antibodies as mentioned in panels (A–E). Proteins bound to agarose beads were boiled in 1X Laemmli sample buffer containing βME and run in SDS-PAGE and then transferred to membranes for WB assays as described before.9 The antibodies used for immunoblotting are mentioned in (A–E). In (A), anti-cyclinD3 and anti-CDK4 antibodies were used to pull down p27(Kip1). The 3 antibodies, anti-Akt (pan), anti-Akt (S473) and anti-Akt (T308), were unable to pull down any detectable levels of p27(Kip1), indicating an inability or low levels of interaction with p27(Kip1). In (B), anti-p27(Kip1) and anti-CDK6 antibodies were used to pull down cyclinD3. The 2 antibodies, anti-cyclinE and anti-CDK2, were used as negative controls. In (C), anti-CDK6 antibody was employed to pull down p27(Kip1). In (D and E), anti-p18(Ink4c) antibodies were used to pull down p18(Ink4c) and CDK4, respectively. Controls without primary antibodies were included to examine any non-specific bindings (data not shown since non-specific bindings were absent). In (E), the band with slower mobility (in duplicate lanes) indicates a phosphorylated from of CDK4 that remains bound with p18(Ink4c). Total cell extracts (CE) from rodent β-cells (INS-1/Ins-1) were applied as positive controls. Here, the abbreviated forms, p27, CD3, CycE and p18, stand for p27(Kip1), cyclinD3, cyclin E, and p18(Ink4c), respectively.
Promoting adult human β-cell proliferation ex vivo
We wanted to examine if treatments with small molecule GSK-3 inhibitor and mitogen/s could promote adult human β-cell proliferation ex vivo. We thus analyzed the combined effects of either 1-AKP, a potent small molecule GSK-3 inhibitor,33,34 and insulin or 1-AKP+insulin+glucagon-like peptide-1 (GLP-1), on β-cell proliferation using isolated adult human islets. The rationale to select these particular combinations were the following: (1) GSK-3 likely plays a critical role in stabilizing the levels of important CDK inhibitors, such as, p27(Kip1), in adult human β-cells, (2) insulin signaling is known to inactivate GSK-3 function,22,23 (3) GLP-1 signaling is known to regulate p27(Kip1) levels,35 (4) GLP-1 analog, exendin-4, elevates rodent β-cell proliferation by downregulating p27(Kip1) levels,36 and (5) our unpublished studies using isolated adult human islets and tritiated thymidine incorporation assay revealed significant increase in islet cell proliferation in the presence of either 1-AKP+insuin or 1-AKP+insuin+GLP-1 (1-AKP+insuin+GLP-1 > 1-AKP+insulin) relative to untreated controls. Here, our immunofluorescence analyses using BrdU incorporation demonstrate that 1-AKP+insulin treatments can promote proliferation of β-cells (Ins+; BrdU+) modestly but significantly compared with untreated controls where we were unable to find any Ins+ and BrdU+ cells (Fig. 8A, Treatment A, Ins+; BrdU+ cells, represented by yellow arrow, and B, Treatment A) vs. (Fig. 8A, No Treatment, Ins-; BrdU+ cells, represented by white arrow, and B, No Treatment). Our data reveal that 1-AKP+insulin is capable of driving about 1% β-cells to proliferate compared with none (0%) in the absence of such treatments (Fig. 8B, Treatment A vs. No Treatment). It is noteworthy that given the quiescent nature of mature differentiated human β-cells,13,37,38 such increase in ex vivo proliferation is a step forward especially since our cocktails lack any viruses to drive replication as used by other investigators.6-8 When we incubated isolated human islets in the presence of 1-AKP+insulin+GLP-1, we found increased β-cell proliferation relative to untreated controls (Fig. 8A and B, Treatment B, Ins+; BrdU+ cells, 0.45%) vs. (Fig. 8A and B, No Treatment, Ins-; BrdU+ cells, 0%). However, to our surprise, we found more than 2-fold reduction in β-cell proliferation in the presence of 1-AKP+insulin+GLP-1 compared with 1-AKP+insulin treatments (Fig. 8B, Treatment B, 0.45% vs. Treatment A, 1.0%), suggesting a likely action of GLP-1 on other islet endocrine cell types and also, on non-islet cells/tissue. Independent studies have reported the expression of GLP-1 receptor in α- and δ-cells, apart from β-cells, and also in pancreatic ducts.39,40 In fact, our further analyses showed more than 2-fold induction in non- β-cell proliferation by 1-AKP+insulin+GLP-1 relative to 1-AKP+insulin (Fig. 8C, Treatment B, 1.9% vs. Treatment A, 0.8%). The data collectively underscore the potential of adult human β-cells in overcoming their quiescence by responding to stimulatory signals, mediated in this case by pharmacological inhibitor of GSK-3 and insulin, to commit to the G1/S-phase transition leading to increased proliferation.

Figure 8. Stimulation of adult human β-cell proliferation ex vivo. Increase in β-cell proliferation, following treatments with pharmacological inhibitor of GSK-3 and mitogen/s, was analyzed in isolated adult human islets by immunofluorescence and confocal microscopy. Isolated adult human islets (purity: 80–90%), immediately following delivery, were cultured for 24–48 h in 10% FBS-containing CMRL medium as described before9 and then on the day of experiment, islets (200) were placed in 2.5% FBS-containing medium and incubated in the absence (DMSO) or presence of 1-AKP (5μM)+insulin (40 nM) or 1-AKP (5μM)+insulin (40 nM)+GLP-1 (40 nM) for 2 d followed by incubation with BrdU (50μM) for overnight (~20 h). Islet sections from No Treatment (DMSO controls), Treatment A (1-AKP+insulin) and Treatment B (1-AKP+insulin+GLP-1) were then subjected to incubation with anti-insulin and anti-BrdU antibodies followed by incubation with the appropriate secondary antibodies. DAPI+ nuclei (blue) as well as Ins+ (green); BrdU+ (red) and Ins-; BrdU+ (red) cells were counted from multiple experiments (n = 3 independent experiments). The numbers of DAPI+ nuclei counted were 3266, 4232 and 4133 for No Tr., Tr. A, and Tr. B, respectively. (A) shows the presence of Ins+; BrdU+ and Ins-; BrdU+ cells that are indicated by yellow and white arrows, respectively. (B) is the graphical representation of %increase in the ratio of proliferating β-cells to total number of DAPI+ cells following No Tr., Tr. A, and Tr. B. (C) is the graphical representation of %increase in the ratio of proliferating non-β-cells to total number of DAPI+ cells following No Tr., Tr. A, and Tr. B. For (B and C): **p < 0.01 (1-AKP+insulin) and **p < 0.01 (1-AKP+insulin+GLP-1) and *p < 0.05 (1-AKP+insulin+GLP-1) indicate significant differences compared with controls (No Tr. [DMSO]). Error bars denote SEM.
Downregulation of the levels of CDK inhibitor in adult human islets
To understand the likely mechanisms for elevated proliferation of adult human β-cells mediated by 1-AKP+insulin (see Fig. 8A and B), we examined whether such combination could downregulate p27(Kip1) levels. We thus treated isolated adult human islets in the absence or presence of 1-AKP+insulin or 1-AKP+insulin+GLP-1. Our WB studies followed by densitometric scanning analyses show that p27(Kip1) levels are reduced by about 40% in the presence of 1-AKP+insulin compared with untreated controls (Fig. 9A, lanes, 2 vs.1 and C, 1-AKP+insulin vs. No Treatment [DMSO]). Our data also demonstrate that addition of GLP-1 leads to further reduction of p27(Kip1) levels, about 53%, relative to untreated controls (Fig. 9A, lanes, 3 vs. 1 and C, 1-AKP+insulin+GLP-1 vs. No Treatment [DMSO]), indicating an ability of GLP-1 in downregulating p27(Kip1) levels in other islet cells, including α- and δ-cells, that have marked amounts of p-p27(S10) as discussed above (also see Fig. 3). The data altogether suggest that modest yet significant increase in human β-cell proliferation in the presence of 1-AKP+insulin is partly mediated due to the reduction in p27(Kip1) levels. Our findings also indicate that efficient downregulation of p27(Kip1) and other CDK inhibitors, such as, p18(Ink4c), specifically in adult human β-cells, is necessary to promote their proliferation much more effectively.
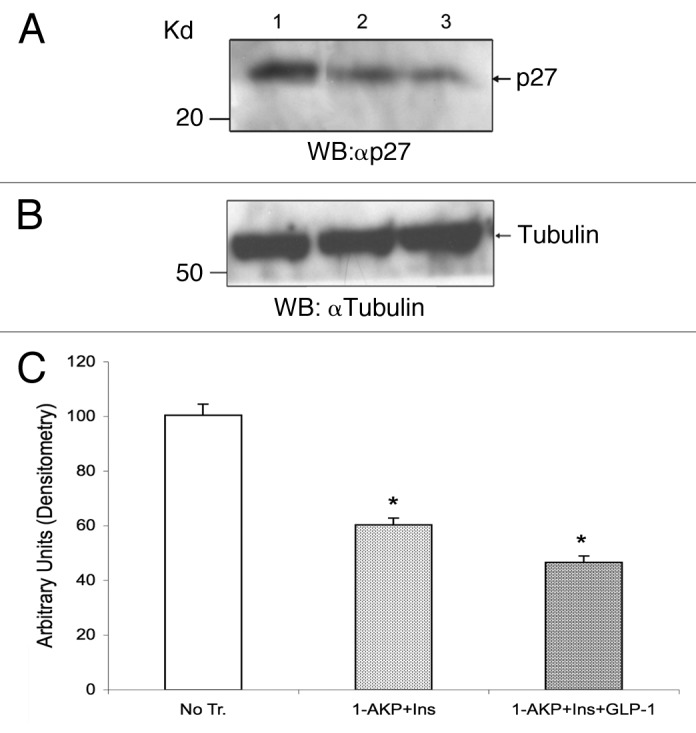
Figure 9. Downregulation of p27(Kip1) protein levels in adult human islets. Levels of p27(Kip1) in isolated adult human islets were analyzed by direct WB assay following treatments without or with small molecule GSK-3 inhibitor and mitogen/s. On the day of each experiment (n = 3 independent experiments), isolated human islets (80–90% purity) were placed in 2.5% FBS-containing medium as described in Figure 8 legend and treated without (DMSO) or with either 1-AKP (5 μM)+insulin (40 nM) or 1-AKP (5 μM) +insulin (40 nM) +GLP-1 (40 nM) for 2–3 d. Thereafter, islets were washed with PBS and total lysates were prepared as described previously.9 Equal amounts of lysates (50–60 μg) for each sample were then separated in SDS-PAGE and following membrane transfer, peptides were immunoblotted with either anti-p27(Kip1) or anti-tubulin antibodies. (A) shows the levels of p27(Kip1) after treatments with DMSO (lane 1), 1-AKP+insulin (lane 2) and 1-AKP+insulin+GLP-1 (lane 3). (B) represents the levels of tubulin as loading controls for lanes 1, 2, and 3, respectively. (C) is the graphical representation of p27(Kip1) band intensities for lanes 1, 2, and 3 following normalization against tubulin as described previously.9 For (C): *p < 0.05 (1-AKP+insulin and 1-AKP+insulin+GLP-1) indicate significant differences compared with controls (DMSO). Error bars denote SEM.
Discussion
The long held notion of the inability of adult differentiated insulin-producing β-cells to enter into the cell cycle and proliferate was first challenged by a seminal study in 2004, which demonstrated via lineage tracing analysis in adult mice that mature insulin-producing β-cells generate from pre-existing β-cells by self-duplication/proliferation.41 In 2004, another independent group revealed that β-cell proliferation is the primary mechanism for maintaining postnatal β-cell mass in mice.42 Subsequently, numerous studies demonstrated using animal models of β-cell ablation, insulin resistance, high fat diet/obesity and pregnancy that β-cell proliferation is a major mechanism for restoring adequate β-cell mass resulting in maintenance of normal glucose homeostasis in both pathological and physiological conditions.2-5,43,44 However, knowledge of human β-cell proliferation was obscure until an elegant study published in 2008.37 The studies demonstrated using cadaveric donors (age range: 2 wk–21 y) that β-cells possess substantial ability to proliferate in the first few years after birth and thereafter, such efficiency is diminished markedly as age progresses. Subsequently, other independent studies revealed that adult human β-cells are mostly refractory to proliferation,13,29,38 suggesting their predominantly quiescent (G0) state without entering into the cell cycle. It is, therefore, relevant and necessary to understand the mechanisms that regulate adult human β-cell quiescence because such knowledge is critical to promote their proliferation ex vivo for β-cell replacement therapy in diabetes and ultimately, in diabetics in future.
In our current study, we have shown that the critical negative cell cycle regulators, p27(Kip1) and p18(Ink4c), key members of the cyclin-dependent kinase (CDK) inhibitor family, and GSK-3, a constitutively active serine-threonine kinase in resting cells, play important role in maintaining adult human β-cell quiescence because of their marked levels in adult human β-cells and also, due to the protein-protein interactions of p27(Kip1) with the critical positive cell cycle regulators, cyclin D3 and its cognate kinase partners, CDK4/6, as well as the binding of p18(Ink4c) with CDK4/6. Such protein interactions likely inactivate cyclin D3-CDK4/6 holoenzyme function resulting in hypophosphorylation of retinoblastoma (Rb) protein that remains bound with the E2F family of transcription factors leading to prevention of the G1/S-phase progression of adult human β-cells. We have demonstrated that combination of small molecule GSK-3 inhibitor and mitogen/s, particularly 1-AKP+insulin, is capable of promoting adult human β-cell proliferation ex vivo, indicating a road map for β-cell mass expansion. Our mechanistic studies suggest that downregulation of the levels of p27(Kip1), mediated by 1-AKP+insulin, partly contributes to increased proliferation of adult human β-cells as measured by BrdU incorporation assay. Additionally, corresponding to our findings, an independent group has recently demonstrated the inability of GLP-1 to increase β-cell proliferation using isolated adult human islets,45 suggesting its efficacy in regulating particularly rodent β-cell proliferation.35 We didn’t find any appreciable changes in human β-cell proliferation when we added insulin-like growth factor-1 (IGF-1) with either of the 2 combinations, 1-AKP+insulin and 1-AKP+insulin+GLP-1, indicating its lack of efficacy at least in our culture conditions. It is worth mentioning though that β-cell specific IGF-1 receptor knockout in mice does not alter β-cell mass and additionally, such mice do not display impaired islet growth when subjected to high-fat diet.46,47 While we were preparing our manuscript, a report was published by Doug Melton’s group demonstrating tremendous proliferative capacity of adult rodent β-cells in response to a secretory protein, betatrophin,48 suggesting the critical importance for adult β-cells to exit their quiescence (G0) stage and enter into the cell cycle via modulation of the levels/function of negative regulators of proliferation.
Thus far, knowledge of the role of 3 negative cell cycle regulators, p27(Kip1), p18(Ink4c), and GSK-3, in controlling adult β-cell proliferation is solely based on the studies using animal models, primary β-cells isolated from rodents and rodent β-cell lines as discussed below. Deletion of the gene encoding p27(Kip1) in animal models of insulin resistance (type 2 diabetes) results in amelioration of hyperglycemia by stimulating pancreatic β-cell proliferation and increasing β-cell mass.2 Studies using Skp2 knockout mice demonstrate the critical importance of p27(Kip1) degradation in adult β-cells to establish adequate β-cell mass for responding to increased metabolic demand associated with insulin resistance.49 Pancreatic β-cells retain the capacity to reenter the cell cycle at a far greater frequency in p27(Kip1) knockout mice after developing streptozotocin-induced diabetes relative to wild-type control littermates.50 Studies using animal model of insulin resistance (IRS2−/− mice) show the efficacy of deletion of an allele of GSK-3β in preserving β-cell mass by promoting β-cell proliferation via downregulation of p27(Kip1).3 We have previously demonstrated using a rodent β-cell line, INS-1, the significance of siRNA-mediated downregulation of p27(Kip1) in stimulating β-cell proliferation.9 Our studies using INS-1 cells and an independent report employing various non-β-cell lines have revealed that pharmacological inactivation of GSK-3 reduces p27(Kip1) levels.9,28 We also have demonstrated that siRNA-mediated downregulation of either of the GSK-3 isoforms significantly promotes rodent β-cell proliferation.9 Additionally, our studies have revealed that siRNA-mediated downregulation of p27(Kip1) along with either of the GSK-3 isoforms does not stimulate INS-1 cell proliferation any further relative to individual knockdown of each of the GSK-3 isoforms, indicating GSK-3 as an upstream modulator of p27(Kip1) in β-cells. Studies have shown that adaptive growth of β-cell mass occurs via increased proliferation of β-cells for compensating elevated insulin demand during pregnancy and also, in response to high fat diet/obesity.43,44,51 Studies have also demonstrated that decreased levels of p27(Kip1) and p18(Ink4c) are required for increased β-cell proliferation during pregnancy and hyperphagic obesity.51 Deletion of GSK-3β in β-cells of adult mice resists fat-induced diabetes by increased proliferation of β-cells.52 Additionally, studies display the importance of p18(Ink4c) and p16(Ink4a) in regulating β-cell proliferation and mass.31,53
Knowledge of the efficacy of several mitogens, such as, insulin and glucagon-like peptide-1 (GLP-1), in promoting β-cell proliferation is also based on the studies using animal models, primary rodent β-cells and rodent β-cell lines.35,54,55 To modulate β-cell proliferation, insulin signaling operates mainly through the 2 pathways, IRS-PI3K-Akt/PKB and IRS-Ras/Raf-MEK-ERK pathways.56 Additionally, it is known that insulin via activation of IRS-PI3K-Akt signaling inhibits GSK-3 function by phosphorylating its S21 and S9 residues of α and β isoforms, respectively.22,23 Moreover, mice lacking β-cell insulin receptors display reduction in β-cell mass because of decreased proliferation of β-cells,47,57 suggesting the critical importance of insulin signaling in controlling adult β-cell replication and mass in rodents.
The studies collectively underscore an important link between insulin-GSK-3-p27(Kip1) wherein insulin signaling through activation of IRS-PI3K-Akt/PKB pathway inhibits the function of GSK-3 and in turn, contributes to p27(Kip1) degradation in adult β-cells. In our current studies, we have found the importance of this link, insulin-GSK-3-p27(Kip1), that plays a critical role in promoting adult human β-cell proliferation ex vivo. Additionally, the IRS-Ras/Raf-MEK-ERK pathway may play an important role in mediating the stimulatory effects on human β-cell proliferation.
Our studies via understanding the mechanisms regulating quiescence (G0) of adult human β-cells and promoting their proliferation thus reveal a road map for expansion of human β-cells ex vivo. Independent groups have demonstrated the ability of mitogen analog or pharmacological inhibitor in elevating (low-moderate levels) adult human β-cell proliferation ex vivo using specific culture conditions.58,59 Broader understanding of the key signaling molecules controlling the levels and function of important CDK inhibitors, such as, p27(Kip1) and its phosphorylated form, p-p27(S10), and p18(Ink4c) and also, GSK-3 in adult human β-cells is thus relevant and necessary for identification and development of effective small molecules/drugs, which can lead to far more efficient and selective degradation or inactivation of these negative cell cycle regulators resulting in greater elevation of adult human β-cell proliferation ex vivo and also, in diabetics in future.
Materials and Methods
Materials
The antibodies used for our immunofluorescence studies are the following: guinea pig anti-insulin (Dako) was used to identify pancreatic islet β-cells; mouse anti-glucagon (Sigma-Aldrich) and rabbit anti-glucagon (Cell Signaling Technology) were used to identify pancreatic islet α-cells; goat anti-somatostatin (Santa Cruz Biotechnology, Inc.) was used to identify pancreatic islet δ-cells. The other antibodies used for immunostaining include mouse anti-p27(Kip1) (BD Biosciences), rabbit anti-p-p27(Ser10) and rabbit anti-p-p27(Thr187) (Santa Cruz Biotechnology, Inc.), mouse anti-p18Ink4c and GSK-3β rabbit monoclonal (Cell Signaling Technology) and mouse anti-p18Ink4c and mouse anti-BrdU (Sigma-Aldrich). Species-specific secondary antibodies conjugated with fluorophores FITC, Cy3, Texas Red or DyLight were from Jackson Immuno Research Laboratories. The antibodies used for immunoprecipitation and/or western blot studies are the following: mouse anti-p27(Kip1), mouse anti-cyclinD3, mouse anti-CDK2 and mouse anti-CDK4 were from BD Biosciences; mouse anti-cyclinE was from two sources, Santa Cruz Biotechnology, Inc. and BD Biosciences; mouse anti-p18Ink4c, mouse anti-CDK6, mouse anti-Akt (pan), phopho-Akt (Ser473) rabbit monoclonal and phospho-Akt (Thr308) rabbit monoclonal were from Cell Signaling Technology; mouse anti-α-tubulin was from Sigma-Aldrich. The chemicals, including mitogens and small molecule inhibitor, used in our studies are the following: insulin, insulin-like growth factor-1 (IGF-1) and BrdU (5-Bromo-2’-deoxyuridine) were from Sigma-Aldrich; glucagon-like peptide-1 (GLP-1) (7–36 amide) was from Bachem; 1-azakenpaullone (1-AKP) was from Calbiochem; protein G-agarose and protein A-agarose were from Roche.
Immunofluorescence and confocal microscopy
For immunofluorescence analysis, we used both adult human and mouse pancreatic tissue that were fixed in formalin and embedded in paraffin. Normal human pancreatic tissue was obtained from donors of 30–50 y of age (n = 5) and normal mouse pancreatic tissue was obtained from 9 mo old animals (n = 4). The Institutional Review Board, University of Chicago, approved the use of human pancreatic tissue for our studies. The immunostaining procedure that we used here was provided by the laboratory of Dr Chris Rhodes at the University of Chicago and briefly discussed below. Formalin-fixed, paraffin embedded tissue were sectioned into slices as thin as 5 μm. Wherever possible, sections were obtained from the tail and head and also, from the body of the pancreata. Tissue sections were deparaffinized in xylene (histological grade) (Sigma-Aldrich) and rehydrated in decreasing concentrations of ethanol (200 proof) (100–60%) followed by washing in deionized H2O and then subjected to heat-induced isotope retrieval using 10 mM sodium citrate buffer (pH 6.0) and heating in the microwave on high power for ~15 min. Following blocking, tissue sections were incubated with the specific primary antibodies (in PBS containing 2% BSA and 0.3% Triton X-100) for overnight (~20 h) at 4 °C followed by incubation with the appropriate secondary antibodies conjugated with fluorophores (in PBS containing 2% BSA) for ~40 min at 25 °C in dark. After each incubation period, tissue sections were washed and following addition of slow fade gold with DAPI (Invitrogen), coverslips were mounted and edges were sealed with fingernail polish. The primary antibody dilutions used for immunofluorescence analyses were: guinea pig anti-insulin – 1:300; mouse anti-glucagon – 1:500; rabbit anti-glucagon – 1:100; goat anti-somatostatin –1:400; mouse anti-p27(Kip1) - 1:50; rabbit anti-p27(S10) – 1:50; rabbit anti-p-p27(T187) – 1:50; monoclonal anti-p18Ink4c – 1:200; GSK-3β rabbit monoclonal - 1:100. Controls without primary antibodies were used for each experiment (negative data not shown). For confocal fluorescence microscopy, we used Olympus DSU “fixed cell” Spinning Disk Confocal equipped with image capture through Slide Book software and CCD camera. The excitation and emission filters used were – Ex: 387/11nm and Em: 440/40 (blue [DAPI]); Ex:485/20 and Em: 525/30 (green [FITC]); Ex:560/25 and Em:607/36 (red [Cy3, Texas Red]) and Ex: 650/13 and Em: 684/24 (far red [DyLight]). All the images shown are of x 40 magnification.
Isolated adult human islet culture
For our studies, isolated adult human islets were received primarily from NIH-supported Integrated Islet Distribution Program (IIDP), City of Hope National Medical Center, and also, from the University of Chicago Islet Isolation Center. Islets used for our experiments were derived from normal donors of various ages (40–70 y). Islets after delivery were cultured as described before.9 The Institutional Review Board, University of Chicago, approved the use of isolated human pancreatic islets for our studies.
BrdU incorporation assay
To examine the rate of proliferation of human β-cells (insulin+ and BrdU+) and also, of non-β-cells (insulin- and BrdU+), isolated adult human islets following treatments without (DMSO) or with small molecule GSK-3 inhibitor (1-AKP) and mitogen (either insulin or insulin+GLP-1) were incubated in the presence of 50 μM BrdU for ~20 h. After incubation, islets were washed, fixed in 4% paraformaldehyde and then processed for paraffin embedding and sectioning (5 μm) following the procedure as described before.60 For immunostaining of insulin and BrdU, guinea pig anti-insulin and monoclonal anti-BrdU antibodies were used at the dilutions of 1:300 and 1:2,500, respectively.
Immunoprecipitation and western blotting
Total lysates from isolated adult human islets, without or with treatments (see Figs. 7 and 9), were prepared according to the procedure as described before.9 Equal amounts of total lysates were then subjected to either immunoprecipitation followed by immunoblotting or direct immunoblotting (see the respective figure legends) following the protocol as described before.9
Statistical analysis
Quantitative data are presented as the mean ± SEM (n = 3 or more). Statistically significant differences were analyzed using the 2-tailed Student t-test. A p value < 0.05 was considered to indicate a significant difference.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
For this work, the authors greatly acknowledge the support provided by the grants, NIH-RO1-DK-013914 and NIH-RO1-CA-090764, to Dr Donald F Steiner of the University of Chicago and Dr Nissim Hay of the University of Illinois at Chicago, respectively. The authors gratefully acknowledge the support of Drs Louis H Philipson and Christopher J Rhodes of the University of Chicago who provided adult human pancreatic tissue and immunohistochemistry protocol and also, some of the antibodies (primary and secondary) and reagents required for this work. Isolated adult human islets were obtained primarily form the NIH-sponsored Integrated Islet Distribution Program (IIDP), City of Hope National Medical Center, Duarte, CA. The authors are also thankful to Dr Piotr Witkowski of the University of Chicago for proving isolated adult human islets. Additionally, the authors like to gratefully acknowledge the assistance provided by the integrated microscopy facility and human tissue resource center of the University of Chicago.
Glossary
Abbreviations:
- 1-AKP
1-azakenpaullone
- GLP-1
glucagon-like peptide-1
- GSK-3
glycogen synthase kinase-3
- CDK
cyclin-dependent kinase
- BrdU
5-bromo-2’-deoxyuridine
References
- 1.Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–8. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 2.Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med. 2005;11:175–82. doi: 10.1038/nm1187. [DOI] [PubMed] [Google Scholar]
- 3.Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Meneur C, et al. Genetic deficiency of glycogen synthase kinase-3β corrects diabetes in mouse models of insulin resistance. PLoS Biol 2008; 6: 0307 – 0318. [DOI] [PMC free article] [PubMed]
- 4.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117:2553–61. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cano DA, Rulifson IC, Heiser PW, Swigart LB, Pelengaris S, German M, et al. Regulated β-cell regeneration in the adult mouse pancreas. Diabetes. 2008;57:958–66. doi: 10.2337/db07-0913. [DOI] [PubMed] [Google Scholar]
- 6.Fiaschi-Taesch NM, Bigatel TA, Sicari B, Takane KK, Salim F, Velazquez-Garcia S, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes. 2009;58:882–93. doi: 10.2337/db08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, et al. Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010;59:1926–36. doi: 10.2337/db09-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takane KK, Kleinberger JW, Salim FG, Fiaschi-Taesch NM, Stewart AF. Regulated and reversible induction of adult human β-cell replication. Diabetes. 2012;61:418–24. doi: 10.2337/db11-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein J, Milewski WM, Hara M, Steiner DF, Dey A. GSK-3 inactivation or depletion promotes β-cell replication via down regulation of the CDK inhibitor, p27 (Kip1) Islets. 2011;3:21–34. doi: 10.4161/isl.3.1.14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab. 2011;300:E221–30. doi: 10.1152/ajpendo.00496.2010. [DOI] [PubMed] [Google Scholar]
- 11.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–20. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 12.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–7. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, et al. Significant human β-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010;95:E234–9. doi: 10.1210/jc.2010-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, et al. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59:2846–53. doi: 10.2337/db10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem. 2012;287:27407–14. doi: 10.1074/jbc.M112.350736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-X. [DOI] [PubMed] [Google Scholar]
- 17.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 18.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 19.Susaki E, Nakayama KI. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015–20. doi: 10.4161/cc.6.24.5087. [DOI] [PubMed] [Google Scholar]
- 20.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 21.Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O’Keefe CL, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939–52. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 22.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 23.Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem Soc Trans. 2004;32:803–8. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem. 2007;282:16989–7001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003;12:381–92. doi: 10.1016/S1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 27.Naderi S, Gutzkow KB, Låhne HU, Lefdal S, Ryves WJ, Harwood AJ, et al. cAMP-induced degradation of cyclin D3 through association with GSK-3β. J Cell Sci. 2004;117:3769–83. doi: 10.1242/jcs.01210. [DOI] [PubMed] [Google Scholar]
- 28.Surjit M, Lal SK. Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle. 2007;6:580–8. doi: 10.4161/cc.6.5.3899. [DOI] [PubMed] [Google Scholar]
- 29.Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle. 2007;6:1053–61. doi: 10.4161/cc.6.9.4142. [DOI] [PubMed] [Google Scholar]
- 31.Ramsey MR, Krishnamurthy J, Pei XH, Torrice C, Lin W, Carrasco DR, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res. 2007;67:4732–41. doi: 10.1158/0008-5472.CAN-06-3437. [DOI] [PubMed] [Google Scholar]
- 32.Noh SJ, Li Y, Xiong Y, Guan KL. Identification of functional elements of p18INK4C essential for binding and inhibition of cyclin-dependent kinase (CDK) 4 and CDK6. Cancer Res. 1999;59:558–64. [PubMed] [Google Scholar]
- 33.Mussmann R, Geese M, Harder F, Kegel S, Andag U, Lomow A, et al. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J Biol Chem. 2007;282:12030–7. doi: 10.1074/jbc.M609637200. [DOI] [PubMed] [Google Scholar]
- 34.Stukenbrock H, Mussmann R, Geese M, Ferandin Y, Lozach O, Lemcke T, et al. 9-cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic β cell protection and replication. J Med Chem. 2008;51:2196–207. doi: 10.1021/jm701582f. [DOI] [PubMed] [Google Scholar]
- 35.Lavine JA, Attie AD. Gastrointestinal hormones and the regulation of β-cell mass. Ann N Y Acad Sci. 2010;1212:41–58. doi: 10.1111/j.1749-6632.2010.05802.x. [DOI] [PubMed] [Google Scholar]
- 36.Song WJ, Schreiber WE, Zhong E, Liu FF, Kornfeld BD, Wondisford FE, et al. Exendin-4 stimulation of cyclin A2 in beta-cell proliferation. Diabetes. 2008;57:2371–81. doi: 10.2337/db07-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57:1584–94. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia. 2010;53:321–30. doi: 10.1007/s00125-009-1562-x. [DOI] [PubMed] [Google Scholar]
- 39.Heller RS, Kieffer TJ, Habener JF. Insulinotropic glucagon-like peptide I receptor expression in glucagon-producing alpha-cells of the rat endocrine pancreas. Diabetes. 1997;46:785–91. doi: 10.2337/diabetes.46.5.785. [DOI] [PubMed] [Google Scholar]
- 40.Tornehave D, Kristensen P, Rømer J, Knudsen LB, Heller RS. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J Histochem Cytochem. 2008;56:841–51. doi: 10.1369/jhc.2008.951319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–6. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 42.Georgia S, Bhushan A. β cell replication is the primary mechanism for maintaining postnatal β cell mass. J Clin Invest. 2004;114:963–8. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hakonen E, Ustinov J, Mathijs I, Palgi J, Bouwens L, Miettinen PJ, et al. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia. 2011;54:1735–43. doi: 10.1007/s00125-011-2153-1. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–8. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tschen SI, Georgia S, Dhawan S, Bhushan A. Skp2 is required for incretin hormone-mediated β-cell proliferation. Mol Endocrinol. 2011;25:2134–43. doi: 10.1210/me.2011-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, Magnuson MA, et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet. 2002;31:111–5. doi: 10.1038/ng872. [DOI] [PubMed] [Google Scholar]
- 47.Okada T, Liew CW, Hu J, Hinault C, Michael MD, Krtzfeldt J, et al. Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A. 2007;104:8977–82. doi: 10.1073/pnas.0608703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell. 2013;153:747–58. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Zhong L, Georgia S, Tschen SI, Nakayama K, Nakayama K, Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic beta cells. J Clin Invest. 2007;117:2869–76. doi: 10.1172/JCI32198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Georgia S, Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. 2006;55:2950–6. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 51.Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, et al. Menin controls growth of pancreatic β-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–9. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Tanabe K, Baronnier D, Patel S, Woodgett J, Cras-Méneur C, et al. Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53:2600–10. doi: 10.1007/s00125-010-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pascoe J, Hollern D, Stamateris R, Abbasi M, Romano LC, Zou B, et al. Free fatty acids block glucose-induced β-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes. 2012;61:632–41. doi: 10.2337/db11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, Garcia-Ocaña A. Growth factors and beta cell replication. Int J Biochem Cell Biol. 2006;38:931–50. doi: 10.1016/j.biocel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Assmann A, Hinault C, Kulkarni RN. Growth factor control of pancreatic islet regeneration and function. Pediatr Diabetes. 2009;10:14–32. doi: 10.1111/j.1399-5448.2008.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson JD, Alejandro EU. Control of pancreatic β-cell fate by insulin signaling: The sweet spot hypothesis. Cell Cycle. 2008;7:1343–7. doi: 10.4161/cc.7.10.5865. [DOI] [PubMed] [Google Scholar]
- 57.Otani K, Kulkarni RN, Baldwin AC, Krutzfeldt J, Ueki K, Stoffel M, et al. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am J Physiol Endocrinol Metab. 2004;286:E41–9. doi: 10.1152/ajpendo.00533.2001. [DOI] [PubMed] [Google Scholar]
- 58.Liu H, Remedi MS, Pappan KL, Kwon G, Rohatgi N, Marshall CA, et al. Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes. 2009;58:663–72. doi: 10.2337/db07-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PLoS One. 2012;7:e35801–4. doi: 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478:349–55. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]