

Abstract
Treatment of patients with relapsed or refractory aggressive non-Hodgkin B-cell lymphoma remains an unmet clinical need, and the progressive myocardial toxicity related to cumulative, dose-dependent damage induced by anthracyclines represents a tricky issue in the planning of therapy. Pixantrone is a promising aza-anthracenedione with reduced cardiotoxicity and significant antineoplastic activity, and has been investigated in solid and hematologic tumors in several Phase I, II, and III trials. The aim of this review is to summarize the data reported so far on pixantrone as a salvage therapy in relapsed/refractory non-Hodgkin B-cell lymphoma.
Keywords: pixantrone, aggressive non-Hodgkin B-cell lymphoma, relapsed/refractory
Introduction
Treatment of patients with relapsed or refractory aggressive non-Hodgkin B-cell lymphoma (NHL) represents a challenge in everyday clinical practice. The standard approach for adult patients not achieving a complete remission or relapsing after anthracycline-based induction and eligible for intensive treatment includes a salvage combination based on the anti-CD20 antibody rituximab (in CD20-positive NHL) in combination with a non-cross-resistant platinum-containing regimen such as DHAP/DHAOX (dexamethasone, cytarabine and cisplatin/oxaliplatin),1,2 ICE (ifosfamide, carboplatin, etoposide),3,4 or ESHAP (methylprednisolone, etoposide, cytarabine, cisplatin).5,6 Responders to reinduction usually undergo consolidation with high-dose therapy and autologous stem cell transplantation;7 in selected cases, allogeneic stem cell transplantation may be considered. The outcome for patients not eligible for intensive treatment because of comorbidities, age, or poor performance status is dismal, and the goal of therapy is palliative; lenalidomide, gemcitabine, or other novel biological drugs within clinical trials are usually proposed, and localized radiation therapy may also be considered in the presence of large symptomatic masses. The management of relapsed/refractory patients remains an unmet clinical need, due to the poor prognosis and lack of therapeutic options. One of the trickiest issues that physicians have to face in the planning of treatment for patients in this setting is progressive myocardial toxicity related to the cumulative, dose-dependent damage induced by anthracyclines, which may lead to congestive heart failure.8 In this review, we investigate the role of pixantrone, an anthraquinone-based inhibitor of topoisomerase II with reduced cardiotoxicity, as a salvage therapy in relapsed/refractory NHL.
Pixantrone
Pixantrone (CTI Life Sciences Limited, Welwyn Garden City, UK) is a compound belonging to the aza-anthracenedione family, and was developed from the molecular structure of anthracyclines nearly 20 years ago to reduce anthracycline-related cardiotoxicity while maintaining similar antineoplastic activity. The molecular structure of pixantrone (Figure 1) is similar to that of mitoxantrone but without the 5,8-dihydroxy substitution pattern. These conformational differences reduce treatment-related cardiotoxicity by decreasing formation of free radicals and production of cardiotoxic alcohol metabolites.9–13 The mechanism of action of pixantrone is partially similar to that of mitoxantrone and doxorubicin in that it involves DNA intercalation and nucleic acid compaction, and has only a weak inhibitory effect on topoisomerase II; moreover, it directly alkylates DNA, forming stable DNA adducts.9,14–17
Figure 1.
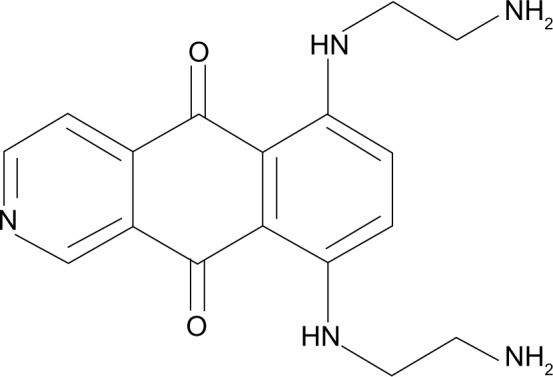
Molecular structure of pixantrone.
Pixantrone is an intravenously administered agent with a median half-life of 21 hours. It is weakly bound to serum protein (approximately 50%) and has a large volume of distribution (nearly 26 L). The pathway of elimination appears to be mainly nonrenal, with biliary excretion of the unmodified drug being the most important clearance mechanism.18,19 At present, clinical data regarding the safety of pixantrone are lacking for patients with impaired hepatic function. No drug interactions have been reported and no studies have been performed in humans; the data obtained in vitro analyzing the most common human cytochrome P450 (CYP) isoforms have shown possible mixed-type inhibition of CYP1A2 and CYP2C8 that may be of clinical relevance, while no other significant interactions with CYP have been observed. Moreover, pixantrone is a substrate for the membrane transport proteins, P-glycoprotein/BRCP and OCT1; agents that inhibit these transporters (ie, cyclosporine A, tacrolimus, ritonavir, saquinavir, or nelfinavir) may decrease hepatic uptake and excretion of the drug, resulting in higher toxicity. In contrast, inducers such as rifampicin, carbamazepine, and glucocorticoids may increase pixantrone excretion with a consequent decrease in efficacy. On the other hand, pixantrone has only a weak capability to inhibit P-glycoprotein, BCRP, and BSEP transport mechanism in vitro. Moreover, it inhibits OCT1-mediated transport in vitro although it seems unable to exert such action in vivo at clinically relevant concentrations. Finally, sex, age, and race seem not to have a significant influence on pharmacokinetics, with clearance being dependent only on body size. For this reason, dosing must be planned according to body surface area.20,21
Preclinical studies
Preclinical models have investigated pixantrone in solid tumors and hematologic malignancies, and reported significant activity. In particular, the drug showed superior activity in hematologic neoplasms when compared with other agents (including anthracyclines) at different dosages, with a lower incidence of cardiotoxicity.22–25 The activity of pixantrone was initially compared with that of doxorubicin in murine lymphomatous and leukemic cell lines and in in vivo models, and was found to be superior.26 Subsequently, toxicology studies were performed in murine models to investigate the maximum tolerated dose, which was found to be 65 mg/kg; the dose-limiting toxicity was mainly hematologic, ie, myelosuppression. Nevertheless, equipotent doses of mitoxantrone induced greater myelotoxicity and caused myocardial damage, which was not induced by pixantrone.24,27 Interestingly, some cases of sudden death were recorded in rodents during bolus infusion or immediately thereafter, probably related to volume and injection rate. Accordingly, slow infusion is recommended.
Phase I studies
Three Phase I studies have investigated the dose and schedule of pixantrone (Table 1), and one included patients affected by NHL or chronic lymphocytic leukemia. Two schedules were tested: Dawson et al20 analyzed data from 24 pretreated patients with solid tumors, mainly colorectal, renal, and pulmonary, who underwent a dose escalation schedule from 20 mg/m2 to 240 mg/m2 administered once every 3 weeks; the other schedule, explored by Borchmann et al in relapsed/refractory NHL or chronic lymphocytic leukemia19 and by Faivre et al21 in solid tumors, escalated the dose from 5 mg/m2 to 84 mg/m2 and to 150 mg/m2, respectively, administered on days 1, 8, and 15 of a 28-day cycle. In all of these studies, the dose-limiting toxicity according to Common Toxicity Criteria was grade IV neutropenia, usually lasting 4–7 days; in contrast, anemia and thrombocytopenia were infrequent. The most common nonhematologic toxicities were nausea and vomiting (Common Toxicity Criteria, grade I–III), and blue discoloration of the skin, veins, and urine. None of the patients treated in these studies experienced cardiotoxic events or symptoms. Cardiac toxicity was carefully monitored in the study by Dawson et al, in which all patients were assessed by echocardiography before starting pixantrone and during treatment. After four cycles, one patient showed a decrease in ejection fraction from 67% to 46%, deemed to be a grade II toxicity according to Common Toxicity Criteria, without clinical sequelae: of note, this patient had a previous history of viral myocarditis. On the basis of these findings, the pixantrone schedule selected for the Phase II trials was 85 mg/m2 on days 1, 8, and 15 of a 28-day cycle.
Table 1.
Phase I studies
| Borchmann et al19 | Dawson et al20 | Faivre et al21 | |
|---|---|---|---|
| Patient characteristics | |||
| Patients | 26 | 24 | 30 |
| Median (range) age, years | 59.5 (33–74) | 56 (27–66) | 56 (29–75) |
| Treatment | |||
| Recommend dose (mg/m2) | 84 | 180 | 112.5 |
| Toxicities | |||
| Grade III–IV hematologic toxicities (n) | Neutropenia (6) | Neutropenia (3) | Neutropenia (12) |
| Anemia (1) | Lymphopenia (5) | Anemia (4) | |
| Thrombocytopenia (1) | Thrombocytopenia (2) | Thrombocytopenia (1) | |
| Grade III–IV nonhematologic toxicities (n) | Diarrhea (1) | Vomiting (1) | None |
| Nausea (1) | |||
| Vomiting (1) | |||
Phase II–III single-agent studies
Pixantrone has been tested as a single agent in two prospective studies. The first, reported by Borchmann et al, was an open-label, nonrandomized, noncomparative Phase II study28 that enrolled 33 adult patients with relapsed aggressive NHL. The patients had mainly diffuse large B-cell lymphoma (73%) or mantle cell lymphoma (21%), their median age was 66 years, and they were previously treated with a median of two cycles; International Prognostic Index (IPI) score was unfavorable (≥3) in 36% of patients. Further patient characteristics are reported in Table 2. Pixantrone was administered at a dosage of 85 mg/m2 on days 1, 8, and 15 of a 28-day cycle, for up to six courses. Most patients (82%) discontinued treatment before the sixth cycle, with a median of two cycles administered per patient; the main reasons for discontinuation were progressive disease (58%) and nonhematologic toxicity (12%). The most relevant toxicity was hematologic (neutropenia, 58%), while nonhematologic toxicity was mild and included hepatobiliary disorders (3%), arthritis (3%), and asthenia (3%), with one toxic death (fatal septicemia) registered. As far as cardiotoxicity is concerned, a significant decrease in left ventricular ejection fraction (LVEF) ≥10% as measured by multigated acquisition scan was noted in three patients, all of whom were previously treated with anthracyclines; two of these patients had a low cardiac output at baseline, and in one treatment had to be discontinued when the patient developed cardiac symptoms and LVEF decreased to 25%. Five patients achieved a complete remission (15%) and nine had a partial response (27%; five partial responses were unconfirmed), with a median progression-free survival of 106 days; all but one of the patients who achieved a confirmed response had diffuse large B-cell or other high-grade B-cell lymphoma, and only one patient (14%) with mantle cell lymphoma responded with a complete remission.
Table 2.
Phase II–III single-agent studies
| Borchmann et al28 | Pettengell et al29 (pixantrone arm only in this table) | |
|---|---|---|
| Patient characteristics | ||
| Patients | 33 | 70 (only 68 treated) |
| Histotypes included, n (%) | DLBCL, 24 (73%) | DLBCL, 53 (76%) |
| MCL, 7 (21%) | Transformed, 10 (14%) | |
| Others, 2 (6%) | PTCL NOS, 3 (4%) Null ALCL, 3 (4%) FL grade III, 1 (1%) |
|
| Median age (range) | 66 (24–81) | 60 (18–80) |
| Sex (M/F) | 18/15 | 24/46 |
| Ann Arbor stage | ||
| I–II (%) | 8 (24%) | 19 (26%) |
| III–IV (%) | 25 (76%) | 51 (74%) |
| Median (range) previous lines of therapy | 2 (0–5) | 3 (2–9) |
| Prior treatment with anti-CD20 (%) | NA | 38 (54%) |
| Prior stem cell transplantation (%) | 2 (6%) | 11 (16%) |
| Median (range) prior equivalent dose of doxorubicin (mg/m2) | 300 (110–600) | 292.9 (51–472) |
| Median (range) time from last prior therapy | 123 days (6 days to 5 years) | 9 months (1–86 months) |
| Response | ||
| Median (range) number of cycles administered | 2 (1–6) | 4 (2–6) |
| Early treatment discontinuations (%) | 27 (82%) | 48 (71%) |
| CR (%) | 5 (15%) | 8 (11%) and 6 CRu (9%) |
| PR (%) | 4 (12%) | 12 (17.1%) |
| Median PFS | 106 days | 5.3 months |
| Median OS | NA | 10.2 months |
| Toxicities | ||
| Grade III–IV hematologic toxicities (%) | Neutropenia (58%) | Neutropenia (41%) |
| Thrombocytopenia (6%) | Thrombocytopenia (12%) | |
| Anemia (6%) | Anemia (6%) | |
| Grade III–IV nonhematologic toxicities (%) | Hepatobiliary disorders (3%) | Abdominal pain (7%) |
| Arthritis (3%) | Pneumonia (6%) | |
| Asthenia (3%) | Dyspnea (6%) | |
| Cardiac toxicity (%) | LVEF-D ≥10% (9%) | LVEF-D (19%) |
| LVEF-D CTC gr III–IV (2%) | ||
Abbreviations: n, number; M, male; F, female; CR, complete response; CRu, complete response unconfirmed; PR, partial response; DLBCL, diffuse large B-cell lymphoma; MCL, mantle cell lymphoma; PTCL NOS, peripheral T-cell lymphoma not otherwise specified; null ALCL, anaplastic large-cell lymphoma T/null-cell type; LVEF-D, left ventricle ejection fraction decrease; CTC gr, Common Toxicity Criteria grade; PFS, progression-free survival; OS, overall survival; NA, not available; FL, follicular lymphoma.
The second report was an international, multicenter, randomized, active-controlled, open-label Phase III study reported by Pettengell et al29 and it led to registration by the European Medicines Agency. This trial was designed to assess the efficacy and safety of pixantrone in comparison with single-agent physician’s choice therapy in patients with aggressive NHL relapsed or refractory to at least two prior regimens. The primary endpoints were complete remission and complete remission unconfirmed evaluated according to the 1999 International Working Group criteria,30 with progression-free survival and overall survival as secondary endpoints. The initial planned sample size was 320 patients, but the study was subsequently closed in September 2007, 3 years after the first patient was enrolled, with only 140 patients randomized (70 per arm) due to very slow accrual. Most of the enrolled patients had diffuse large B-cell lymphoma (76% in the pixantrone arm versus 73% in comparator arm) or transformed indolent lymphoma (14% versus 13%), with a median age of 60 years versus 58 years, mainly unfavorable Ann Arbor stage and International Prognostic Index scores; baseline patients characteristics were well balanced in experimental and control arm. In the experimental arm, patients received pixantrone at a dosage of 85 mg/m2 weekly (days 1, 8, and 15 of a 28-day cycle) for up to six courses, while in the comparator arm physicians could choose between six single-agent courses of vinorelbine, oxaliplatin, ifosfamide, etoposide, mitoxantrone, or gemcitabine at prespecified standard doses and schedules. Seventy-one percent of patients in the experimental arm and 76% in the comparator arm did not complete the six planned cycles because of disease progression or relapse (41% in the pixantrone group versus 58% in the comparator group) or adverse events (22% versus 13%, respectively). The response analysis, based on the intention to treat population, showed a benefit in terms of complete remission/complete remission unconfirmed rate and overall response rate for pixantrone (20% versus 5.7%, P=0.021; 37.1% versus 14.3%, P=0.003, respectively). Median progression-free survival was longer in the experimental arm (5.3 months versus 2.6 months, P=0.005) and a trend towards longer median overall survival was observed with pixantrone, but this was not statistically significant (10.2 months versus 7.6 months, P=0.251). An exploratory analysis was performed to investigate if any favorable factor predicting a better outcome was recognizable; an absence of prior anti-CD20 treatment or stem cell transplantation, less than three prior chemotherapy regimens, age ≥65 years, and female sex were identified as favorable prognostic factors, but prior use of rituximab did not influence the benefit on progression-free survival. Unfortunately, no data are available regarding the relationship between response and specific biological subgroups such as germinal center B-cell-like or activated B-cell, as defined on the basis of on immunohistochemistry or gene expression profiling. In terms of toxicity, patients in the experimental arm experienced more grade III and IV adverse events (76.5% versus 52.2%); however, the overall proportion of complications was similar in the two groups (97.1% versus 91%). In particular, grade III–IV neutropenia was more common in patients treated with pixantrone (41.2% versus 19.4%), as was febrile neutropenia (7.4% versus 3.0%), while the rate of thrombocytopenia was similar and that of anemia was lower (11.8% versus 10.4% and 5.9% versus 13.4%, respectively). Cardiotoxicity was more common in the pixantrone group (35.3% versus 20.9%), and the most common adverse cardiac event was a decrease in LVEF without symptoms; the median decrease in LVEF from baseline to the end of treatment was 4% (range −25% to +21%) and 0% (range −13% to +10%) in the experimental arm and comparator arm, respectively. No evidence of cumulative, dose-related cardiotoxicity was reported, and decreases in LVEF were not associated with clinical evidence of cardiac impairment. Moreover, patients were previously treated with anthracyclines in both arms (Table 2), and five patients in the experimental arm had a previous history of congestive heart failure or cardiomyopathy at the time of enrollment whereas none of the patients in the comparator group had a history of these conditions; this might partially explain the higher frequency of cardiac adverse events in the pixantrone group.
Combination therapy studies
Three studies have investigated pixantrone as part of a polychemotherapeutic regimen (Table 3). Two of these studies were conducted in the relapsed/refractory setting, while the third investigated pixantrone as a first-line treatment. The first study, published by Lim et al31 in 2007, evaluated the safety, efficacy, and pharmacokinetics of a combination derived from ESHAP in which pixantrone at a single dose of 80 mg/m2 was substituted for etoposide in patients with aggressive relapsed or refractory NHL. In Phase I, myelosuppression was the dose-limiting toxicity, occurring at the first dose level tested (80 mg/m2), so this was the dose adopted for the Phase II. Nineteen patients with diffuse large B-cell, anaplastic large B-cell, or follicular large cell lymphoma, all previously treated with doxorubicin, were enrolled. Median age was 50 years (range 35 to 75), performance status was good (0 to 1 in all patients), and the patients previously received a median of one line of therapy (range 1 to 7). A total of 70 cycles were administered (median four cycles), and 26 (37%) had to be delayed, mainly because of hematologic toxicity. Thrombocytopenia, neutropenia, and anemia were the most common grade III–IV toxicities, occurring in 95%, 84%, and 53% of patients, respectively; other nonhematologic toxicities were metabolic and gastrointestinal. With regard to cardiotoxicity, seven patients (37%) experienced a 10%–20% decrease in LVEF from baseline or to a LVEF lower than 50%. The overall response rate was 58%, with 37% achieving a complete remission and 21% having a partial response. Six of the eleven responders (55%) underwent stem cell transplantation. Median time to progression and overall survival were 5.7 months and 14.5 months, respectively.
Table 3.
Combination therapy studies
| Lim et al31 | Borchmann et al32 | Herbrecht et al33 (pixantrone arm) | |
|---|---|---|---|
| Response | |||
| Median number of cycles administered (range) | 4 (1–6) | 6 (1–6) | 8 (1–8) |
| CR (%) | 7 (37%) | 14 (47%) | 46 (75%) |
| PR (%) | 4 (21%) | 8 (26%) | 4 (7%) |
| Median PFS | TTP 5.7 months (I and II) | 8.2 months (II) | Not reached |
| Median OS | 14.7 months (I and II) | 17.9 months (II) | Not reached |
| Toxicities | |||
| Grade III–IV hematologic toxicities (%) | Neutropenia (68% gr. IV) | Neutropenia (I: 89%; II: 97%) | Neutropenia |
| Anemia (53% I and II) | Anemia (I: 14%; II: 30%) | Anemia | |
| Thrombocytopenia (95% I and II) | Thrombocytopenia (I: 14%; II: 20%) | Febrile neutropenia | |
| Febrile neutropenia (5% gr. IV) | Febrile neutropenia (I: 11%; II: 20%) | Thrombocytopenia | |
| Grade III–IV nonhematologic toxicities (%) | Metabolic (16% I and II) | Metabolic (I: 0%; II: 10%) | Metabolic (20%) |
| Gastrointestinal (11% I and II) | Gastrointestinal (I: 3%; II: 10%) | Gastrointestinal (17%) | |
| Fatigue (5% I and II) | Infectious (17%) | ||
| Deep vein thrombosis (5% I and II) | |||
| Cardiac toxicity (%) | LVEF-D ≥10% (37% I and II) | Any disorder (I: 37%; II: 27%) | LVEF-D ≥15% (17%) |
| LVEF-D ≥10% (I: 29%; II: 13%) | LVEF-D ≥20% (2%) | ||
| Troponin T increase (7%) | |||
Abbreviations: CRu, complete response unconfirmed; PR, partial response; TTP, time to progression; LVEF-D, left ventricle ejection fraction decrease; I, Phase I; II, Phase II; PFS, progression-free survival; OS, overall survival; gr., grade.
Another Phase I/II trial, reported by Borchmann et al, investigated pixantrone as a substitute for doxorubicin in the classic CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) regimen to treat relapsed aggressive NHL, including diffuse large B-cell, mantle cell, and follicular grade III lymphomas.32 Overall, 65 patients with a median age of 62 years (range 26 to 81), mostly with a favorable IPI score (0 to 1 in 69%) and previously treated with a median with one line of therapy (range 1 to 7, including stem cell transplantation in eleven patients), were enrolled. In Phase I of the study, 35 patients underwent dose escalation from 80 mg/m2 to 180 mg/m2, defining 150 mg/m2 as the dose for Phase II. In the second part of the trial, 20 (67%) of 30 patients received all six planned cycles and all patients were assessed for response. The complete remission rate (including complete remission unconfirmed) was 47% and median overall survival was 17.9 months. The most common toxicity was hematologic in both Phase I and Phase II, with grade III–IV neutropenia in 89% and 97%, anemia in 14% and 30%, thrombocytopenia in 14% and 20%, and febrile neutropenia in 11% and 20% of patients, respectively. Overall, LVEF decreases of ≥10% from baseline occurred in 14 patients (22%), but seemed to be transient and unrelated to dose. Symptomatic cardiac failure occurred in four patients, but pre-existing cardiac conditions might have confounded causality.
Most recently reported was an open-label, multicenter, comparative Phase II trial enrolling previously untreated patients;33 this paper is mentioned because it is the only comparative trial available in which pixantrone was given as part of a combination regimen. A total of 124 patients with newly diagnosed diffuse large B-cell lymphoma were randomized 1:1 to receive either a regimen of cyclophosphamide, pixantrone, vincristine, prednisone, and rituximab (CPOP-R) or CHOP-rituximab (CHOP-R) for up to six to eight cycles in patients in complete remission or in partial response after four cycles, respectively. In experimental arm where 61 patients were included, median age was 68 years (range 38 to 88) and 46% of the patients had an unfavorable IPI score (≥3). In the CPOP-R arm, the complete remission/complete remission unconfirmed rate was 75% versus 84% for CHOP-R, and the criteria for noninferiority of CPOP-R were not met. Of note, the trial was closed before reaching the planned enrollment of 240 patients, which might explain the failure with regard to this endpoint. The overall response rate was 82% for the CPOP-R arm and 90% for the CHOP-R arm, and median progression-free survival was not reached in the CPOP-R arm and was 40 months in the CHOP-R arm; median overall survival was not reached in either treatment arm. Overall survival rates were lower for patients treated with CPOP-R (hazard ratio 2.37, P=0.029), with more deaths occurring in the CPOP-R arm (30% versus 14%). According to the investigators, it is unlikely that treatment-related toxic effects were responsible for the discrepancy in deaths between the two study arms, given that the most common grade III–IV drug-related adverse events had similar incidence both in experimental and in control arm (neutropenia in 61% versus 62%, febrile neutropenia in 15% versus 16%, anemia in 12% versus 5% and thrombocytopenia in 5% versus 6%, respectively). Serious cardiac events were more common in the CHOP-R arm, and included clinical congestive heart failure, reductions of more than 15% in LVEF from baseline, and elevations in serum troponin T.
Ongoing trials
At the time of publication of this paper, two relevant trials with ongoing enrollment are registered on the ClinicalTrials.gov website, both concerning the treatment of NHL. The first study investigates relapsed diffuse large B-cell lymphoma, transformed from indolent lymphoma, and follicular grade III lymphoma not eligible for stem cell transplantation. This Phase III, multicenter, randomized trial is comparing a pixantrone-rituximab regimen with a gemcitabine-rituximab regimen, with a planned sample size of 350 patients. The second study is a Phase I/II trial enrolling patients with relapsed/refractory B-cell NHL in whom a combination of pixantrone, bendamustine, and rituximab will be investigated, with a planned accrual of 36 patients. No data are available for these studies at present, but preliminary data are expected in August, 2015.
Conclusion
Anthracyclines are still considered to be the cornerstone of treatment for the majority of patients with aggressive B-cell NHL. Unfortunately, the cumulative dose-related cardiac toxicity of these drugs often limits their use in previously treated patients. For this reason, the development of an analog with reduced cardiotoxicity would respond to a relevant medical need. Pixantrone has resulted in a better or similar outcome in terms of response rate and survival when compared with single-agent and combination chemotherapeutic regimens. Obviously, given the period during which these studies were performed, no prospective data are available for comparison with biological agents such as lenalidomide, bortezomib, or ibrutinib, which were recently introduced for the treatment of relapsed/refractory NHL. Probably the most interesting setting in which pixantrone could be useful is the treatment of chemosensitive relapses. As previously reported, prior treatment with rituximab or more than three prior lines of chemotherapy seem to reduce the efficacy of pixantrone; this observation is also common for other agents in several studies, such as rituximab in the Collaborative Trial in Relapsed Aggressive Lymphoma,4 and in the case of pixantrone, the power of these observations may be at least partially reduced by the relatively small series investigated. Further studies may be useful to define better the ideal subset of patients and the best time point at which this drug should be administered. Moreover, some studies have reported significant differences in terms of response, depending on the diverse histotypes of NHL enrolled. Accurate histologic selection would be advisable in upcoming studies, including gene expression profiling or immunohistochemical discrimination of diffuse large B-cell lymphoma in germinal center B cell-like and activated B-cell. Finally, a biological study analyzing the potential of pixantrone to overcome the multidrug resistance typically seen with anthracycline could be interesting, considering the relapse/refractory setting in which this drug is actually used.
On the other hand, pixantrone appears to be a safe drug. Its hematologic toxicity is mainly represented by neutropenia, with anemia and thrombocytopenia usually mild to moderate. Moreover, the incidence of severe systemic infection is low, even if the overall risk of infectious complications is relatively high. The susceptibility to infection is at least in part mediated by the deficient immune competence typical of a relapse/refractory patient. One of the principal benefits of the chemical conformation of pixantrone, in which the 5,8-dihydroxy substitution has been removed, is a reduction in treatment-related cardiac toxicity. For this reason, cardiotoxicity was closely monitored in all of the studies reported thus far. In one Phase III trial, in which pixantrone was administered as a single drug, a higher incidence of cardiac events was seen in the experimental arm, even if these events were apparently less severe than those reported with anthracenediones such as mitoxantrone and anthracyclines such as doxorubicin. This observation was not confirmed in the other Phase III trial where pixantrone was substituted for doxorubicin in a first-line CHOP-like regimen, ie, a substantial benefit in the cardiotoxicity profile was observed in the cohort of patients treated with the experimental combination. Moreover, no relationship between cumulative pixantrone dose and symptomatic decrease in LVEF or congestive heart failure was observed. The studies cited in this review have reported cardiotoxicity rates ranging from 7% to 19%; however, grade III–IV toxicities were limited (2%–3%) and usually manageable. Notably, almost all of the patients included had previously received a median cumulative doxorubicin-equivalent dose ranging from 292.9 mg/m2 to 315 mg/m2, below the limit of 400–500 mg/m2 conventionally considered the safety limit to prevent anthracycline-related cardiotoxicity,34 even if several studies have shown a linear correlation between doses higher than 200 mg/m2 and cardiac damage.35,36 Further, in recent years, many studies have investigated combinatorial strategies, including immunotherapy and chemotherapy, and shown the efficacy of this approach; however, side effects such as cardiac and immunosuppressive toxicities may limit the use of these combinations.37,38 Pixantrone, with its activity and favorable safety profile, could represent a promising agent for novel combination regimens aiming to improve progression-free survival. In conclusion, the availability of an effective and apparently less toxic compound such as pixantrone may represent a therapeutic option in the treatment of relapsed/refractory NHL.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Velasquez WS, Cabanillas F, Salvador P, et al. Effective salvage therapy for lymphoma with cisplatin in combination with high-dose Ara-C and dexamethasone (DHAP) Blood. 1988;71:117–122. [PubMed] [Google Scholar]
- 2.Witzig TE, Geyer SM, Kurtin PJ, et al. Salvage chemotherapy with rituximab DHAP for relapsed non-Hodgkin lymphoma: a phase II trial in the North Central Cancer Treatment Group. Leuk Lymphoma. 2008;49:1074–1080. doi: 10.1080/10428190801993470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moskowitz CH, Bertino JR, Glassman JR, et al. Ifosfamide, carboplatin, and etoposide: a highly effective cytoreduction and peripheral-blood progenitor-cell mobilization regimen for transplant-eligible patients with non-Hodgkin’s lymphoma. J Clin Oncol. 1999;17:3776–3785. doi: 10.1200/JCO.1999.17.12.3776. [DOI] [PubMed] [Google Scholar]
- 4.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–4190. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velasquez WS, McLaughlin P, Tucker S, et al. ESHAP: an effective chemotherapy regimen in refractory and relapsing lymphoma: a 4-year follow-up study. J Clin Oncol. 1994;12:1169–1176. doi: 10.1200/JCO.1994.12.6.1169. [DOI] [PubMed] [Google Scholar]
- 6.Harting R, Venugopal P, Gregory SA, O’Brien T, Bogdanova E. Efficacy and safety of rituximab combined with ESHAP chemotherapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Clin Lymphoma Myeloma. 2007;7:406–412. doi: 10.3816/CLM.2007.n.019. [DOI] [PubMed] [Google Scholar]
- 7.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333:1540–1545. doi: 10.1056/NEJM199512073332305. [DOI] [PubMed] [Google Scholar]
- 8.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 9.De Isabella P, Palumbo M, Sissi C, et al. Topoisomerase II DNA cleavage stimulation, DNA binding activity, cytotoxicity, and physicochemical properties of 2-aza- and 2-aza-oxide-anthracenedione derivatives. Mol Pharmacol. 1995;48:30–38. [PubMed] [Google Scholar]
- 10.Krapcho AP, Maresch MJ, Hacker MP, et al. Aza and diaza bioisosteric anthracene-9,10-diones and aza bioisosteres as antitumor agents. Curr Med Chem. 1995;2:803–824. [PubMed] [Google Scholar]
- 11.Singal KS, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 12.Mordente A, Meucci E, Martorana GE, Giardina B, Minotti G. Human heart cytosolic reductases and anthracycline cardiotoxicity. IUBMB Life. 2001;52:83–88. doi: 10.1080/15216540252774829. [DOI] [PubMed] [Google Scholar]
- 13.Mordente A, Meucci E, Silvestrini A, Martorana GE, Giardina B. New developments in anthracycline-induced cardiotoxicity. Curr Med Chem. 2009;16:1656–1672. doi: 10.2174/092986709788186228. [DOI] [PubMed] [Google Scholar]
- 14.Hazlehurst LA, Krapcho AP, Hacker MP. Comparison of aza-anthracenedione-induced DNA damage and cytotoxicity in experimental tumor cells. Biochem Pharmacol. 1995;50:1087–1094. doi: 10.1016/0006-2952(95)00246-v. [DOI] [PubMed] [Google Scholar]
- 15.Hazlehurst LA, Krapcho AP, Hacker MP. Correlation of DNA reactivity and cytotoxicity of a new class of anticancer agents: aza-anthracenediones. Cancer Lett. 1995;91:115–124. doi: 10.1016/0304-3835(95)91035-5. [DOI] [PubMed] [Google Scholar]
- 16.Smith PJ, Morgan SA, Fox ME, Watson JV. Mitoxantrone-DNA binding and the induction of topoisomerase II associated DNA damage in multi-drug resistant small cell lung cancer cells. Biochem Pharmacol. 1990;40:2069–2078. doi: 10.1016/0006-2952(90)90237-f. [DOI] [PubMed] [Google Scholar]
- 17.Zwelling LA, Mayes J, Altschuler E, Satitpunwaycha P, Tritton TR, Hacker MP. Activity of two novel anthracene-9,10-diones against human leukemia cells containing intercalator-sensitive or -resistant forms of topoisomerase II. Biochem Pharmacol. 1993;46:265–271. doi: 10.1016/0006-2952(93)90413-q. [DOI] [PubMed] [Google Scholar]
- 18.Péan E, Flores B, Hudson I, et al. The European Medicines Agency review of pixantrone for the treatment of adult patients with multiply relapsed or refractory aggressive non-Hodgkin’s B-cell lymphomas: summary of the Scientific Assessment of the Committee for Medicinal Products for Human Use. Oncologist. 2013;18:625–633. doi: 10.1634/theoncologist.2013-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borchmann P, Schnell R, Knippertz R, et al. Phase I study of BBR 2778, a new aza-anthracenedione, in advanced or refractory non-Hodgkin’s lymphoma. Ann Oncol. 2001;12:661–667. doi: 10.1023/a:1011139016294. [DOI] [PubMed] [Google Scholar]
- 20.Dawson LK, Jodrell DI, Bowman A, et al. A clinical phase I and pharmacokinetic study of BBR 2778, a novel anthracenedione analogue, administered intravenously, 3 weekly. Eur J Cancer. 2000;36:2353–2359. doi: 10.1016/s0959-8049(00)00342-7. [DOI] [PubMed] [Google Scholar]
- 21.Faivre S, Raymond E, Boige V, et al. A phase I and pharmacokinetic study of the novel aza-anthracenedione compound BBR 2778 in patients with advanced solid malignancies. Clin Cancer Res. 2001;7:43–50. [PubMed] [Google Scholar]
- 22.Von Hoff DD, Degen D. Final Report of Institute for Drug Development. San Antonio: Institute for Drug Development at the Cancer Therapy and Research Center; 1995. Investigation of the effects of BBR2778 on human tumor colony forming units. [Google Scholar]
- 23.Manzotti C, Pezzoni G, Giuliani FG. BBR 2778 (6,9-bis{[(2-amino) ethyl]amino}benzo [g] isoquinoline-5,10-dione dimaleate): in vivo antitumor activity. BM Italy Report. 1994 03/ONC. [Google Scholar]
- 24.Beggiolin G, Crippa L, Menta E, et al. BBR 2778, an aza-anthracenedione endowed with preclinical anticancer activity and lack of delayed cardiotoxicity. Tumori. 2001;87:407–416. doi: 10.1177/030089160108700611. [DOI] [PubMed] [Google Scholar]
- 25.Salvatorelli E, Menna P, Paz OG, et al. The novel anthracenedione, pixantrone, lacks redox activity and inhibits doxorubicinol formation in human myocardium: insight to explain the cardiac safety of pixantrone in doxorubicin-treated patients. J Pharmacol Exp Ther. 2013;344:467–478. doi: 10.1124/jpet.112.200568. [DOI] [PubMed] [Google Scholar]
- 26.Pezzoni G, Beggiolin G, Manzotti C. BBR 2778: a novel aza-analog of anthracenediones endowed with preclinical anticancer activity. Proc Annu Meet Am Assoc Cancer Res. 1993;34:A2226. [Google Scholar]
- 27.Cavalletti E, Crippa L, Melloni E. BBR 2778, a novel aza-anthracenedione: preclinical toxicological evaluation. Proc Annu Meet Am Assoc Cancer Res. 1993;34:A2227. [Google Scholar]
- 28.Borchmann P, Morschhauser F, Parry A, et al. Phase-II study of the new aza-anthracenedione, BBR 2778, in patients with relapsed aggressive non-Hodgkin’s lymphomas. Haematologica. 2003;88:888–894. [PubMed] [Google Scholar]
- 29.Pettengell R, Coiffier B, Narayanan G, et al. Pixantrone dimaleate versus other chemotherapeutic agents as a single-agent salvage treatment in patients with relapsed or refractory aggressive non-Hodgkin lymphoma: a phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2012;13:696–706. doi: 10.1016/S1470-2045(12)70212-7. [DOI] [PubMed] [Google Scholar]
- 30.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 31.Lim ST, Fayad L, Tulpule A, et al. A phase I/II trial of pixantrone (BBR2778), methylprednisolone, cisplatin, and cytosine arabinoside (PSHAP) in relapsed/refractory aggressive non-Hodgkin’s lymphoma. Leuk Lymphoma. 2007;48:374–380. doi: 10.1080/10428190601060496. [DOI] [PubMed] [Google Scholar]
- 32.Borchmann P, Herbrecht R, Wilhelm M, et al. Phase I/II study of pixantrone in combination with cyclophosphamide, vincristine, and prednisone in patients with relapsed aggressive non-Hodgkin lymphoma. Leuk Lymphoma. 2011;52:620–628. doi: 10.3109/10428194.2010.546016. [DOI] [PubMed] [Google Scholar]
- 33.Herbrecht R, Cernohous P, Engert A, et al. Comparison of pixantrone-based regimen (CPOP-R) with doxorubicin-based therapy (CHOP-R) for treatment of diffuse large B-cell lymphoma. Ann Oncol. 2013;24:2618–2623. doi: 10.1093/annonc/mdt289. [DOI] [PubMed] [Google Scholar]
- 34.Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 35.Horenstein MS, Van der Heide RS, L’Ecuyer TJ. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol Genet Metab. 2000;71:436–444. doi: 10.1006/mgme.2000.3043. [DOI] [PubMed] [Google Scholar]
- 36.Limat S, Demesmay K, Voillat L, et al. Early cardiotoxicity of the CHOP regimen in aggressive non-Hodgkin’s lymphoma. Ann Oncol. 2003;14:277–281. doi: 10.1093/annonc/mdg070. [DOI] [PubMed] [Google Scholar]
- 37.Itchaki G, Gafter-Gvili A, Lahav M, et al. Anthracycline-containing regimens for treatment of follicular lymphoma in adults. Cochrane Database Syst Rev. 2013;7:CD008909. doi: 10.1002/14651858.CD008909.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanneman MI, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]