

Abstract
Vibrio cholerae, the microorganism responsible for the diarrheal disease cholera, is able to sense and respond to a variety of changing stimuli in both its aquatic and human gastrointestinal environments. Here we present a review of research efforts aimed toward understanding the signals this organism senses in the human host. V. cholerae’s ability to sense and respond to temperature and pH, bile, osmolarity, oxygen and catabolite levels, nitric oxide, and mucus, as well as the quorum sensing signals produced in response to these factors will be discussed. We also review the known quorum sensing regulatory pathways and discuss their importance with regard to the regulation of virulence and colonization during infection.
Keywords: V. cholerae, mucus, bile, pH, quorum sensing, virulence, colonization, environmental signals
Introduction
Vibrio cholerae, the causative agent of the disease cholera, is spread by fecal contamination of water and food. It has been implicated in disease outbreaks as far back as 460 BC, and through history, there have been a series of pandemics from which countless deaths can be attributed.1V. cholerae has successfully transitioned from the ancient to the modern world by taking advantage of breakdowns in sanitation and health structure, especially after natural disasters and in disadvantaged countries.2 Cholera is an acute diarrheal disease that is characterized by profuse, watery diarrhea, vomiting, and leg cramps. A rapid loss of fluids can lead to dehydration and shock, and without prompt rehydration, death can occur within hours.3 Although cholera can be easily treated with oral rehydration, it still causes an enormous human health burden; globally, there are an estimated 3 to 5 million cases per year and 100 000 to 120 000 deaths.4 Although vaccines are available, efficacy is moderate at best.5
V. cholerae is a gram-negative bacterium that primarily persists in aquatic environments around the world. It is able to survive in freshwater lakes and rivers, a characteristic that increases the likelihood of the bacterium to come into contact with people, its natural host. What makes V. cholerae an interesting pathogen from a biological standpoint is its ability to survive a variety of environments, from persisting in aquatic environments in a viable but non-culturable state, to active colonization of the human gastrointestinal tract. This trait is conferred by the bacterium’s ability to rapidly alter its gene expression profile in response to changing environmental stimuli; this phenomenon remains the subject of continued intensive research.2
Upon human colonization, virulence is mitigated primarily by cholera toxin (CT), which causes cell damage and diarrhea, and the toxin coregulated pilus (TCP), which is important for binding to the intestinal mucosa. Expression of these genes is highly regulated by a variety of factors that are able to respond to environmental signals to coordinately time expression. Factors within the gastrointestinal system, many of which remain unknown, are able to induce genes responsible for colonization, survival, virulence, and subsequent dissipation. This review will focus on current research investigating the use of quorum sensing and virulence gene regulation by V. cholerae in response to the vast array of intestinal signals in the human host.
Pathogenesis and Regulation of Virulence in V. Cholerae
In the transition from the aquatic environment to the human body, V. cholerae cells are exposed to a series of environmental changes (see below). The ability of V. cholerae to colonize and cause disease in hosts requires production of a number of virulence factors during infection. The two major virulence determinants of V. cholerae are encoded by two separate genetic elements. Cholera toxin, which causes the diarrhea characteristic of cholera, is encoded by ctxAB genes on the lysogenic CTXΦ bacteriophage.6 V. cholerae also produces toxin-coregulated pili (TCP), a type IV pilus required for intestinal colonization both in animal models and in human volunteers.7,8 TCP is thought to be a polymer of the main structural subunit, TcpA, and serves as the receptor for the CTXΦ bacteriophage.6,9 The genes required for TCP synthesis, as well as accessory colonization factor genes and the genes encoding the virulence transcriptional activators, are located on a 40-kb Vibrio pathogenicity island (VPI).10 Coordinate expression of V. cholerae virulence genes results from the activity of a cascading system of regulatory factors.11 The primary direct transcriptional activator of V. cholerae virulence genes is ToxT, a member of the AraC family.12 To activate transcription, ToxT recognizes and binds to a degenerate 13-bp DNA sequence, the toxbox.13 The expression of ToxT is under control of a complex regulatory pathway (Fig. 1). The ToxR protein was identified as the first positive regulator of V. cholerae virulence genes; thus the genes involved in the transcriptional cascade resulting in toxT expression and consequent virulence gene activation is sometimes referred to as the “ToxR regulon.”14
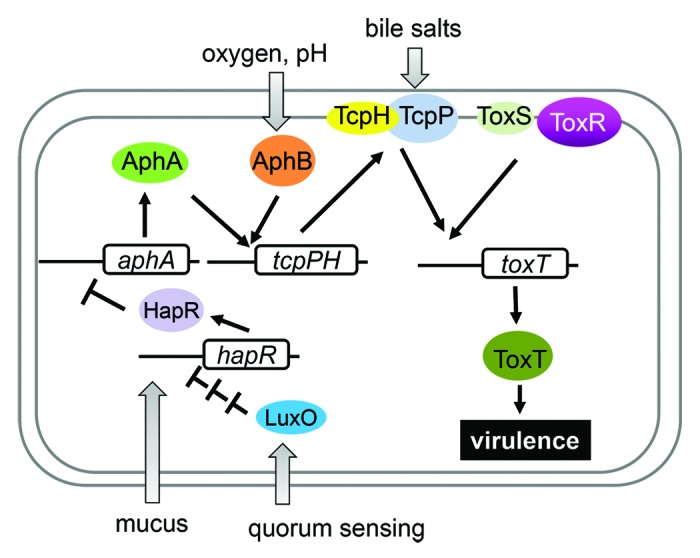
Figure 1. Environmental signals alter V. cholerae virulence. A variety of environmental signals, including pH, temperature, bile, mucus, nitric oxide, and quorum sensing, alter the transcriptional program of V. cholerae, ensuring that virulence factors are produced at the optimal point of the infectious cycle.
ToxR is a membrane protein containing a cytoplasmically localized DNA-binding domain, a transmembrane domain, and a periplasmic domain. ToxR activity requires the presence of another protein, ToxS, which is also localized to the inner membrane.12 To regulate expression of toxT, ToxR must act in conjunction with a second transcriptional activator, TcpP, which is also membrane-localized.15 TcpP, like ToxR, requires the presence of a membrane-bound effector protein, TcpH, which interacts with TcpP.16 TcpP is degraded by a protease in the absence of TcpH and during conditions unfavorable for virulence gene activation.17 ToxR and TcpP bind nearby sites in the toxT promoter, with ToxR binding more tightly than TcpP.18 In addition, two activators encoded by unlinked genes, AphA and AphB, regulate transcription of tcpPH. AphA is a dimer with an N-terminal winged-helix DNA binding domain that is structurally similar to those of the MarR family transcriptional regulators.19 AphA cannot activate transcription of tcpPH alone, but requires interaction with the LysR-type regulator AphB, which binds downstream of the AphA binding site.20 This interaction is thought to stabilize AphB binding to its recognition site and result in activation of the tcpPH promoter.
The Role of Quorum Sensing in V. Cholerae Pathogenesis
Quorum sensing (QS) enables bacterial cells to monitor their population density through the constitutive expression of small molecules called autoinducers (AIs). The QS regulatory circuit (Fig. 2) in V. cholerae is initiated at low cell densities by the membrane associated receptors LuxPQ and CqsS, each of which recognizes a specific AI molecule. CqsS and LuxQ act as kinases to transfer phosphate to LuxU to LuxO, two response regulators. Phosphorylated LuxO, in conjunction with the σ54 subunit of RNA polymerase, serves as a transcription factor for four small RNAs, termed qrr1–4 (Quorum Regulatory RNA). These small RNAs base pair with the 5′ UTR of hapR mRNA to promote its degradation, resulting in repression of HapR, the master transcriptional regulator of QS at high cell density.21 HapR is responsible for the negative regulation of both virulence and biofilm gene expression, as well as induction of the hemagluttinin/protease HapA.22-26 Alternatively, at high cell densities, the AIs bind their cognate sensors, which now act as phosphatases, effectively reversing the flow of phosphate through the regulatory circuit. LuxO becomes deactivated, qrr1–4 expression becomes repressed, and HapR is expressed. HapR regulates a variety of genes involved in protease production, repression of biofilm formation and virulence factor production, in addition to its own expression.22,27-29 The reciprocal expression of hapR and aphA through the quorum sensing circuit is paramount to the coordinate expression of genes necessary for survival at low and high cell densities.30

Figure 2. Quorum sensing circuits in V. cholerae. At low cell density, the membrane proteins CqsS and LuxQ act as kinases to initiate transfer of phosphate through LuxU to LuxO. LuxO-P, with σ54 of RNA polymerase, serve as a transcription factor to produce qrr1–4. Qrr1–4 and Hfq inhibit HapR. At high cell density,21-26 CqsS and LuxO, through the binding of AI molecules, act as phosphatases to reverse the flow of phosphate through the regulatory circuit. LuxO becomes deactivated, qrr1–4 expression becomes repressed, and HapR is expressed.22,27-29
QS enhances survival in response to environmental stresses like nutrient limitation, oxidative stress, salinity and temperature; this response is mediated at least in part by expression of the RNA polymerase sigma factor RpoS.In particular, RpoS is known to be important for V. cholerae resistance to oxidative and nutrient stresses.31,32 In the gut, QS is important for establishing colonization of both commensal and pathogenic bacteria, and may be responsible for specific reactions to other stimuli like pH and bile. The ability to sense bacterial cell numbers allows the population to coordinate or time the release of toxins or enzymes necessary for survival of the group. It may also allow bacteria to form and disperse from colonies when necessary. Furthermore, QS is thought to aid in colonization and pathogenesis in the gastrointestinal tract by facilitating interspecies communication or by modulating host responses to favor survival.33
Gastrointestinal Factors Alter Gene Expression
In its transition from an aquatic environment to the human gastrointestinal tract, V. cholerae must sense and respond to a variety of unique environmental signals, some of which include bile, differences in osmolarity, oxygen availability, changes in pH as it travels from the stomach to the intestines, and the innate immune system. An abundance of research has demonstrated that this bacterium has evolved to rapidly respond to these signals, both individually and in coordinate groups, to colonize, proliferate, and escape back to its natural environment. Research on the response of V. cholerae to these signals has been ongoing, and we are only now beginning to elucidate the intricate cellular responses of the bacterium to these signals and the role they play in the survival and proliferation of V. cholerae in the gut.
pH and Temperature
Upon ingestion, V. cholerae first encounters the extremely acidic pH of the stomach in addition to a shift in temperature from ambient temperature to 37 °C. As briefly mentioned earlier, temperature and pH have been identified as factors that affect gene expression in order to enhance survival and promote the production of virulence factors. One example is the protein PepA, which has been identified as a negative transcriptional regulator of virulence determinants; disruption of the pepA gene causes elevated expression of CT, tcpA, toxT, and tcpP specifically in response to variations in pH and temperature.34 Although the exact function of PepA is yet to be elucidated, it is known to be an aminopeptidase that is orthologous to PepA in E. coli (which is important for the generation of amino acids for cell metabolism). E. coli PepA has also been shown to be involved in Xer site-specific recombination.35,36 The exact mechanism by which pH or temperature affects PepA activity is unknown, but the DNA binding activity of this protein suggests that it functions as a direct regulator of transcription.34 PepA dependent ToxT expression activates TcpI and AcfB, two methyl-accepting chemoreceptors that contribute to colonization by promoting chemotaxis toward the correct intestinal location.37 tcpPH activation by AphA-AphB has been linked to variations in pH and temperature as well. Expression is strongly repressed at high pH and to a lesser extent by temperature, which results in downregulation of the entire virulence operon.38,39 Kovacikova’s 2010 study also identified five other operons that were responsive to pH under aerobic growth conditions and anaerobiosis at neutral pH.40 The implications of this finding with respect to V. cholerae colonization are clear; the organism first encounters low pH in the stomach after ingestion, followed by a neutralization of the pH and a further reduction in oxygen availability upon entering the small intestine. Thus, the ability to sense these environmental signals not only facilitates survival early in colonization, but also serves as an indicator for the correct timing of virulence gene expression.
AphB controls the induction of many genes, and can respond to both pH when grown aerobically, and anaerobiosis at neutral pH.40,41 In fact, the crystal structure of AphB has revealed that specific residues in the ligand-binding pocket of this protein contribute to its ability to respond to these signals.40,41 Interestingly, AphB is known to induce the expression of a number of genes involved in pH homeostasis, which are undoubtedly important for the survival of the organism as it passes from the stomach into the small intestine. For example, AphB induces the expression of cadC and a number of CIC family integral membrane proteins, which enable the organism to respond and adapt to acid stress.40,42-44
The capacity of V. cholerae for survival in the acid environment of the stomach is also facilitated by the protective nature of biofilms since biofilm-associated cells are over 1000 times more resistant to acid shock than planktonic cells.24 V. cholerae is an efficient biofilm producer and can change from a smooth (non-biofilm forming) to rugose (biofilm-forming) phenotype in response to cyclic di-guanylic acid, or c-di-GMP.45,46 Recent reports have demonstrated that V. cholerae regulates biofilm formation by monitoring c-di-GMP levels together with QS signals.47,48 Repression of HapR, the master QS regulator at high cell density, causes an increase in the expression of VPS (Vibrio polysaccharide synthesis) genes and biofilm.49 Our lab has added to this body of evidence by showing that the timing of HapR expression is critical in controlling biofilm formation; we have found that late expression of HapR results in the formation of thick biofilms that makes detachment difficult, resulting in a greater than 10-fold decrease in colonization efficiency in infant mice.50 We have also shown that AphA can directly stimulate biofilm formation by binding to vps genes and serving as a vital connection between virulence factor production and biofilm formation to promote colonization and resist antimicrobial agents in the host.51 ToxR also seems to play a role in the regulation of biofilm formation, however, the exact mechanism remains undiscovered.52
Another interesting observation is that V. cholerae cells grown in biofilms are hyperinfective and have enhanced colonization efficiency compared with planktonic cells, two facts that further strengthen the link between biofilm formation and pathogenesis.53 A comparison of V. cholerae grown in biofilms vs. planktonic cell culture revealed the upregulation of several proteins, including a phosphate uptake system. The authors of this work demonstrated that the Pst2 phosphate uptake system contributes to host colonization. This finding allowed them to propose a model in which V. cholerae uses the Pst2 system to generate phosphate stores to either (1) enhance fitness after dispersal, or (2) to prepare the bacterium to take up phosphate shortly after dispersal from the biofilm since phosphate may be scarce in the host.54 These works have clearly demonstrated the fact that biofilms play an important role in the colonization and dissemination of V. cholerae in the host, however, additional research is needed to gain a better understanding of their role in resistance to the immune system and how they might promote adherence in the gastrointestinal tract.
Bile
Once V. cholerae has entered the small intestine, a number of new factors come into play that may affect survival and colonization. One major factor is the presence of a high concentration of bile, a digestive secretion that is primarily involved in the emulsification and solubilization of lipids. The detergent-like effects of bile, which is composed of bile acids, cholesterol, phospholipids, and IgA, can destabilize membranes and disrupt cellular homeostasis, as well as to prevent bacterial growth and adherence.55 The ability to sense and respond to bile provides such a serious advantage to pathogenic bacteria that many organisms have evolved resistance mechanisms to respond to this important intestinal signal.
Survival of enteric bacteria in the presence of bile is generally mediated through the use of efflux pumps and by altering the fluidity of the outer membrane.56 Bile may also serve as a spatiotemporal cue, advising the organism that they are located in the intestinal lumen. As bile components enter the cell, they begin to initiate different responses, from increasing motility to decreasing virulence factor production.57 In V. cholerae, transcriptional activation of CT and TCP by ToxT was first reported to be negatively regulated in response to crude bile and temperature, presumably to facilitate movement toward the intestinal surface where bile concentrations are predicted to be low.58 However, Hung and Mekalanos later reported that purified bile acids actually activate CT in a ToxT-independent manner. They went on to propose that while crude bile may inhibit the ToxT branch of virulence factor expression, individual bile acids activate an alternative regulatory branch that is instead modulated by the activity of ToxRS.59 Furthermore, another report showed that the unsaturated fatty acid component of bile negatively regulates expression of ctxAB and tcpA in an H-NS-dependent manner. Fatty acids and cholesterol also both seem to promote motility by activating flrA (the transcriptional activator of the motility regulatory cascade) expression.60 A recent report by our lab has also demonstrated that host-derived factors, one of which was identified as the bile salt taurocholate, can directly stimulate virulence gene expression by inducing dimerization of TcpP through intermolecular disulfide bond formation.61 Lastly, bile acids also stimulate biofilm formation, which may help the bacterium survive some of the antibiotic or emulsifying effects of bile.62
Secretory IgA (S-IgA) is an important antibacterial component of bile that facilitates bacterial clearance through agglutination. Thus, the ability to evade the effects of S-IgA would provide an advantage to any organism colonizing the intestines. Experimental evidence from our lab has demonstrated that V. cholerae has indeed developed mechanisms for evasion of S-IgA, primarily through the repression of a type IV mannose-sensitive hemagglutinin (MSHA) pilus. Repression of this structure, which is regulated by ToxT, completely prevents binding by S-IgA and allows the bacterium to efficiently colonize the intestinal epithelium. Conversely, bacteria that are unable to repress MSHA are dramatically impaired in their ability to colonize the host. We believe that this response is critical in the early stages of infection.63
Anaerobiosis
The oxygen content of the intestinal microenvironment is also important for efficient colonization. Anaerobiosis is a stress V. cholerae encounters in the upper intestine (i.e., early in infection) and transcriptional patterns of in vivo grown bacteria have confirmed the expression of metabolic genes responsible for anaerobic energy metabolism.64 It has long been known that V. cholerae is a facultative anaerobe, so it can efficiently grow in low oxygen, however it seems to also use this information as a signal to stimulate both virulence and colonization factor production. When grown in low oxygen conditions, the transcriptional regulators ToxR, TcpP, and ToxT are produced, TcpA is synthesized, and ctxAB is repressed, with H-NS being responsible for the repression.65 These findings corroborated an earlier study that demonstrated the spatiotemporal differences in gene expression during infection, specifically that tcpA expression was detected throughout infection, while ctxA expression was only detected late in infection.66 A new report also links the production of CT to anaerobic respiration of trimethylamine N-oxide (TMAO); when infant mice infected with V. cholerae were given TMAO, they exhibited more severe pathogenic symptoms.67
Anaerobiosis has also been shown to induce the production of the colonization factors TcpC, TcpQ, TcpS, TcpF, AcfA, and TagD, further suggesting that low oxygen conditions are an early signal for colonization in vivo. This same study also identified the production of biofilms during microaerobiosis, implying that oxygen availability may also be a signal for exopolysaccharide and biofilm production in vivo. The authors hypothesize that the microaerobic environment experienced by V. cholerae in the stomach and proximal intestine would favor biofilm formation to enhance survival in the presence of stomach acid and bile, while expression of colonization factors would be favored in the ileum where oxygen concentrations are at their lowest.68
AphB, the key regulator of TcpP, has also been shown to influence the expression of a large subset of genes in response to anaerobic environmental conditions. Some of these genes include a Na+/H+ antiporter, a carbonic anhydrase, and a member of the CIC family of chloride channels.40 Indeed, we have been able to corroborate these results by demonstrating that anaerobiosis enhances dimerization and activity of AphB; specifically, one key cysteine residue is required for sensing oxygen concentration. Thus, under low oxygen conditions, like those encountered in the body during colonization, AphB becomes reduced, which promotes dimerization and subsequently makes AphB capable of activating virulence gene expression. We believe that this thiol-based regulatory switch facilitates survival in both its aquatic (aerobic) and intestinal (anaerobic) environments.69
Osmolarity
V. cholerae experiences variable levels of salinity and osmolarity as it leaves its natural aquatic habitat and enters the gastrointestinal system. In fact, the osmolarity of the intestinal lumen is quite high due to the presence of bile and other sodium salts.57 The primary means of response to this environmental signal is believed to be mediated by ToxRS, which is known to be stimulated by changes in osmolarity to induce expression of TcpA, OmpT, and OmpU.70,71 Furthermore, mutations in tcpR (a gene in the same operon as the pilus-colonizing factor TCP) results in a loss of tolerance to high osmolarity, defects in colonization, and attenuation of virulence, suggesting that this gene may be at least partially responsible for virulence in vivo.72
A more recent study looking for changes in the transcriptional profile of bacteria grown in various NaCl concentrations revealed that large classes of genes (including those involved in pathogenesis, biofilm formation, exopolysaccharide synthesis, and solute transport) are differentially regulated in response to salt. A novel transcriptional regulator, OscR, whose expression is downregulated in response to increasing NaCl levels, was also identified in this study. OscR was shown to negatively regulate vps genes under low-salinity conditions in parallel with HapR. This evidence suggests that motility and biofilm formation are inversely regulated in an osmolarity dependent manner.73 Additionally, TolC, which is important for bile resistance and colonization, modulates TcpPH protein activity through ToxT in a way that is protective against the negative effects of high osmolarity.74,75
V. cholerae is also able to directly sense changes in sodium levels and respond with changes in its transcriptional program. In fact, Hase and Mekalanos observed that in an nqr mutant (which is a respiration-linked Na+ pump believed to be important for generating an electrochemical gradient of sodium ions across the membrane), virulence gene expression (using a toxT::lacZ reporter) was markedly increased. The authors believe that the nqr deletion prevents removal of excess Na+ from the cell. Thus, virulence gene expression is intimately linked to extracellular Na+ levels, a factor present in both the intestinal and aquatic environments where V. cholerae thrives. The authors further hypothesize that since the activity of CT in the intestines increases electrolyte levels, which may favor growth in the gut, the increased NaCl levels may serve as a negative feedback loop for CT production.76
Mucus
The epithelial surface of the colon is covered by a viscoelastic mucus gel that is extremely important for protecting the host against adhesion and invasion by microorganisms, deleterious effects of bacterial toxins, and other antigens. Some microorganisms, like V. cholerae, can invade the mucosal layer and even use mucin, a component of mucus, as a nutrient or energy source. Sialic acids, which are conjugated to mucin oligosaccharides to prevent digestion of mucus by the host, can also be used by microorganisms as an energy source or as a source of bacterial mimicry to evade the host immune system.77,78 In fact, experimental evidence suggests that deletion of genes used for metabolism of sialic acids in V. cholerae significantly reduces its capacity for colonization. The authors of this work believe that the ability to metabolize sialic acid provides a growth advantage for V. cholerae in the intestines, and may even help “jump-start” infection, especially in the intestine where competition for resources is high.79
Furthermore, V. cholerae has been shown to bind to mucin, and binding is mediated at least in part by a protein called GbpA. Interestingly, mucin stimulates increased production of GbpA in V. cholerae, and the subsequent increased secretion of GbpA leads to increased mucus production by the host. The authors of this study propose a model in which initial binding of V. cholerae to intestinal mucin by GbpA increases intestinal mucus production, which in turn attracts more bacteria to the area to promote colonization. Simultaneously, the host response enhances binding of the bacteria to the intestinal surface to perpetuate infection. This work is a good example of the interplay between host factors and the response by V. cholerae in promoting colonization and infection.80
Another interesting aspect of mucus, with respect to its role as an environmental factor, is the inherent increased viscosity that the bacterium experiences as it swims. V. cholerae seems to be able to sense these increases in viscosity, which subsequently leads to a dramatic increase in virulence factor gene expression (specifically ToxT). Hase and Mekalanos hypothesized that this sensing might be due to changes in drag experienced by the flagellum as it swims through mucus, as is the case in V. parahemolyticus, or possibly through a mechanosensitive ion channel.76 A follow-up study by Hase reinforced the link between environmental viscosity levels and virulence gene expression, but was unable to identify the sensor. It is hypothesized that it is likely an as yet unidentified mechanosensitive ion channel.81 Work in our lab has further reinforced the importance of flagellum and mucus in the intestinal mucosa. Our research has revealed that the vast majority of bacteria lose their flagella as they move through mucus, and this breakage is a signal for the bacterium that they have reached the site of colonization and that expression of virulence genes should be initiated. Our work has also shown that the act of swimming through mucus causes secretion of FlgM (an anti-σ28 factor), which in turn leads to derepression of the σ28 factor fliA and subsequent repression of hapR (through the activation of fliA).82 This process ensures that HapR is repressed at the time of colonization so that optimal expression of virulence factors may commence at a critical time in the infectious cycle.83
Finally, it is important to discuss the role of mucus in bacterial escape from the host. In order for V. cholerae to be an effective pathogen, it needs to coordinately repress virulence gene expression and initiate motility to escape from the host before being killed by host adaptive immunity. This phenomenon is called the “mucosal escape response” and is driven by the alternative sigma factor RpoS. At approximately 12 h post-inoculation, there is a transition from rapidly replicating surface attached bacteria to bacteria in a state of stasis within the fluid filled intestinal lumen. This environmental change, from highly viscous mucus to thinner, more watery mucus, occurs upon damage to the intestinal lumen resulting from the expression of CT by the large number of V. cholerae now present in the intestines. Genetically, there is a transition to a metabolic state which favors the production of chemotaxis and motility genes, while at the same time repressing production of CT and TCP. RpoS is responsible for the inhibition of luxO, which together with QS-mediated dephosphorylation of LuxO-P, relieves repression of hapR. HapR negatively regulates virulence gene expression and promotes expression of chemotaxis and flagellar genes.84 Rapid repression of virulence gene expression is also mediated through proteolysis of both ToxT and TcpP.17,85 Although the exact environmental signal that initiates RpoS activity remains unknown, it is interesting to imagine a model in which the reduced viscosity of mucus, caused by the CT dependent release of fluids into the intestinal lumen, serves as an environmental signal for the bacterium to alter its genetic program toward survival outside the host.
Nitric oxide
Nitric oxide (NO) is a toxic radical produced by the host during infection that disrupts the function of bacterial proteins containing cysteine residues, as well as enzymes involved in catalyzing iron-dependent reactions or the electron transport chain. Epithelial cells are known to produce NO, and patients infected by V. cholerae have increased NO metabolite levels in their serum and urine, suggesting that V. cholerae likely encounters NO during infection. Therefore, one would expect that the microorganism has evolved mechanisms for sensing and responding to this stress in the host. Indeed, like many host-associated bacteria, V. cholerae encodes an NO sensor in its genome, called NorR, as well as the detoxifying enzyme HmpA. Research in our lab has confirmed the importance of these genes for infection by demonstrating that hmpA and norR deletion mutants are unable to sustain long-term colonization in mice.86 Davies et. al. have also shown that reactive nitrogen species, which are known to cause DNA damage, cause the expression of a variety of genes responsible for detoxifying this stress and repairing damage to DNA. V. cholerae cells deficient in DNA repair or reactive nitrogen species defense are unable to colonize infant mice, in addition to gaining increasing numbers of potentially deleterious mutations.87
Catabolite sensing
The cAMP-CRP system is a global regulator of catabolite repression in enteric bacteria, allowing them to sense the levels of carbon and energy sources in their environment. Intracellular cAMP levels are high in nutrient limiting environments and low in nutrient rich environments, including the gastrointestinal tract. cAMP is also responsible for the dramatic efflux of ions and water indicative of cholera disease; the ADP-ribosylating action of cholera toxin on the G protein Gsα continually stimulates adenylate cyclase to produce cAMP, which activates the CFTR receptor on epithelial cells. It is no surprise then that cAMP-CRP regulation is also used by V. cholerae to coordinately time expression of virulence factors. Early scientific evidence demonstrated that mutations in crp, for example, increased the production of both cholera toxin and TCP under growth conditions that are normally not permissive for their expression, suggesting that cAMP-CRP may be involved in the negative regulation of tcpA and ctx. In fact, both classical and El Tor biotypes have putative CRP binding sites overlapping the -35 site of tcpA.88 Follow-up studies revealed that in addition to tcpA, negative regulation by cAMP-CRP also seem to be involved in the bifurcated control of virulence genes including ctxA, and toxT, such that a heterogeneous population of V. cholerae exists during infection. This model is feasible since intracellular cAMP-CRP levels would be high early in infection when nutrient levels are still quite low.89
cAMP-CRP also negatively regulates the tcpPH promoter by competing for binding at an upstream consensus which overlaps the AphA/B binding sites.90 Carbon catabolite levels can also influence cAMP-CRP mediated expression of other factors, like Hap protease, which is important for detachment of adherent bacteria in vivo, and Hcp, a component of the type VI secretion system.91,92 cAMP has also been recognized as a quorum sensing modulator. cAMP-CRP negatively regulates cqsA at the post-transcriptional level, thereby preventing the synthesis of autoinducers and preventing activation of HapR (since HapR is induced by the accumulation of autoinducers in the local environment).93 In addition, microarray analyses revealed that CRP is critical for regulation of biofilm production, CT expression, motility and chemotaxis genes (including flaC, flaA, flaD, flgL, cheV, flaB, and fliN) and genes important for intestinal colonization (ompT, ompW, and ompU).94 These works demonstrate how cAMP-CRP-mediated negative gene regulation is critical for many aspects of V. cholerae survival, proliferation, motility, colonization, and virulence during infection.
Conclusion
V. cholerae is a clinically important pathogen, and while research into the clinical aspects of its pathogenesis is interesting and important, an equally interesting and important research question relates to its ability to thrive in two very unique environments: the human gastrointestinal tract and aquatic reservoirs. This fascinating microorganism has evolved to respond to an ever changing environmental milieu, and in doing so has become one of the world’s most important microbial pathogens. The works presented here are demonstrative of the fact that V. cholerae is able to integrate many different environmental signals into its colonization and virulence programs, thereby allowing the microorganism to coordinately time expression and repression of genes at each stage in the infectious cycle. However, our understanding of the complex interplay between V. cholerae and its host is still in its infancy because of a lack of ideal animal models to study V. cholerae infection. For example, no animal models are known to have a gut microbiota that matches the complexity of that present in humans; thus what and how commensal bacteria and host signals integrate to control intricately regulated V. cholerae gene expression after bacteria enter the small intestine and during colonization is still largely unknown. More work is needed to understand the complexities of how this organism is able to identify and respond to such a vast array of environmental signals. Characterizing bacterial gene regulation in the host environment provides further understanding of the basic biology of pathogenic microbes and promises to pave the way for developing virulence-targeted drugs and vaccines.
Acknowledgments
The work on V. cholerae pathogenesis study in our laboratory is supported by NIH/NIAID R01 AI080654, R01 AI072479, and R21 AI 088172.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- 1.Global epidemics and impact of cholera [Internet]. World Health Organization; c2013 [cited 2013 March 19]. Available from: http://www.who.int/topics/cholera/impact/en/index.html
- 2.Morris JG., Jr. Cholera--modern pandemic disease of ancient lineage. Emerg Infect Dis. 2011;17:2099–104. doi: 10.3201/eid1711.111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Vibrio cholerae general information. 2-24-2011.
- 4.Ali M, Lopez AL, You YA, Kim YE, Sah B, Maskery B, Clemens J. The global burden of cholera. Bull World Health Organ. 2012;90:209–218A. doi: 10.2471/BLT.11.093427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Cholera vaccines: WHO position paper.3-26-2010;85:117-128. 3-25-2013.
- 6.Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–4. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 7.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168:1487–92. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller VL, Taylor RK, Mekalanos JJ. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell. 1987;48:271–9. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 9.Shaw CE, Taylor RK. Vibrio cholerae O395 tcpA pilin gene sequence and comparison of predicted protein structural features to those of type 4 pilins. Infect Immun. 1990;58:3042–9. doi: 10.1128/iai.58.9.3042-3049.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, 2nd, Peterson KM. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166:175–6. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 11.DiRita VJ. Co-ordinate expression of virulence genes by ToxR in Vibrio cholerae. Mol Microbiol. 1992;6:451–8. doi: 10.1111/j.1365-2958.1992.tb01489.x. [DOI] [PubMed] [Google Scholar]
- 12.DiRita VJ, Mekalanos JJ. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell. 1991;64:29–37. doi: 10.1016/0092-8674(91)90206-E. [DOI] [PubMed] [Google Scholar]
- 13.Matson JS, Withey JH, DiRita VJ. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun. 2007;75:5542–9. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skorupski K, Taylor RK. Control of the ToxR virulence regulon in Vibrio cholerae by environmental stimuli. Mol Microbiol. 1997;25:1003–9. doi: 10.1046/j.1365-2958.1997.5481909.x. [DOI] [PubMed] [Google Scholar]
- 15.Häse CC, Mekalanos JJ. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A. 1998;95:730–4. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck NA, Krukonis ES, DiRita VJ. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J Bacteriol. 2004;186:8309–16. doi: 10.1128/JB.186.24.8309-8316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matson JS, DiRita VJ. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc Natl Acad Sci U S A. 2005;102:16403–8. doi: 10.1073/pnas.0505818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krukonis ES, Yu RR, Dirita VJ. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol. 2000;38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 19.De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. Crystal structure of the virulence gene activator AphA from Vibrio cholerae reveals it is a novel member of the winged helix transcription factor superfamily. J Biol Chem. 2005;280:13779–83. doi: 10.1074/jbc.M413781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovacikova G, Lin W, Skorupski K. Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol Microbiol. 2004;53:129–42. doi: 10.1111/j.1365-2958.2004.04121.x. [DOI] [PubMed] [Google Scholar]
- 21.Svenningsen SL, Waters CM, Bassler BL. A negative feedback loop involving small RNAs accelerates Vibrio cholerae’s transition out of quorum-sensing mode. Genes Dev. 2008;22:226–38. doi: 10.1101/gad.1629908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jobling MG, Holmes RK. Characterization of hapR, a positive regulator of the Vibrio cholerae HA/protease gene hap, and its identification as a functional homologue of the Vibrio harveyi luxR gene. Mol Microbiol. 1997;26:1023–34. doi: 10.1046/j.1365-2958.1997.6402011.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A. 2002;99:3129–34. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell. 2003;5:647–56. doi: 10.1016/S1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]
- 25.Hammer BK, Bassler BL. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol. 2003;50:101–4. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 26.Kovacikova G, Skorupski K. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol Microbiol. 2002;46:1135–47. doi: 10.1046/j.1365-2958.2002.03229.x. [DOI] [PubMed] [Google Scholar]
- 27.Tsou AM, Liu Z, Cai T, Zhu J. The VarS/VarA two-component system modulates the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiology. 2011;157:1620–8. doi: 10.1099/mic.0.046235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell. 2002;110:303–14. doi: 10.1016/S0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 29.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Rutherford ST, van Kessel JC, Shao Y, Bassler BL. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 2011;25:397–408. doi: 10.1101/gad.2015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joelsson A, Liu Z, Zhu J. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect Immun. 2006;74:1141–7. doi: 10.1128/IAI.74.2.1141-1147.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joelsson A, Kan B, Zhu J. Quorum sensing enhances the stress response in Vibrio cholerae. Appl Environ Microbiol. 2007;73:3742–6. doi: 10.1128/AEM.02804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swift S, Vaughan E, de Vos W. Quorum sensing within the gut ecosystem. Microb Ecol Health Dis. 2000;2:81–92. [Google Scholar]
- 34.Behari J, Stagon L, Calderwood SB. pepA, a gene mediating pH regulation of virulence genes in Vibrio cholerae. J Bacteriol. 2001;183:178–88. doi: 10.1128/JB.183.1.178-188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reijns M, Lu Y, Leach S, Colloms SD. Mutagenesis of PepA suggests a new model for the Xer/cer synaptic complex. Mol Microbiol. 2005;57:927–41. doi: 10.1111/j.1365-2958.2005.04716.x. [DOI] [PubMed] [Google Scholar]
- 36.Lutfullah G, Azhar N, Amin F, Khan Z, Azim MK, Shouqat K, Noor S, Ali R. Structural bioinformatics of Vibrio cholerae aminopeptidase A (PepA) monomer. Protein Pept Lett. 2009;16:36–45. doi: 10.2174/092986609787049484. [DOI] [PubMed] [Google Scholar]
- 37.Chaparro AP, Ali SK, Klose KE. The ToxT-dependent methyl-accepting chemoreceptors AcfB and TcpI contribute to Vibrio cholerae intestinal colonization. FEMS Microbiol Lett. 2010;302:99–105. doi: 10.1111/j.1574-6968.2009.01835.x. [DOI] [PubMed] [Google Scholar]
- 38.Kovacikova G, Skorupski K. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol. 1999;181:4250–6. doi: 10.1128/jb.181.14.4250-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skorupski K, Taylor RK. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol. 1999;31:763–71. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- 40.Kovacikova G, Lin W, Skorupski K. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J Bacteriol. 2010;192:4181–91. doi: 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor JL, De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol Microbiol. 2012;83:457–70. doi: 10.1111/j.1365-2958.2011.07919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrell DS, Camilli A. Regulation of vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J Bacteriol. 2000;182:5342–50. doi: 10.1128/JB.182.19.5342-5350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding Y, Waldor MK. Deletion of a Vibrio cholerae ClC channel results in acid sensitivity and enhanced intestinal colonization. Infect Immun. 2003;71:4197–200. doi: 10.1128/IAI.71.7.4197-4200.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dutzler R. The ClC family of chloride channels and transporters. Curr Opin Struct Biol. 2006;16:439–46. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 45.Beyhan S, Yildiz FH. Smooth to rugose phase variation in Vibrio cholerae can be mediated by a single nucleotide change that targets c-di-GMP signalling pathway. Mol Microbiol. 2007;63:995–1007. doi: 10.1111/j.1365-2958.2006.05568.x. [DOI] [PubMed] [Google Scholar]
- 46.Beyhan S, Odell LS, Yildiz FH. Identification and characterization of cyclic diguanylate signaling systems controlling rugosity in Vibrio cholerae. J Bacteriol. 2008;190:7392–405. doi: 10.1128/JB.00564-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava D, Harris RC, Waters CM. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J Bacteriol. 2011;193:6331–41. doi: 10.1128/JB.05167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, Koestler BJ, Waters CM, Hammer BK. Post-transcriptional activation of a diguanylate cyclase by quorum sensing small RNAs promotes biofilm formation in Vibrio cholerae. Mol Microbiol. 2013;89:989–1002. doi: 10.1111/mmi.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang W, Silva AJ, Benitez JA. The cyclic AMP receptor protein modulates colonial morphology in Vibrio cholerae. Appl Environ Microbiol. 2007;73:7482–7. doi: 10.1128/AEM.01564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Z, Stirling FR, Zhu J. Temporal quorum-sensing induction regulates Vibrio cholerae biofilm architecture. Infect Immun. 2007;75:122–6. doi: 10.1128/IAI.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang M, Frey EM, Liu Z, Bishar R, Zhu J. The virulence transcriptional activator AphA enhances biofilm formation by Vibrio cholerae by activating expression of the biofilm regulator VpsT. Infect Immun. 2010;78:697–703. doi: 10.1128/IAI.00429-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valeru SP, Wai SN, Saeed A, Sandström G, Abd H. ToxR of Vibrio cholerae affects biofilm, rugosity and survival with Acanthamoeba castellanii. BMC Res Notes. 2012;5:33. doi: 10.1186/1756-0500-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamayo R, Patimalla B, Camilli A. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect Immun. 2010;78:3560–9. doi: 10.1128/IAI.00048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mudrak B, Tamayo R. The Vibrio cholerae Pst2 phosphate transport system is upregulated in biofilms and contributes to biofilm-induced hyperinfectivity. Infect Immun. 2012;80:1794–802. doi: 10.1128/IAI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29:625–51. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Gunn JS. Mechanisms of bacterial resistance and response to bile. Microbes Infect. 2000;2:907–13. doi: 10.1016/S1286-4579(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 57.Gupta S, Chowdhury R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun. 1997;65:1131–4. doi: 10.1128/iai.65.3.1131-1134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuhmacher DA, Klose KE. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J Bacteriol. 1999;181:1508–14. doi: 10.1128/jb.181.5.1508-1514.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hung DT, Mekalanos JJ. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc Natl Acad Sci U S A. 2005;102:3028–33. doi: 10.1073/pnas.0409559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chatterjee A, Dutta PK, Chowdhury R. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun. 2007;75:1946–53. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A. 2013;110:2348–53. doi: 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol Microbiol. 2006;59:193–201. doi: 10.1111/j.1365-2958.2005.04846.x. [DOI] [PubMed] [Google Scholar]
- 63.Hsiao A, Liu Z, Joelsson A, Zhu J. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci U S A. 2006;103:14542–7. doi: 10.1073/pnas.0604650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu Q, Dziejman M, Mekalanos JJ. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc Natl Acad Sci U S A. 2003;100:1286–91. doi: 10.1073/pnas.0337479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnan HH, Ghosh A, Paul K, Chowdhury R. Effect of anaerobiosis on expression of virulence factors in Vibrio cholerae. Infect Immun. 2004;72:3961–7. doi: 10.1128/IAI.72.7.3961-3967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–34. doi: 10.1016/S0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 67.Lee KM, Park Y, Bari W, Yoon MY, Go J, Kim SC, Lee HI, Yoon SS. Activation of cholera toxin production by anaerobic respiration of trimethylamine N-oxide in Vibrio cholerae. J Biol Chem. 2012;287:39742–52. doi: 10.1074/jbc.M112.394932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marrero K, Sánchez A, Rodríguez-Ulloa A, González LJ, Castellanos-Serra L, Paz-Lago D, Campos J, Rodríguez BL, Suzarte E, Ledón T, et al. Anaerobic growth promotes synthesis of colonization factors encoded at the Vibrio pathogenicity island in Vibrio cholerae El Tor. Res Microbiol. 2009;160:48–56. doi: 10.1016/j.resmic.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci U S A. 2011;108:810–5. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peterson KM. Expression of Vibrio cholerae virulence genes in response to environmental signals. Curr Issues Intest Microbiol. 2002;3:29–38. [PubMed] [Google Scholar]
- 71.Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–83. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mishra A, Srivastava R, Pruzzo C, Srivastava BS. Mutation in tcpR gene (Vc0832) of Vibrio cholerae O1 causes loss of tolerance to high osmolarity and affects colonization and virulence in infant mice. J Med Microbiol. 2003;52:933–9. doi: 10.1099/jmm.0.05171-0. [DOI] [PubMed] [Google Scholar]
- 73.Shikuma NJ, Yildiz FH. Identification and characterization of OscR, a transcriptional regulator involved in osmolarity adaptation in Vibrio cholerae. J Bacteriol. 2009;191:4082–96. doi: 10.1128/JB.01540-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bina JE, Mekalanos JJ. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun. 2001;69:4681–5. doi: 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minato Y, Siefken RL, Häse CC. TolC affects virulence gene expression in Vibrio cholerae. J Bacteriol. 2011;193:5850–2. doi: 10.1128/JB.05222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Häse CC, Mekalanos JJ. Effects of changes in membrane sodium flux on virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A. 1999;96:3183–7. doi: 10.1073/pnas.96.6.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corazziari ES. Intestinal mucus barrier in normal and inflamed colon. J Pediatr Gastroenterol Nutr. 2009;48(Suppl 2):S54–5. doi: 10.1097/MPG.0b013e3181a117ea. [DOI] [PubMed] [Google Scholar]
- 78.Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 2004;68:132–53. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Almagro-Moreno S, Boyd EF. Sialic acid catabolism confers a competitive advantage to pathogenic vibrio cholerae in the mouse intestine. Infect Immun. 2009;77:3807–16. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhowmick R, Ghosal A, Das B, Koley H, Saha DR, Ganguly S, Nandy RK, Bhadra RK, Chatterjee NS. Intestinal adherence of Vibrio cholerae involves a coordinated interaction between colonization factor GbpA and mucin. Infect Immun. 2008;76:4968–77. doi: 10.1128/IAI.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Häse CC. Analysis of the role of flagellar activity in virulence gene expression in Vibrio cholerae. Microbiology. 2001;147:831–7. doi: 10.1099/00221287-147-4-831. [DOI] [PubMed] [Google Scholar]
- 82.Liu Z, Miyashiro T, Tsou A, Hsiao A, Goulian M, Zhu J. Mucosal penetration primes Vibrio cholerae for host colonization by repressing quorum sensing. Proc Natl Acad Sci U S A. 2008;105:9769–74. doi: 10.1073/pnas.0802241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsou AM, Frey EM, Hsiao A, Liu Z, Zhu J. Coordinated regulation of virulence by quorum sensing and motility pathways during the initial stages of Vibrio cholerae infection. Commun Integr Biol. 2008;1:42–4. doi: 10.4161/cib.1.1.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nielsen AT, Dolganov NA, Otto G, Miller MC, Wu CY, Schoolnik GK. RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2006;2:e109. doi: 10.1371/journal.ppat.0020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abuaita BH, Withey JH. Termination of Vibrio cholerae virulence gene expression is mediated by proteolysis of the major virulence activator, ToxT. Mol Microbiol. 2011;81:1640–53. doi: 10.1111/j.1365-2958.2011.07798.x. [DOI] [PubMed] [Google Scholar]
- 86.Stern AM, Hay AJ, Liu Z, Desland FA, Zhang J, Zhong Z, Zhu J. The NorR regulon is critical for Vibrio cholerae resistance to nitric oxide and sustained colonization of the intestines. MBio. 2012;3:e00013–12. doi: 10.1128/mBio.00013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davies BW, Bogard RW, Dupes NM, Gerstenfeld TA, Simmons LA, Mekalanos JJ. DNA damage and reactive nitrogen species are barriers to Vibrio cholerae colonization of the infant mouse intestine. PLoS Pathog. 2011;7:e1001295. doi: 10.1371/journal.ppat.1001295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skorupski K, Taylor RK. Cyclic AMP and its receptor protein negatively regulate the coordinate expression of cholera toxin and toxin-coregulated pilus in Vibrio cholerae. Proc Natl Acad Sci U S A. 1997;94:265–70. doi: 10.1073/pnas.94.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog. 2010;6:e1001102. doi: 10.1371/journal.ppat.1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kovacikova G, Skorupski K. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol. 2001;41:393–407. doi: 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- 91.Benitez JA, Silva AJ, Finkelstein RA. Environmental signals controlling production of hemagglutinin/protease in Vibrio cholerae. Infect Immun. 2001;69:6549–53. doi: 10.1128/IAI.69.10.6549-6553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishikawa T, Rompikuntal PK, Lindmark B, Milton DL, Wai SN. Quorum sensing regulation of the two hcp alleles in Vibrio cholerae O1 strains. PLoS One. 2009;4:e6734. doi: 10.1371/journal.pone.0006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang W, Sultan SZ, Silva AJ, Benitez JA. Cyclic AMP post-transcriptionally regulates the biosynthesis of a major bacterial autoinducer to modulate the cell density required to activate quorum sensing. FEBS Lett. 2008;582:3744–50. doi: 10.1016/j.febslet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liang W, Pascual-Montano A, Silva AJ, Benitez JA. The cyclic AMP receptor protein modulates quorum sensing, motility and multiple genes that affect intestinal colonization in Vibrio cholerae. Microbiology. 2007;153:2964–75. doi: 10.1099/mic.0.2007/006668-0. [DOI] [PubMed] [Google Scholar]